

Abstract
In sepsis and systemic inflammation, increased microvascular permeability and consecutive breakdown of microcirculatory flow significantly contribute to organ failure and death. Evidence points to a critical role of cAMP levels in endothelial cells to maintain capillary endothelial barrier properties in acute inflammation. However, approaches to verify this observation in systemic models are rare. Therefore we tested here whether systemic application of the phosphodiesterase-4-inhibitors (PD-4-Is) rolipram or roflumilast to increase endothelial cAMP was effective to attenuate capillary leakage and breakdown of microcirculatory flow in severe lipopolysaccharide (LPS)-induced systemic inflammation in rats. Measurements of cAMP in mesenteric microvessels demonstrated significant LPS-induced loss of cAMP levels which was blocked by application of rolipram. Increased endothelial cAMP by application of either PD-4-I rolipram or roflumilast led to stabilization of endothelial barrier properties as revealed by measurements of extravasated FITC-albumin in postcapillary mesenteric venules. Accordingly, microcirculatory flow in mesenteric venules was significantly increased following PD-4-I treatment and blood gas analyses indicated improved metabolism. Furthermore application of PD-4-I after manifestation of LPS-induced systemic inflammation and capillary leakage therapeutically stabilized endothelial barrier properties as revealed by significantly reduced volume resuscitation for haemodynamic stabilization. Accordingly microcirculation was significantly improved following treatment with PD-4-Is. Our results demonstrate that inflammation-derived loss of endothelial cAMP contributes to capillary leakage which was blocked by systemic PD-4-I treatment. Therefore these data suggest a highly clinically relevant and applicable approach to stabilize capillary leakage in sepsis and systemic inflammation.
Key points
A specific therapy to treat capillary leakage in systemic inflammation and sepsis is not available at present.
Recent studies demonstrated that reduced cAMP levels in endothelial cells contribute to inflammation-induced breakdown of the endothelial barrier.
The present study demonstrates that systemically applied phosphodiesterase-4 inhibitors to increase endothelial cAMP are effective to prevent and to treat capillary leakage followed by improved microcirculation in a rodent model of systemic inflammation.
These data suggest a highly clinically relevant and applicable approach to stabilize capillary leakage in sepsis and systemic inflammation.
Introduction
Despite continuing efforts to improve sepsis therapy, most septic patients develop single or multiple organ failure which results in an unacceptably high mortality rate of up to 70% (Russell, 2006). A typical sign of sepsis and systemic inflammation is the development of progressive subcutaneous and body-cavity oedema, which is caused by breakdown of endothelial barrier functions leading to a massive increase in vascular permeability (Lee & Slutsky, 2010). It is increasingly recognized that microvascular leakage predisposes for microvascular thrombosis, breakdown of microcirculatory flow and organ failure, which are common events preceding death in patients with severe sepsis and systemic inflammation (Cinel & Dellinger, 2007; Lee & Slutsky, 2010). A specific therapy to address this problem is not available at present.
The endothelial barrier is sealed by tight and adherens junctions, both of which are targeted during acute inflammation, which results in the formation of intercellular gaps with consecutive extravasation of fluid (Mehta & Malik, 2006; Vandenbroucke et al. 2008; Spindler et al. 2010). Meanwhile it is well established that cAMP regulates the stability of intercellular junctions by PKA- or EPAC/Rap1-dependent activation of small GTPase Rac1 (Adamson et al. 1998; Patterson et al. 2000; Birukova et al. 2004; Waschke et al. 2004c; Cullere et al. 2005; Birukova et al. 2008; Spindler et al. 2010).
Accordingly, accumulating evidence points to a significant role of the cAMP/Rac1-signalling pathway in acute inflammation. Bacterial cell wall component lipopolysaccharide (LPS), which is known as a key molecule in the onset of septic inflammation (Medzhitov, 2001), and tumour necrosis factor-α (TNF-α) induce endothelial barrier breakdown by dramatically decreasing intracellular cAMP levels (Koga et al. 1995; Seybold et al. 2005; Schlegel et al. 2009; Schlegel & Waschke, 2009a). Consistently, increased cAMP levels in endothelial cells were effective to prevent increased endothelial permeability by inflammatory mediators in vitro and in single postcapillary venules in vivo (Adamson et al. 2003; Schlegel et al. 2009; Schlegel & Waschke, 2009a; Beckers et al. 2010). Similarly, the use of phosphodiesterase inhibitors (PDIs) to increase cAMP in models of lung injury in isolated lungs was effective to significantly reduce pulmonary oedema (Schmidt et al. 2008; Witzenrath et al. 2009).
Because all these data suggest a potential role for cAMP-increasing agents to overcome the problem of capillary leakage in sepsis and acute hyperinflammation, we conducted extensive in vivo studies in adult anaesthetized rats. We induced severe systemic inflammation by intravenous injection of LPS and used this model to test the hypothesis that increased cAMP would stabilize endothelial barrier properties and thereby improve microcirculatory flow leading to a decreased mortality rate. Because phosphodiesterase-4 is the most highly expressed cAMP-hydrolysing phosphodiesterase in ordinary endothelium (Netherton & Maurice, 2005; Lugnier, 2006), we used the phosphodiesterase-4 inhibitors (PD-4-Is) rolipram, which is a common substance used in vitro and in animal experiments, and roflumilast, which is a highly potent selective PD-4-I and newly clinically approved for COPD treatment, to increase endothelial cAMP (Bundschuh et al. 2001). To enable a close comparison to the clinical situation we applied a detailed intensive care set-up including controlled ventilation, haemodynamic monitoring such as blood pressure, heart rate and cardiac output, and took continuous samples for blood gas analyses. In parallel, we continuously monitored mesenteric microcirculatory flow and assessed changes in microvascular permeability by measurements of FITC-albumin extravasation across postcapillary venules.
In this model measurements of cAMP levels in mesenteric microvessels revealed a significant loss of cAMP in LPS-treated animals. A first set of experiments demonstrated that systemic application of PD-4-Is in animals simultaneously treated with LPS was effective to stabilize endothelial barrier properties followed by significantly improved microcirculatory flow, while injection of PD-4-Is did not appear to have direct effects on macrohaemodynamic parameters. In a second step application of PD-4-Is after manifestation of capillary leakage in LPS-treated animals proved to be effective for therapeutic stabilization of endothelial barrier properties and microcirculation.
Taken together, these data may carefully be used as a basis to suggest a clinically applicable approach to stabilize capillary leakage in sepsis and systemic inflammation.
Methods
Ethical approval
After animal care committee approval (laboratory animal care and use committee of the district of Unterfranken, Germany), experiments were performed on male Sprague–Dawley rats (355 ± 34 g BW; Harlan Winkelmann, NM Horst, Netherlands). Animals were kept under conditions that conformed to the National Institutes of Health's Guide for the Care and Use of Laboratory Animals, and were approved by the Regierung of Unterfranken. All rats were maintained on a standard diet and water ad libitum at 12 h day and night cycles. Animals were not fasted prior to the procedure. Anaesthesia of all animals was always performed by a consultant anaesthesiologist who had been specially trained also for rodent anaesthesia.
Test reagents
LPS from Escherichia coli (Sigma, L2630; Deisenhofen, Germany) was admininistered intravenously at 0.5 and 0.25 mg per 100 g body weight (BW) in vivo (Coimbra et al. 2006). PD-4-Is were administered intravenously to increase cAMP levels: (a) rolipram (Sigma-Aldrich, Taufkirchen, Germany), single intravenous (i.v.) bolus of 3 mg kg−1 followed by continuous intravenous administration of 0.04 μg (100 g)−1 h−1; (b) roflumilast (LGM Pharma, FL, USA), single i.v. bolus of 240 μg kg−1 followed by continuous i.v. administration of 4 μg (100 g)−1 h−1 in experimental set-up A (see below) or by repeated boli of 240 μg kg−1 in experimental set-up B. Fluorescein isothiocyanate (FITC)–albumin was applied i.v. at 5 mg (100 g)−1 (Sigma, Schnelldorf, Germany).
Experimental set-up
We established a model of severe LPS-induced systemic inflammation in adult rats to mimic the macro- and microhaemodynamic situation in patients with severe sepsis or systemic inflammation. To achieve this we combined established experimental set-ups to allow continuous measurements of microcirculatory flow and capillary leakage using intravital microscopy as well as the simultaneous assessment of macrohaemodynamic parameters and metabolic state using blood gas analyses (Fig. 1) (Sielenkamper et al. 2001; Wunder et al. 2002; Waschke et al. 2004b; Schlegel et al. 2009; Schick et al. 2010).
Figure 1. Experimental set-ups.
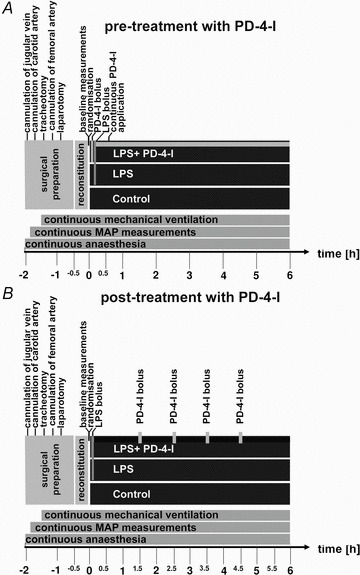
Schematic presentation of the experimental set-ups. A shows the detailed set-up in the experiments where simulataneous treatment of PD-4-Is and LPS was performed and B shows experimental procedures when PD-4-I s were applied 1.5 h after LPS was applied. In both set-ups continuous monitoring of microcirculatory flow in mesenteric postcapillary venules was performed. Measurements of FITC-albumin extravasation were performed at 0, 0.5, 1 and 2 h in set-up A. Cardiac output and blood gas analyses were analysed at 0, 0.5 and 1 h and then each hour in both set-ups. After 6 h of experiments animals were killed by barbiturate overdose. LPS, lipopolysaccharide; PD-4-I, phosphodiesterase-4-inhibitor; MAP, mean arterial pressure.
Animal Preparation for assessment of macrohaemodynamic and metabolic changes
Animals were anaesthetized using isoflurane 1.5–2.5% (v/v) (Forene, Abbott, Wiesbaden, Germany)–nitrous oxide inhalation 50% v/v (Forene, Abbott) (Fig. 1). To apply i.v. medication the right jugular vein was cannulated. Additionally the left carotid artery was cannulated for continuous blood pressure and heart rate measurements (Hewlett-Packard Model 88S, Hamburg, Germany) as well as for taking arterial blood samples for repeated blood gas analyses. For cardiac output (CO) measurements, the right femoral artery was cannulated and a thermocatheter (MLT1402 T-type Ultra Fast Thermocouple, ADInstruments, Spechbach, Germany) was installed. CO was measured by the thermodilution technique using PowerLab software, Chart 5.0 (ADInstruments, Spechbach, Germany).
Tracheotomy was performed and rats were mechanically ventilated with 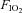 0.28 using a rodent ventilator (Type: 7025, Hugo Sachs Elektronic KG, March-Hugstetten, Germany) in a constant ventilation regime. After tracheotomy the anaesthesia regime was changed using midazolam (Midazolam-ratiopharm, Ratiopharm, Ulm, Germany) 0.7 mg (100 g BW)−1 h−1, Fentanyl 7 μg (100 g BW)−1 h−1 and for a sufficient anaesthetic depth 0.7% (v/v) isoflurane (without N2O) was applied. Animals’ body temperature was always kept at 37°C using a heating plate. Sufficient depth of anaesthesia under the latter conditions had been tested in prelimary experiments and by estimation of heart rates, MAP, leg movement in response to painful stimuli (which was possible because muscle relaxants were not applied) and eye-lid closing reflex.
0.28 using a rodent ventilator (Type: 7025, Hugo Sachs Elektronic KG, March-Hugstetten, Germany) in a constant ventilation regime. After tracheotomy the anaesthesia regime was changed using midazolam (Midazolam-ratiopharm, Ratiopharm, Ulm, Germany) 0.7 mg (100 g BW)−1 h−1, Fentanyl 7 μg (100 g BW)−1 h−1 and for a sufficient anaesthetic depth 0.7% (v/v) isoflurane (without N2O) was applied. Animals’ body temperature was always kept at 37°C using a heating plate. Sufficient depth of anaesthesia under the latter conditions had been tested in prelimary experiments and by estimation of heart rates, MAP, leg movement in response to painful stimuli (which was possible because muscle relaxants were not applied) and eye-lid closing reflex.
Animal preparation for monitoring of the microcirculation and capillary leakage
Thereafter median laparotomy was performed, and the mesentery was gently taken out and spread over a pillar as has been described elsewhere (Sielenkamper et al. 2001; Waschke et al. 2004a). The whole experimental set-up was then carefully placed under a modified inverted Zeiss microscope (Axiovert 200, Carl Zeiss, Göttingen, Germany) equipped with different lenses (Achroplan ×10 NA 0.25/×20 NA0.4/×40 NA 0.6). This allowed continuous observation of microcirculatory flow within postcapillary venules in the mesenteric windows as well as observation of capillary leakage following i.v. injection of FITC-albumin at 5 mg (100 g)−1 as described below. Images and videos were captured using a digital camera, ColorSnap CF (Photometrics, Tucson, USA), driven by Metamorph analysis software (Molecular Devices GmbH, Biberach a.d. Riss, Germany) and digitally recorded for off-line analysis as described below. In the time course of experiments the upper surface of the mesentery was continuously superperfused with 37.5°C crystalloid solution (Sterofundin, B. Braun Melsung AG, Melsung, Germany).
Randomization of animals into experimental groups
After the preparation procedure animals were allowed to recover for 30 min and then samples for the first blood gas analyses were taken using an ABL505 blood gas analyser (Radiometer, Copenhagen, Denmark). Each collected amount of blood was replaced by an equal volume of 0.9% sodium chloride (Fresenius Kabi, Bad Homburg, Germany). The respiratory set-up was then changed if needed after the standard protocol to achieve normocapnia and was then fixed until the end of the experiment. Before randomization of animals into different groups, the following criteria for exclusion were applied: MAP <70 mmHG, blood loss with Hb <14 g dl−1 or 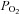 <90 mmHg during preparation procedures; on this basis, five animals had to be excluded from further experimental procedures.
<90 mmHg during preparation procedures; on this basis, five animals had to be excluded from further experimental procedures.
After baseline analyses of blood gas samples and haemodynamic parameters, rats were randomized into experimental groups.
Experimental set-up A: PD-4-I application to prevent capillary leakage (Fig. 1A)
While the control group (n = 6) was left untreated, systemic inflammation was induced by intravenous injection of LPS over 5 min in the other groups (n = 6 each). Additionally, two of the latter groups of animals were treated by i.v. injection with PD-4-Is. One group received a starting bolus of 0.3 mg (100 g BW)−1 rolipram 5 min before LPS application followed by continuous administration of 0.04 μg (100 g BW)−1 h−1 of rolipram (rolipram+LPS). The second group received roflumilast (roflumilast+LPS) with a 24 μg (100 g BW)−1 bolus 5 min before LPS administration followed by continuous application at a dose of 4 μg (100 g BW)−1 h−1.
Experimental set-up B: PD-4-I application to treat capillary leakage (Fig. 1B)
To test whether PD-4-I application was effective for therapeutic treatment of inflammation-induced capillary leakage, we modified our model for an additional set of experiments. Because of the high and rapid mortality rate observed with 0.5 mg (100 g BW)−1 LPS i.v. (see below), LPS doses were reduced to 0.25 mg (100 g BW)−1i.v. For cAMP enhancement we used a rolipram (3 mg kg−1) and roflumilast (0.24 mg kg−1) i.v. bolus at 1.5 h, 2.5 h, 3.5 h and 4.5 h after LPS application. In order to maintain mesenteric microcirculation until PD-4-I treatment was started, we additionally applied the following infusion regime: all groups received 2 ml h−1 of the balanced crystalloid solution Sterofundin Iso (Iso). If MAP decreased below 60 mmHg, an additional 1 ml Iso bolus was given intravenously. If MAP did not show fluid responsiveness, if there was no systolic pulse variation (as a sign of hypovolaemia), or if MAP decreased after volume therapy (e.g. due to septic cardiomyopathy), fluid administration was stopped.
In both experimental set-ups, experiments were terminated after 6 h. Animals that had survived the experimental procedure to this time point were killed by an overdose of barbiturate. Tissue samples were taken from several organs of each animal followed by processing of the tissue for histological analyses as described below.
Macrohaemodynamic monitoring and evaluation
Mean arterial pressure (MAP) and heart rate were continuously measured throughout the experiments. Cardiac index (CI), stroke volume index (SVI), total peripheral resistance index (TPRI) and delivery oxygen index (DO2-I) were calculated as recently published using the following equations (Shinoda et al. 1991): cardiac index (CI) [ml min−1 kg−1] = cardiac output/BW; stroke volume index (SVI) [ml beat−1 kg−1] = CI/heart rate; total peripheral resistance (TPRI) [mmHg ml−1 min−1 kg−1] = MAP/CI; delivery of oxygen index (DO2-I) = (CI × (((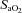 [%]/100) × Hb [g dl−1]× 1.34) +
[%]/100) × Hb [g dl−1]× 1.34) +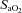 [%]× 0.0031)/100), where
[%]× 0.0031)/100), where 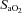 is arterial oxygen saturation and Hb is haemoglobin concentration.
is arterial oxygen saturation and Hb is haemoglobin concentration.
Intravital measurement of capillary endothelial barrier properties
To assess changes of microvascular permeability during the experimental procedures, digital fluorescence images were taken after single i.v. injection of FITC-albumin 5 mg (100 g BW)−1 (Sigma, Deisenhofen, Germany). Fluorescence images were taken using a 100 W mercury lamp and a filter set consisting of a 450–490 nm excitation and a 520 nm emission filter within an inverted microscope (Zeiss Axiovert 200, Carl Zeiss, Göttingen, Germany). Images were taken before randomization and then each hour dependent on the survival of the respective animal.
Microvascular permeability was then estimated by determining the extravasation of FITC-albumin by measurements of integrated optical intensity as described previously (Bekker et al. 1989). The labelled FITC-albumin represented relative changes in permeability: ΔI = 1 − (Ii−Io)/Ii, where ΔI is the change in light intensity, Ii is the light intensity inside the vessel, and Io is the light intensity outside the vessel. Grey scale values were measured in the postcapillary venules and in the extravascular space around the venules per unit area throughout the experiments and at selected times using ImageJ software. For each time point 10 randomly selected intravascular and intersititial areas near the postcapillary venules were selected for the measurements by a blinded observer.
Evaluation of microcirculatory flow
Analysis of microvascular blood flow was carried out in straight segments of venular microvessels (20–35 μm in diameter). Velocity of erythrocytes was measured by a blinded investigator using the software MetaMorph v. 6.1r4. For each time point the velocities of 27 randomly chosen erythrocytes within postcapillary venules were measured for each animal. For each time point six to nine vessels per animal were analysed. For each of these vessels, diameter was measured and from these data volumetric flow was calculated as described previously (Gross & Aroesty, 1972): volumetric flow (QV) was derived from the average velocity of free passing red blood cells (VRBC; μm ms−1) of each vessel assuming a cylindrical vessel geometry (QV[pl s−1] = [VRBC/1.6]×[phi]×r2).
Tissue preparation and immunostaining of mesenteric windows
To detect alterations of the endothelial adherens junction protein VE-cadherin in postcapillary mesenteric venules, we prepared rat mesenteric windows as has been described previously with some modifications (Nehls & Drenckhahn, 1991). In brief, after completion of experimental procedures the mesenteric window of the small intestine containing the postcapillary venules were stretched over Teflon rings and removed by cutting the edges with a scalpel blade. Thereafter samples were fixed in 2% formaldehyde in phosphate-buffered saline (PBS; pH 7.4) for 10 min and washed three times in PBS. Subsequently, the mesothelial cell layer was removed by digestion in 0.5% w/v Dispase II (Sigma-Aldrich, Taufkirchen, Germany) in PBS for 15 min at RT. Proteases were washed out with PBS and then mesenterial were treated with 0.1% Triton X-100 in PBS for 5 min.
Thereafter, immunostaining for VE-cadherin was performed as described in detail previously (Schlegel et al. 2008). After preincubation for 30 min with 10% normal donkey serum (NDS) and 1% bovine serum albumin (BSA) at RT, we applied a polyclonal goat anti-human VE-cadherin antibody (1:100) in PBS as the primary antibody overnight at 4°C. As secondary antibody we used a Cy3-labelled donkey anti-goat IgG (Dianova, Hamburg, Germany, diluted 1:600 in PBS). Mesenterial windows were then carefully transferred on glass slides and covered with 60% glycerol in PBS, containing 1.5%n-propyl gallate (Serva, Heidelberg, Germany) as an anti-fading compound.
Histopathological analyses of lungs
Lungs were removed after experimental procedures for histopathological studies. The tissues were fixed in formaldehyde 3.5% (Otto Fischar, Germany) for more than 24 h. Tissues were then embedded in paraffin and subsequently, sections were stained with H&E for analyses of morphological alterations within the tissues as described previously (Schick et al. 2010). ImageJ morphometric software was used to measure thickness of alveolar septa. In each of the animals, three sections were analysed and in each section thickness of 15 randomly chosen alveolar septa were measured by a blinded observer.
cAMP measurements
For quantification of cAMP levels, 12 additional animals (n = 4 for each condition) were treated as described above without abdominal surgery. After 3 h the mesentery of the ileum was taken out and the microvessels were gently prepared from the mesentery, extracted from the surrounding tissues and snap frozen using liquid nitrogen. Thereafter cAMP measurements (in pmol mg−1) were performed using the cAMP enzyme immunoassay (Biomol, Hamburg, Germany) according to the manufacturer's recommendations as described previously (Schlegel & Waschke, 2009b).
Statistical analysis
Values are expressed as means ± SEM. SPSS 19.0 statistical software was used to analyse possible differences and statistical significance was determined by analysis of variance (one way ANOVA followed by a post hoc Duncan's test). Due to the high mortality rate in the 5 mg kg−1 LPS group, valid statistical tests could not be applied at 2 h in the LPS group. Survival was analysed by the Kaplan–Meier method with Log Rank analysis as described elsewhere (Bland & Altman, 1998). Statistical significance is assumed for P < 0.05.
Results
cAMP levels were significantly reduced in mesenteric microvessels following LPS application
First we performed cAMP measurements in mesenteric microvessels to test whether LPS-induced loss of endothelial cAMP which we previously described in vitro (Schlegel et al. 2009) was also evident in our model of systemic inflammation. In control animals the cAMP level was 5.3 ± 1.0 pmol mg−1 (Fig. 2). This was significantly reduced to 1.8 ± 0.9 pmol mg−1 after 3 h of LPS treatment. After systemic application of PD-4-I, no significant difference from controls was observed (3.8 ± 0.9 pmol mg−1).
Figure 2. cAMP levels in mesenteric microvessels were reduced after systemic LPS application.
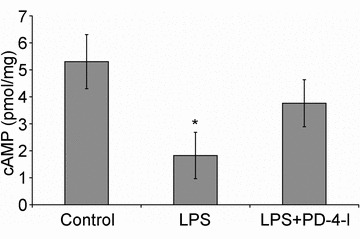
LPS treatment of animals (n = 4) resulted in significantly reduced cAMP levels of 1.8 ± 0.9 pmol mg−1 compared to controls (n = 4; 5.3 ± 1.0 pmol mg−1) whereas treatment with PDI-4-I did not change cAMP levels significantly (n = 4; 3.8 ± 0.9 pmol mg−1). *P < 0.05 compared to controls.
Capillary endothelial barrier properties were stabilized by application of PD-4-Is
The next set of experiments was conducted to test our hypothesis that systemic application of PD-4-Is would result in stabilization of endothelial barrier properties. To investigate this we used two different types of PD-4 inhibitors, namely rolipram and roflumilast. We analysed extravasation of intravenously applied FITC-albumin at different time points in postcapillary mesenteric venules. Under baseline conditions, i.e. before animals were randomized in different groups, FITC-albumin was constantly observed in postcapillary venules demonstrating an intact microvascular endothelial barrier at the beginning of the experimental procedures (Fig. 3Aa, c, e and d). LPS application resulted in significant extravasation of FITC-albumin to 132 ± 6% after 1 h and to 136 ± 3% after 2 h compared to baseline conditions (Fig. 3Ad and B). In contrast such extravasation was not observed in controls and in LPS+rolipram- and LPS+roflumilast-treated animals. Extravasated FITC-albumin amounted to 103 ± 6% after 1 h and 106 ± 3% after 2 h after rolipram treatment and 96 ± 6% after 1 h and 103 ± 2% after 2 h of roflumilast treatment, respectively (Fig. 3B).
Figure 3. PD-4-Is blocked capillary leakage in postcapillary venules.
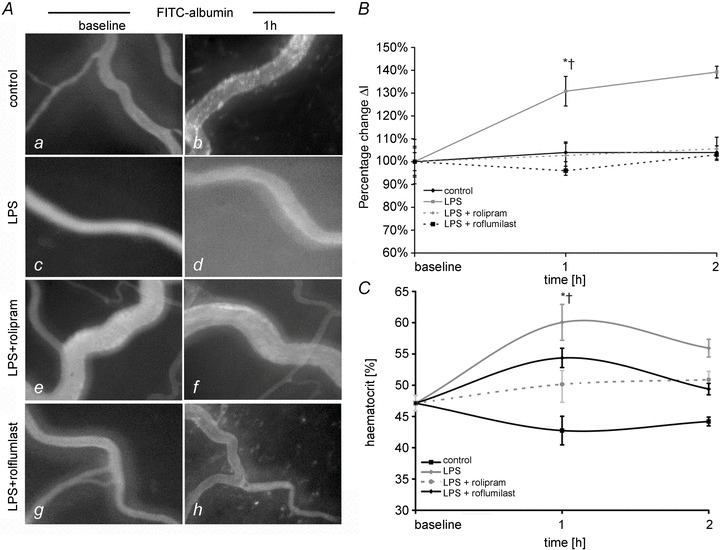
A, postcapillary mesenteric venules following i.v. application of FITC-albumin are shown under baseline conditions (a, d, g) and after 1 h (b, e, h) of experimental procedures. B, quantifications of extravasated FITC-albumin in postcapillary venules are shown. ΔI is the change in light intensity measured inside and outside the vessels. C, changes of haematocrit values in the different groups are shown. *P < 0.05 vs. control; †P < 0.05 vs. LPS+PD-4-I.
Because capillary leakage and consecutive loss of intravascular plasma fluid results in increased heamatocrit values (hct) we used this as an indirect parameter of endothelial barrier dysfunction (Fig. 3C). Baseline values of hct before randomization of animals into different experimental groups amounted 47.9 ± 1.2%. In controls, hct slightly decreased after 1 h and 2 h of measurements to 42.8 ± 2.3% and 44.2 ± 0.7%, respectively (Fig. 3C). This may be explained by repeated application of NaCl i.v. boli after taking blood samples for blood gas analyses or CO measurement. In spite of this effect we found significantly increased hct in the LPS group (60.1 ± 2.2% after 1 h and 56.0 ± 1.3% after 2 h). In the LPS+PD-4-I-treated animals, no significant increase of hct compared to controls was observed (50.1 ± 2.2% (rolipram) and 51.3 ± 0.9% (roflumilast) after 1 h; 51.0 ± 1.3% (rolipram) and 48.8 ± 1.2% (roflumilast) after 2 h). Thus, hct values were significantly lower compared to the LPS group suggesting reduced loss of plasma volume under these conditions.
In immunostaining, the endothelial adherens junction protein VE-cadherin appeared regularly distributed at the margins of endothelial cells within the postcapillary mesenteric venules under control conditions (Fig. 4A). After LPS treatment we observed in most vessels fragmentation and loss of VE-cadherin at endothelial cell borders (Fig. 4B). In contrast, in postcapillary venules of LPS+rolipram-treated animals we constantly observed regular distribution of VE-cadherin at the cell borders of endothelial cells comparable to control conditions (Fig. 4C).
Figure 4. LPS-induced fragmentation of VE-cadherin in endothelial microvessels is abolished by PD-4-I.
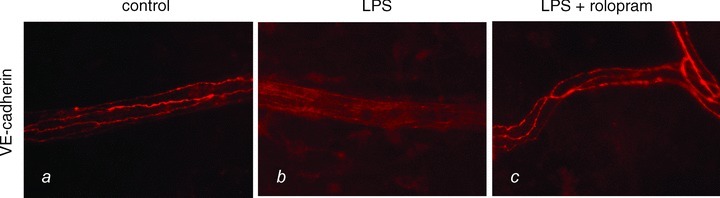
VE-cadherin immunostaining in mesenteric postcapillary venules is demonstrated under the different experimental conditions.
Since it is known that systemic hyperinflammation can also result in acute lung injury (ALI) with severe pulmonary oedema (Ware & Matthay, 2000) we additionally analysed H&E-stained lung tissue sections. After LPS treatment all lungs of this experimental group showed severe lung oedema as revealed by dramatic thickening of alveolar septa from 6.8 ± 0.6 μm in controls to 23.8 ± 3.8 μm after LPS treatment (Fig. 5Aa and b and B). This was accompanied with considerable interstitial cellular infiltration of neutrophils and hyperaemia suggesting microvascular dysfunction. In contrast, LPS+rolipram-treated animals only showed a slight difference compared to controls (alveolar septa were 8.3 ± 1.2 μm; Fig. 5B) indicating that pulmonary oedema was attenuated under these experimental conditions. Additionally, cellular infiltration and hyperaemia were dramatically reduced. Accordingly, the oxygen delivery index (DO2-I), as an overall parameter of oxygen supply, was significantly reduced in LPS-treated animals compared to the LPS+rolipram group (Fig. 5C).
Figure 5. PD-4-Is attenuated LPS-induced lung oedema.
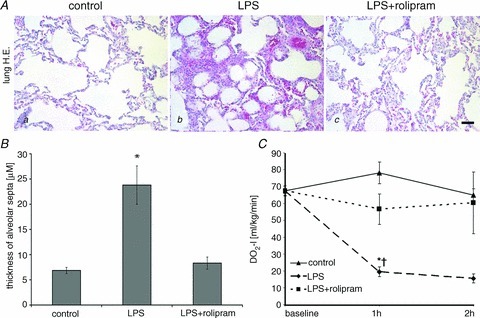
A, representative images of H&E stained sections of the lung demonstrate that LPS (b) induced severe lung oedema and cellular infiltration whereas these changes were not observed in the LPS+PD-4-I groups (b). Controls (a) displayed normal morphology; scale bar is 20 μm. B, quantification of thickness alveolar septa in the different experimental groups are shown. C, oxygen delivery index (DO2-I) as an overall parameter of oxygen supply was reduced in LPS-treated animals compared to the LPS+PD-4-I group whereas controls remained unchanged. *P < 0.05 vs. control; †P < 0.05 vs. LPS+PD-4-I.
In summary these data demonstrate that systemic application of PD-4-Is in LPS-induced systemic inflammation was effective to stabilize endothelial barrier properties.
Microcirculatory flow was improved by PD-4-I treatment
Thereafter we tested whether stabilization of endothelial barrier properties was associated with improved microcirculatory flow. We measured volumetric flow in postcapillary mesenterial venules. Within 0.5 h after LPS was applied, mesenteric volumetric flow decreased dramatically to 12 ± 2% of baseline levels. In the following time course, volumetric flow remained reduced and tended to even be suspended completely (Fig. 6). PD-4-I application resulted in significantly improved microcirculatory flow. At later time points, i.e. after 2 h, volumetric flow also dropped in rolipram-treated animals, which was still 236% higher than in the LPS group. Roflumilast-treated animals showed also an initial drop of volumetric flow, which recovered to stable values within 2 h of measurements. In controls volumetric flow remained largely unchanged in this time course.
Figure 6. Microcirculatory flow was improved following PD-4-I treatment.
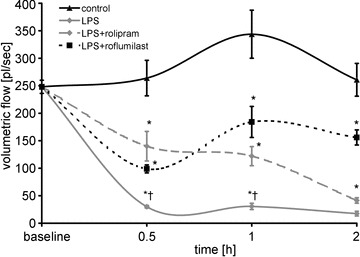
LPS induced dramatic breakdown of microcirculatory flow. LPS+PD-4-I had no significant changes within the 1 h compared to controls. Volumetric flow in controls remained unaltered throughout the experiments. *P < 0.05 vs. control; †P < 0.05 vs. LPS+PD-4-I.
In order to test whether PD-4-I treatment would lead to vasodilatation at least at the levels of postcapillary venules, we measured diameters of blood vessels within the mesenteric windows at different time points (Table 1). At the beginning of PD-4-I treatment, when vasodilatation would be expected to be most pronounced, we observed no differences between the experimental groups. In contrast, after 2 h we observed a significant vasoconstriction in the LPS+rolipram group.
Table 1.
Diameter of postcapillary mesenteric venules: LPS-treated animals and controls displayed no changes in capillary diameter
| Baseline | 0.5 h | 1 h | 2 h | |
|---|---|---|---|---|
| Control | 25.61 ± 0.57 | 26.84 ± 1.30 | 27.88 ± 1.68 | 27.83 ± 1.50 |
| LPS | 25.61 ± 0.57 | 27.43 ± 0.88 | 25.33 ± 1.2 | 24.58 ± 1.20 |
| Rolipram+LPS | 25.61 ± 0.57 | 27.08 ± 2.03 | 31.38 ± 1.47 | 22.9 ± 1.0* |
| Roflumilast+LPS | 25.61 ± 0.57 | 24.27 ± 0.66 | 28.07 ± 1.66 | 28.44 ± 1.11 |
PD-4-I treatment showed no consistent effect on microvascular vessels;
P < 0.05 vs. control.
Blood gas analyses indicated improved metabolism by PD-4-I treatment
Blood gas analyses were used to test for metabolic differences in the different experimental groups. It is important to note that respiratory set-up was unchanged after randomization. LPS and LPS+PD-4-I groups showed acidosis as revealed by decreased pH levels after 1 h and 2 h (Table 2). Accordingly, lactate was significantly increased, HCO3− and SBE (standard base excess) were dramatically decreased, whereas 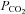 was unchanged in LPS-treated animals after 1 h and 2 h. In the LPS+rolipram group,
was unchanged in LPS-treated animals after 1 h and 2 h. In the LPS+rolipram group, 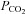 was significantly increased compared to controls and LPS-treated animals, which may be interpreted as improved metabolism compared to the LPS group. As revealed by HCO3− and SBE, metabolic acidosis was significantly less pronounced in the LPS+rolipram group. Accordingly, lactate did not further augment after 2 h in this group. Roflumilast application did not change
was significantly increased compared to controls and LPS-treated animals, which may be interpreted as improved metabolism compared to the LPS group. As revealed by HCO3− and SBE, metabolic acidosis was significantly less pronounced in the LPS+rolipram group. Accordingly, lactate did not further augment after 2 h in this group. Roflumilast application did not change 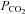 within 2 h of measurements and lactate did not augment after the first hour of experiments (Table 2).
within 2 h of measurements and lactate did not augment after the first hour of experiments (Table 2).
Table 2.
Arterial blood gas analyses
| PCO2[mmHg] | pH | HCO3−[mmol l−1] | SBE [mmol l−1] | Lactate [mmol l−1] | ||
|---|---|---|---|---|---|---|
| 1 h | Baseline | 38.9 ± 1 | 7.44 ± 0 | 26.4 ± 0.4 | 2.6 ± 0.4 | 1.3 ± 0.1 |
| Control | 39.0 ± 1.7# | 7.38 ± 0.01 | 22.5 ± 1.4 | −1.9 ± 1.4 | 1 ± 0.3 | |
| LPS | 36.5 ± 4.8# | 7.18 ± 0.05*† | 14.1 ± 2.5* | −12.7 ± 3.1* | 4.0 ± 0.8* | |
| Rolipram+LPS | 51.5 ± 1.9* | 7.23 ± 0.02* | 20.5 ± 0.9 | −5.8 ± 1.0 | 4.7 ± 0.4* | |
| Roflumilast+LPS | 43.1 ± 1.2 | 7.28 ± 0.02* | 19.56 ± 1.1 | −6.0 ± 1.3 | 3.0 ± 0.4* | |
| 2 h | Control | 40.4 ± 1.7 | 7.39 ± 0.01 | 24.3 ± 1.3 | 0.2 ± 1.3 | 1.1 ± 0.4 |
| LPS | 37.6 ± 9.2 | 7.02 ± 0.04*#† | 9.6 ± 3.0*#† | −19.3 ± 3.5*#† | 8.6 ± 2*#† | |
| Rolipram+LPS | 49.7 ± 2.4 | 7.23 ± 0.03* | 20.7 ± 0.3† | −7.1 ± 0.6* | 5.1 ± 0.8* | |
| Roflumilast+LPS | 41.9 ± 2.0 | 7.18 ± 0.02* | 15.3 ± 1.3*# | −10.8 ± 2.1* | 3.6 ± 0.5 |
Blood gas analyses were obtained after reconstitution (baseline) and after 1 h and 2 h. After baseline correction (see Methods) respiratory set-up remained unchanged. LPS induced metabolic acidosis in both groups whereas PD-4-I improved metabolism
P < 0.05 vs. control;
P < 0.05 vs. LPS+Roflumilast;
P < 0.05 vs. Rolipram+LPS vs. baseline SBE (standard base excess).
Evaluation of macrohaemodynamic parameters
To determine the macrohaemodynamic state and possible side-effects of i.v.-administered PD-4-Is, we performed invasive continuous measurements of blood pressure (MAP), of cardiac output and calculated cardiac index (CI), stroke volume index (SVI) and total peripheral vascular resistance index (TPRI) as previously published for rodent models (Shinoda et al. 1991) (Fig. 7A–D). While macrohaemodynamics remained constant in control experiments, LPS-treated animals showed rapid and significant loss of MAP indicating the manifestation of severe hyperinflammation (Fig. 7A). In LPS+PD-4-I-treated animals, loss of MAP was lower than in controls, but was significantly higher than in LPS-treated animals.
Figure 7. Macrohaemodynamic parameters.
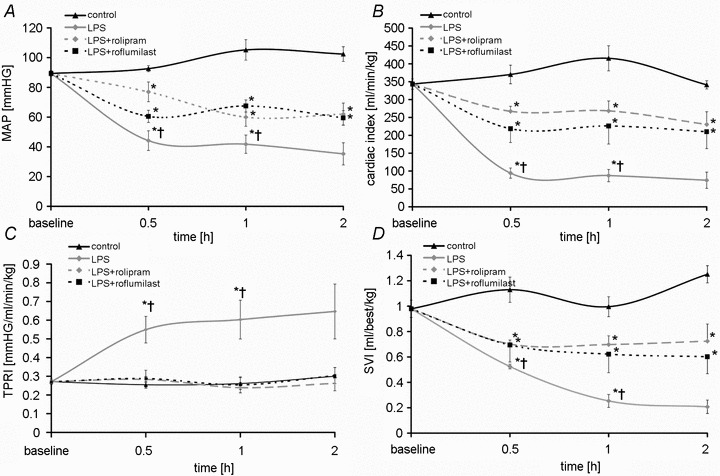
Results of measurements of macrohaemodynamic parameters are shown. A, mean arterial pressure (MAP); B, cardiac index; C, total peripheral resistance index (TPRI); D, stroke volume index SVI. *P < 0.05 vs. control; †P < 0.05 vs. LPS+PD-4-I.
Cardiac index decreased within 30 min in LPS- and LPS+PD-4-I-treated animals compared to controls (Fig. 7B). Comparable to MAP measurements CI in LPS+PD-4-I-treated animals was significantly higher than after LPS treatment. Calculation of TPRI revealed no differences between controls and LPS+PD-4-I-treated animals whereas TPRI was significantly increased in the LPS group (Fig. 7C). Stroke volume index significantly decreased in both LPS- and LPS+PD-4-I-treated animals compared to controls (Fig. 7D). However SVI remained significantly higher in the LPS+PD-4-I compared to the LPS group.
PD-4-I application significantly increased survival in septic rats
Finally, we analysed survival of the animals in the different experimental groups. In controls, all animals survived the experimental procedures and were sacrificed after 6 h (Fig. 8). Application of LPS resulted in a severe systemic inflammation which induced rapid multiple organ failure and death. Thus, 2 h after LPS was applied, 50% of the animals deceased and none of the animals survived over 6 h due to septic hyperinflammation. Animals that were treated with LPS+PD-4-I inhibitor also showed clinical signs of severe systemic inflammation. However, in these groups all animals had survived 2 h after LPS was applied. After 6 h 50% of the rolipram- and 66% of the roflumilast-treated animals had survived, which demonstrated that systemic application of PD-4-I significantly increased survival in LPS-induced systemic inflammation.
Figure 8. Survival of animals is increased by PD-4-I treatment in systemic inflammation.
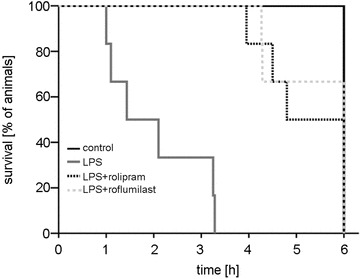
Kaplan-Meier method was used to analyse survival of animals in the different experimental groups. Mortality rate was 50% after 2 h and 100% after 6 h LPS (n = 6) had been applied. All animals survived 2 h and 50% survived 6 h in the rolipram and 66% in the roflumilast group (n = 6). All control animals survived (n = 5).
Application of PD-4-I was effective for therapeutic improvement of LPS-induced capillary leakage and loss of microcirculatory flow
Because all these experiments revealed that PD-4-I application could be a promising approach for effective treatment of capillary leakage, we performed experiments where PD-4-I was applied after manifestation of systemic inflammation and capillary leakage.
According to our preliminary experiments microcirculation was maintained by MAP-dependent application of fluid to maintain MAP at 60 mmHg.
As an indirect measure of possible endothelial barrier stabilization following PD-4-I application, we analysed fluid demand to maintain MAP of 60 mmHg after 1.5 h of experiments (Fig. 9B). LPS-treated animals required 26.6 ± 2.2 ml (100 g BW)−1 in the time course of experiments whereas animals treated with rolipram or roflumilast required significantly less fluid, i.e. 15.1 ± 1.5 ml (100 g BW)−1 and 21.2 ± 0.3 ml (100 g BW)−1, respectively. Controls received 6.5 ml (100 g BW)−1 of fluid.
Figure 9. PD-4-Is therapeutically stabilized endothelial barrier functions after LPS-induced capillary leakage.
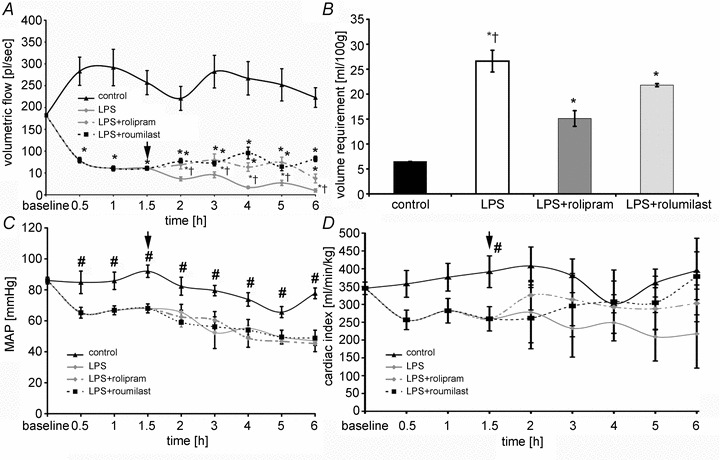
A, volumetric flow significantly decreased after LPS application despite of volume resuscitation to maintain MAP. After starting i.v. application of PD-4-I (arrow) microcirculation improved compared to LPS alone. B, all animals received a fixed volume application (control) and additional i.v. boli to maintain MAP > 60 mmHg if needed. Significant less volume was applied in PD-4-I-treated animals compared to LPS alone. C and D, CI (C) and MAP (D) showed no significant changes between LPS and LPS+PD-4-I groups. *P < 0.05 vs. control, #P < 0.05 vs. LPS and LPS+PD-4-I, †P < 0.05 vs. PD-4-I.
Despite fluid administration microcirculatory flow decreased significantly within the first 1.5 h to 40 ± 6% after LPS was applied (Fig. 9A). After that time animals were treated by boli of PD-4-I rolipram or roflumilast. This resulted in augmented microcirculatory flow and was maintained 1.4- to 3.8-fold (rolipram) and 1.8- to 8.3-fold (roflumilast) higher than in untreated animals over time.
While microcirculation showed significant improvements, MAP and cardiac output (Fig. 9C and D) were not significantly different when LPS and PD-4-I-treated groups were compared. However, CI tended to increase after PD-4-I application. Similar to our observations of the groups that were simultaneously treated with LPS and PD-4-I, diameters of the vessels did not increase when treated with rolipram or roflumilast (data not shown).
Discussion
Capillary leakage and endothelial dysfunction are increasingly recognized to significantly contribute to organ failure and death in sepsis and systemic inflammation (Hack & Zeerleder, 2001; Cinel & Dellinger, 2007; Lee & Slutsky, 2010). Therefore therapeutic targeting of capillary leakage in sepsis and systemic inflammation is considered a clinically highly relevant approach which cannot be established a moment too soon (Jacobson & Garcia, 2007; Lee & Slutsky, 2010). Previous research predominantly focused on modulation of the immune response in sepsis in which endothelial barrier protection occurred as a secondary effect. The present study extends our previous investigations and thereby focuses on capillary leakage as a primary pathophysiological problem. In a rodent model of LPS-induced systemic hyperinflammation, which mimics the situation in septic patients, we demonstrated in a first step that LPS led to decreased cAMP levels in mesenteric microvessels. Accordingly we found that systemic intravenous application of PD-4-Is to increase cAMP was effective to attenuate LPS-induced endothelial barrier breakdown without severe side-effects. This was associated with reduced fluid loss and increased microcirculatory flow leading to improved overall metabolism in the PD-4-I-treated animals. The consecutive haemodynamic stabilization prevented multiple organ failure. Thus, mortality was reduced in PD-4-I-treated animals. Additional experiments showed that PD-4-I application was effective to stabilize endothelial barrier properties because fluid requirement to maintain MAP was dramatically reduced up to 76% compared to LPS-treated animals.
PD-4-I treatment blocked LPS-induced breakdown of endothelial barrier functions
It is meanwhile well established that increased cAMP levels effectively stabilize endothelial barrier properties under resting conditions and in acute inflammation (Adamson et al. 1998, 2008; Birukova et al. 2004, 2008; Schlegel et al. 2008; Baumer et al. 2009; Schlegel et al. 2009; Schlegel & Waschke, 2009a; Spindler et al. 2010; Lin et al. 2011, 2012). However, because most of these studies were carried out in cell cultures, single vessels in vivo or in isolated organs (Schmidt et al. 2008; Schlegel et al. 2009; Schlegel & Waschke, 2009a; Witzenrath et al. 2009), it remained unclear whether the detailed knowledge on cAMP-induced effects on endothelial barrier functions would finally lead to therapeutic benefits to stabilize capillary leakage in sepsis and acute inflammation. On the other hand the clinical use of cAMP-increasing agents in sepsis, such as the PD-3-I inhibitor enoximon or β-adrenoreceptor mimetica such as dobutamine, appeared to be limited by the fact that the latter agents induced severe side-effects such as excessive vasodilatation, tachyarrhythmia and loss of blood pressure (Lehtonen et al. 2004).
In a previous study we showed that LPS treatment of microvascular endothelial cells led to significant reduction of cAMP levels (Schlegel et al. 2009). This is now substantiated by our present data where we demonstrate that systemic application of LPS reduced cAMP levels in mesenteric microvessels. Consequently, in vitro PD-4-I inhibition alone was previously shown to be effective to prevent LPS-induced loss of cAMP in microvascular endothelium and thereby prevented intercellular gap formation and loss of barrier function (Bogatcheva et al. 2009; Schlegel et al. 2009). In line with that the intraperitoneal use of PD-4-Is, rolipram in vivo has previously been successful in preventing ANP-induced loss of plasma fluid (Lin et al. 2011, 2012). Because of these data and since phosphodiesterase-4 (PDE-4) is the most highly expressed cAMP-hydrolysing phosphodiesterase in ordinary endothelium (Netherton & Maurice, 2005; Lugnier, 2006), we decided to use PD-4 inhibitors in this study.
Our present data clearly show that systemic simultaneous intravenous application of PD-4-Is resulted in reduction of lung oedema and attenuated extravasation of FITC-albumin in mesenteric postcapillary venules. This is substantiated by the fact that haematocrit values, which were used in previous studies as an indirect parameter to detect loss of intravascular plasma (Lin et al. 2011, were not significantly increased in the LPS+PD-4-I group compared to controls. In a second line of evidence, immunostaining of adherens junction protein VE-cadherin, which is required for maintenance of the endothelial barrier (Mehta & Malik, 2006), was regularly distributed at endothelial cell borders in postcapillary venules after PD-4-I treatment, which was not the case in LPS-treated animals.
Our data support previous studies in which pulmonary oedema was reduced by PDI application in isolated lungs (Schmidt et al. 2008; Witzenrath et al. 2009). In general the beneficial effects of application of PDIs such as pentoxifyllin or rolipram in models of systemic hyperinflammation or acute lung injury were attributed to a significant reduction of levels of proinflammatory cytokines and of matrix metalloproteinases (Tofovic et al. 2000; Coimbra et al. 2006). Similarly the clinical improvement of patients treated with the PDIs amrinone and milrinone after cardiac reperfusion were explained by possible antiinflammatory effects of PDIs (Chanani et al. 2002), while possible stabilization of endothelial barrier functions remained unexplored in all these studies. Consequently, it must be considered that protective effects of rolipram on endothelial barrier properties may be secondary effects following attenuated cytokine release by PDI application. The fact that application of PD-4-I after manifestation of systemic inflammation in our present study was also effective in stabilizing endothelial barrier properties argues against a dramatic contribution of such anti-inflammatory effects of PD-4-Is. However, although we cannot rule out that this effect of PDI may play a role in our study it must be emphasized that LPS alone leads to endothelial activation and barrier disruption (Dauphinee & Karsan, 2006; Schlegel et al. 2009). In view of the fact that activated endothelial cells represent a relevant source for cytokines in acute inflammation (Hack & Zeerleder, 2001), it may be assumed that LPS-induced activation of endothelial cells and barrier disruption may lead to excessive release of endothelium-derived cytokines. Therefore, inhibition of endothelial barrier disruption by PD-4-Is may prevent such release of cytokines. However, additional studies will be required to test this hypothesis.
Endothelial barrier stabilization resulted in improved microcirculatory flow and metabolism
It is well known that the extravasation of plasma fluid can impair organ function by increasing the distance required for the diffusion of metabolic supply (Lee & Slutsky, 2010). Fluid loss results in changes of blood rheology and vasoconstriction leading to breakdown of microcirculatory flow, which is a hallmark of the onset of organ failure. Our data show that the volumetric flow in these vessels was dramatically decreased following LPS injection indicating severe breakdown of microcirculatory flow. Simultaneous PD-4-I treatment significantly attenuated this effect. This supports a previous study in which application of dobutamine, which also increases cAMP levels, significantly improved the microcirculatory flow of the tongue without affecting macrohaemodynamic parameters (De Backer et al. 2006). The beneficial effects of dobutamine, however, were believed to result in redistribution of blood flow and improvement of the microcirculatory flow in response to β-adrenergic stimulation whereas effects on endothelial barrier stabilization were not assessed (De Backer et al. 2006; Nacul et al. 2010). From the present data it can be concluded that this effect can be explained by effective stabilization of endothelial barrier functions.
As a consequence of enhanced volumetric flow, metabolism was improved in PD-4-I+LPS-treated animals as revealed by blood gas analyses. Predictors of mortality in critically ill patients such as lactate and standard base excess were also evident in our experiments (Rixen et al. 2001; Trzeciak et al. 2007). LPS alone induced severe metabolic acidosis over time, which correlated with high mortality. PD-4-I was able to significantly enhance metabolic supply seen in stabilization of pH (despite of higher 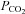 levels), lactate as well as SBE compared to the LPS group. It can be postulated that the significant decrease of HCO3− in LPS-treated rats compared to PD-4-I may be a sign of acute kidney injury (AKI) with acute loss of tubule cell function and impaired reabsorption of filtered HCO3−. In this respect experimental and clinical studies suggested potential protective renal effects of PDI by endothelium-dependent vascular relaxation (Groesdonk et al. 2009). Although this was not specifically addressed in our study we suggest that PD-4-I application may prevent prerenal AKI by improvement of fluid balance in septic shock in our model.
levels), lactate as well as SBE compared to the LPS group. It can be postulated that the significant decrease of HCO3− in LPS-treated rats compared to PD-4-I may be a sign of acute kidney injury (AKI) with acute loss of tubule cell function and impaired reabsorption of filtered HCO3−. In this respect experimental and clinical studies suggested potential protective renal effects of PDI by endothelium-dependent vascular relaxation (Groesdonk et al. 2009). Although this was not specifically addressed in our study we suggest that PD-4-I application may prevent prerenal AKI by improvement of fluid balance in septic shock in our model.
Because of a fixed mechanical respiratory protocol after randomization in the groups in which PD-4-I was simultaneously applied, the increased level of 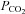 can be interpreted as an enhanced aerobic cell metabolism in our experiments. Despite massive hyperinflammation, LPS-treated animals showed no changes in
can be interpreted as an enhanced aerobic cell metabolism in our experiments. Despite massive hyperinflammation, LPS-treated animals showed no changes in 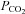 levels, which can be assessed as a critical stage of severe septic shock with malperfusion. In contrast PD-4-I-treated animals appeared to be able to adapt septic cell metabolism as revealed by increased levels of
levels, which can be assessed as a critical stage of severe septic shock with malperfusion. In contrast PD-4-I-treated animals appeared to be able to adapt septic cell metabolism as revealed by increased levels of 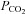 .
.
Taking these data together, PD-4-I treatment improved cell metabolism by an enhanced microcirculatory flow due to reduced plasma loss.
Macrohaemodynamic parameters and side-effects of PD-4-I
It is well known, that macroheamodynamic parameters such as MAP and CI do not necessarily correlate with microcirculation (Hiltebrand et al. 2004). PDIs such as enoximon are commonly used because of their inotropic, lusitropic and vasodilating properties (Chanani et al. 2002). Because vasodilatation and tachyarrhythmia represent possible side-effects of PDI in systemic inflammation or sepsis, we decided to start PDI application in the groups that were simultaneously treated with PD-4-I and LPS using a bolus followed by a low-dose continuous PD-4-I application compared to previously applied doses of rolipram in animal models (Begany et al. 1996). To test for potential side-effects we continuously analysed haemodynamic parameters. As expected, LPS induced rapid loss of MAP, as a sign of severe septic shock in the LPS group. PD-4-Is clearly did not potentiate this effect but even led to increased MAP compared to LPS-treated animals. In line with that, PD-4-I+LPS-treated animals showed no differences in TPRI as a marker of vasodilatation compared to controls. The significantly increased TPRI in the LPS group can be interpreted as a consequence of breakdown of the peripheral microcirculatory flow and stasis. Furthermore no vasodilatation of the microvessels was observed.
The improved CI and SVI in the LPS+PD-4-I-treated animals compared to LPS-treated animals may be explained as a secondary effect of stabilized endothelial barrier properties and improved fluid balance. This is underlined by significantly increased SVI in the PD-4-I+LPS group compared to LPS-treated animals as a marker of enhanced left ventricular restoration. The anti-inflammatory effect of PD-4-I may have an additional synergistic effect to the myocard, but this was not the focus in this trial. In summary macrohaemodynamic parameters demonstrated that PD-4-I treatment did not lead to severe side-effects in our model.
PDI treatment dramatically reduced mortality rate
Finally, the most striking result of our experiments is the dramatic increase of survival in septic rats. In general the early and rapid onset of systemic inflammation in our model is a known characteristic following LPS application in rodent models (Deitch, 2005). In our present study mortality rate amounted to 50% after 2 h and 100% after 6 h LPS had been applied. Although no adjuvant therapy was applied, PD-4-I treatment resulted in 100% survival after 2 h and 50–66% after 6 h.
PDI application after manifestation of systemic inflammation and capillary leakage improved microcirculation by stabilization of endothelial barrier properties
In the clinical situation, therapy of sepsis and systemic inflammation starts after its clinical manifestation. Therefore, in an attempt to approximate our model to the clinical situation we conducted additional experiments where animals received PD-4-I after 1.5 h of ongoing severe systemic inflammation. Apparent systemic inflammation was verified by the fact that volume resuscitation to maintain microcirculation was required. This was especially important because our preliminary experiments had demonstrated that once microcirculation is stopped in the mesenteric microvessels, there was no possibility to recirculate them. In this trial animals treated with PD-4-I required significantly less volume compared to LPS alone, and this effect was more pronounced over time. This can be interpreted as a therapeutic stabilization of the endothelial barrier because fluid loss was obviously dramatically reduced.
Interestingly and similar to the animals that were simultaneously treated with PD-4-I, no severe side-effects of either rolipram or roflumilast were detected in these experiments indicating that its effect may largely be restricted to the microvessels. Furthermore the suspected side-effect of PD-4-I-induced vasodilatation, which represents a main argument against the use of PD-4-I in sepsis, was also absent in the groups that were treated with PD-4-I. However we cannot exclude the occurrence of side-effects after longer times of treatment. This, however, will have to be tested in more detailed analyses which were not in the scope of the present study.
Taken together to our knowledge we show for the first time that capillary leakage can be treated effectively by PD-4-I in inflammation-induced disruption of the endothelial barrier in vivo. Therefore these data demonstrate a clinically applicable approach to stabilize endothelial barrier properties in systemic inflammation.
Conclusion
In summary, intravenously applied PD-4-I was effective to prevent and to treat capillary leakage, to improve fluid balance, to maintain microcirculatory flow and to ameliorate overall metabolism. This was paralleled by haemodynamic stabilization with increased survival in our model. Therefore our data show for the first time a novel therapeutic approach to treat capillary leakage in systemic inflammation. In conclusion these findings warrant further studies to carefully enable the use of PD-4 inhibitors in sepsis therapy for patients.
Acknowledgments
We are grateful to Alexia Witchen and Veronika Heimbach for skilful technical assistance. These studies were supported by a grant from the Interdisziplinäre Zentrum für Klinische Forschung (IZKF) A102 and Z3/2 and from the Deutsche Forschungsgemeinschaft SCHL 1962/2-1.
Glossary
Abbreviations
- CI
cardiac index
- LPS
lipopolysaccharide
- MAP
mean arterial pressure
- PD-4-I
phosphodiesterase-4 inhibitor
- PDI
phosphodiesterase inhibitor
- SBE
Stabdard base axcess
- SVI
stroke volume index
- TPRI
peripheral vascular resistance index
- SBE
standard base excess
Author contributions
All experiments were performed in the laboratories of the departments of general, visceral, vascular and pediatric surgery and department of anaesthesia and critical care (Zentrum Operative Medizin, Oberdürrbacher 6, 97080 Würzburg). M.A.S.: conception and design of the experiments, collection, analysis and interpretation of data, drafting the article and revising it critically for important intellectual content. C.W.: drafting the article or revising it critically for important intellectual content. J.W.: Collection, analysis and interpretation of data. N.R.: revising the article critically for important intellectual content. J.W.: revising the article critically for important intellectual content. C.T.G.: revising the article critically for important intellectual content. N.S.: conception and design of the experiments, collection, analysis and interpretation of data, drafting the article and revising it critically for important intellectual content. All authors approved the final version of the manuscript. The authors declare that they have no conflicts of interest.
References
- Adamson RH, Liu B, Fry GN, Rubin LL, Curry FE. Microvascular permeability and number of tight junctions are modulated by cAMP. Am J Physiol Heart Circ Physiol. 1998;274:H1885–1894. doi: 10.1152/ajpheart.1998.274.6.H1885. [DOI] [PubMed] [Google Scholar]
- Adamson RH, Ly JC, Sarai RK, Lenz JF, Altangerel A, Drenckhahn D, Curry FE. Epac/Rap1 pathway regulates microvascular hyperpermeability induced by PAF in rat mesentery. Am J Physiol Heart Circ Physiol. 2008;294:H1188–1196. doi: 10.1152/ajpheart.00937.2007. [DOI] [PubMed] [Google Scholar]
- Adamson RH, Zeng M, Adamson GN, Lenz JF, Curry FE. PAF- and bradykinin-induced hyperpermeability of rat venules is independent of actin-myosin contraction. Am J Physiol Heart Circ Physiol. 2003;285:H406–417. doi: 10.1152/ajpheart.00021.2003. [DOI] [PubMed] [Google Scholar]
- Baumer Y, Spindler V, Werthmann RC, Bunemann M, Waschke J. Role of Rac 1 and cAMP in endothelial barrier stabilization and thrombin-induced barrier breakdown. J Cell Physiol. 2009;220:716–726. doi: 10.1002/jcp.21819. [DOI] [PubMed] [Google Scholar]
- Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb Haemost. 2010;103:40–55. doi: 10.1160/TH09-06-0403. [DOI] [PubMed] [Google Scholar]
- Begany DP, Carcillo JA, Herzer WA, Mi Z, Jackson EK. Inhibition of type IV phosphodiesterase by Ro 20–1724 attenuates endotoxin-induced acute renal failure. J Pharmacol Exp Ther. 1996;278:37–41. [PubMed] [Google Scholar]
- Bekker AY, Ritter AB, Duran WN. Analysis of microvascular permeability to macromolecules by video-image digital processing. Microvasc Res. 1989;38:200–216. doi: 10.1016/0026-2862(89)90028-9. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Liu F, Garcia JG, Verin AD. Protein kinase A attenuates endothelial cell barrier dysfunction induced by microtubule disassembly. Am J Physiol Lung Cell Mol Physiol. 2004;287:L86–93. doi: 10.1152/ajplung.00441.2003. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Zagranichnaya T, Alekseeva E, Bokoch GM, Birukov KG. Epac/Rap and PKA are novel mechanisms of ANP-induced Rac-mediated pulmonary endothelial barrier protection. J Cell Physiol. 2008;215:715–724. doi: 10.1002/jcp.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland JM, Altman DG. Survival probabilities (the Kaplan-Meier method) BMJ. 1998;317:1572. doi: 10.1136/bmj.317.7172.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogatcheva NV, Zemskova MA, Kovalenkov Y, Poirier C, Verin AD. Molecular mechanisms mediating protective effect of cAMP on lipopolysaccharide (LPS)-induced human lung microvascular endothelial cells (HLMVEC) hyperpermeability. J Cell Physiol. 2009;221:750–759. doi: 10.1002/jcp.21913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundschuh DS, Eltze M, Barsig J, Wollin L, Hatzelmann A, Beume R. In vivo efficacy in airway disease models of roflumilast, a novel orally active PDE4 inhibitor. J Pharmacol Exp Ther. 2001;297:280–290. [PubMed] [Google Scholar]
- Chanani NK, Cowan DB, Takeuchi K, Poutias DN, Garcia LM, del Nido PJ, McGowan FX., Jr Differential effects of amrinone and milrinone upon myocardial inflammatory signalling. Circulation. 2002;106:I284–289. [PubMed] [Google Scholar]
- Cinel I, Dellinger RP. Advances in pathogenesis and management of sepsis. Curr Opin Infect Dis. 2007;20:345–352. doi: 10.1097/QCO.0b013e32818be70a. [DOI] [PubMed] [Google Scholar]
- Coimbra R, Melbostad H, Loomis W, Porcides RD, Wolf P, Tobar M, Hoyt DB. LPS-induced acute lung injury is attenuated by phosphodiesterase inhibition: effects on proinflammatory mediators, metalloproteinases, NF-κB, and ICAM-1 expression. J Trauma. 2006;60:115–125. doi: 10.1097/01.ta.0000200075.12489.74. [DOI] [PubMed] [Google Scholar]
- Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood. 2005;105:1950–1955. doi: 10.1182/blood-2004-05-1987. [DOI] [PubMed] [Google Scholar]
- Dauphinee SM, Karsan A. Lipopolysaccharide signalling in endothelial cells. Lab Invest. 2006;86:9–22. doi: 10.1038/labinvest.3700366. [DOI] [PubMed] [Google Scholar]
- De Backer D, Creteur J, Dubois MJ, Sakr Y, Koch M, Verdant C, Vincent JL. The effects of dobutamine on microcirculatory alterations in patients with septic shock are independent of its systemic effects. Crit Care Med. 2006;34:403–408. doi: 10.1097/01.ccm.0000198107.61493.5a. [DOI] [PubMed] [Google Scholar]
- Deitch EA. Rodent models of intra-abdominal infection. Shock. 2005;24(Suppl 1):19–23. doi: 10.1097/01.shk.0000191386.18818.0a. [DOI] [PubMed] [Google Scholar]
- Groesdonk HV, Bauer A, Kreft B, Heringlake M, Paarmann H, Pagel H. Urodilatin and pentoxifylline prevent the early onset of Escherichia coli-induced acute renal failure in a model of isolated perfused rat kidney. Kidney Blood Press Res. 2009;32:81–90. doi: 10.1159/000209378. [DOI] [PubMed] [Google Scholar]
- Gross JF, Aroesty J. Mathematical models of capillary flow: a critical review. Biorheology. 1972;9:225–264. doi: 10.3233/bir-1972-9402. [DOI] [PubMed] [Google Scholar]
- Hack CE, Zeerleder S. The endothelium in sepsis: source of and a target for inflammation. Crit Care Med. 2001;29:S21–S27. doi: 10.1097/00003246-200107001-00011. [DOI] [PubMed] [Google Scholar]
- Hiltebrand LB, Krejci V, Sigurdsson GH. Effects of dopamine, dobutamine, and dopexamine on microcirculatory blood flow in the gastrointestinal tract during sepsis and anaesthesia. Anesthesiology. 2004;100:1188–1197. doi: 10.1097/00000542-200405000-00022. [DOI] [PubMed] [Google Scholar]
- Jacobson JR, Garcia JG. Novel therapies for microvascular permeability in sepsis. Curr Drug Targets. 2007;8:509–514. doi: 10.2174/138945007780362719. [DOI] [PubMed] [Google Scholar]
- Koga S, Morris S, Ogawa S, Liao H, Bilezikian JP, Chen G, Thompson WJ, Ashikaga T, Brett J, Stern DM, et al. TNF modulates endothelial properties by decreasing cAMP. Am J Physiol Cell Physiol. 1995;268:C1104–C1113. doi: 10.1152/ajpcell.1995.268.5.C1104. [DOI] [PubMed] [Google Scholar]
- Lee WL, Slutsky AS. Sepsis and endothelial permeability. N Engl J Med. 2010;363:689–991. doi: 10.1056/NEJMcibr1007320. [DOI] [PubMed] [Google Scholar]
- Lehtonen LA, Antila S, Pentikainen PJ. Pharmacokinetics and pharmacodynamics of intravenous inotropic agents. Clin Pharmacokinet. 2004;43:187–203. doi: 10.2165/00003088-200443030-00003. [DOI] [PubMed] [Google Scholar]
- Lin YC, Adamson RH, Clark JF, Reed RK, Curry FR. Phosphodiesterase 4 inhibition attenuates plasma volume loss and transvascular exchange in volume-expanded mice. J Physiol. 2012;590:309–322. doi: 10.1113/jphysiol.2011.213447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Samardzic H, Adamson RH, Renkin EM, Clark JF, Reed RK, Curry FR. Phosphodiesterase 4 inhibition attenuates atrial natriuretic peptide-induced vascular hyperpermeability and loss of plasma volume. J Physiol. 2011;589:341–353. doi: 10.1113/jphysiol.2010.199588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol Ther. 2006;109:366–398. doi: 10.1016/j.pharmthera.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- Nacul FE, Guia IL, Lessa MA, Tibirica E. The effects of vasoactive drugs on intestinal functional capillary density in endotoxemic rats: intravital video-microscopy analysis. Anesth Analg. 2010;110:547–554. doi: 10.1213/ANE.0b013e3181c88af1. [DOI] [PubMed] [Google Scholar]
- Nehls V, Drenckhahn D. Demonstration of actin filament stress fibers in microvascular endothelial cells in situ. Microvasc Res. 1991;42:103–112. doi: 10.1016/0026-2862(91)90078-p. [DOI] [PubMed] [Google Scholar]
- Netherton SJ, Maurice DH. Vascular endothelial cell cyclic nucleotide phosphodiesterases and regulated cell migration: implications in angiogenesis. Mol Pharmacol. 2005;67:263–272. doi: 10.1124/mol.104.004853. [DOI] [PubMed] [Google Scholar]
- Patterson CE, Lum H, Schaphorst KL, Verin AD, Garcia JG. Regulation of endothelial barrier function by the cAMP-dependent protein kinase. Endothelium. 2000;7:287–308. doi: 10.3109/10623320009072215. [DOI] [PubMed] [Google Scholar]
- Rixen D, Raum M, Bouillon B, Lefering R, Neugebauer E. Base deficit development and its prognostic significance in posttrauma critical illness: an analysis by the trauma registry of the Deutsche Gesellschaft fur unfallchirurgie. Shock. 2001;15:83–89. doi: 10.1097/00024382-200115020-00001. [DOI] [PubMed] [Google Scholar]
- Russell JA. Management of sepsis. Minerva Med99. 2006:431. [Google Scholar]
- Schick M, Isbary T, Schlegel N, Brugger J, Waschke J, Muellenbach R, Roewer N, Wunder C. The impact of crystalloid and colloid infusion on the kidney in rodent sepsis. Intensive Care Med. 2010;36:541–548. doi: 10.1007/s00134-009-1704-0. [DOI] [PubMed] [Google Scholar]
- Schlegel N, Baumer Y, Drenckhahn D, Waschke J. Lipopolysaccharide-induced endothelial barrier breakdown is cAMP dependent in vivo and in vitro. Crit Care Med. 2009;37:1735–1743. doi: 10.1097/CCM.0b013e31819deb6a. [DOI] [PubMed] [Google Scholar]
- Schlegel N, Burger S, Golenhofen N, Walter U, Drenckhahn D, Waschke J. The role of VASP in regulation of cAMP- and Rac 1-mediated endothelial barrier stabilization. Am J Physiol Cell Physiol. 2008;294:C178–C188. doi: 10.1152/ajpcell.00273.2007. [DOI] [PubMed] [Google Scholar]
- Schlegel N, Waschke J. Impaired cAMP and Rac 1 signaling contribute to TNF-α-induced endothelial barrier breakdown in microvascular endothelium. Microcirculation. 2009a;16:521—533. doi: 10.1080/10739680902967427. [DOI] [PubMed] [Google Scholar]
- Schlegel N, Waschke J. VASP is involved in cAMP-mediated Rac 1 activation in microvascular endothelial cells. Am J Physiol Cell Physiol. 2009b;294:C178–C188. doi: 10.1152/ajpcell.00360.2008. [DOI] [PubMed] [Google Scholar]
- Schmidt EP, Damarla M, Rentsendorj O, Servinsky LE, Zhu B, Moldobaeva A, Gonzalez A, Hassoun PM, Pearse DB. Soluble guanylyl cyclase contributes to ventilator-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2008;295:L1056–L1065. doi: 10.1152/ajplung.90329.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seybold J, Thomas D, Witzenrath M, Boral S, Hocke AC, Brger A, Hatzelmann A, Tenor H, Schudt C, Krll M, Schtte H, Hippenstiel S, Suttorp N. Tumor necrosis factor-α-dependent expression of phosphodiesterase 2: role in endothelial hyperpermeability. Blood. 2005;105:3569–3576. doi: 10.1182/blood-2004-07-2729. [DOI] [PubMed] [Google Scholar]
- Shinoda T, Smith CE, Khairallah PA, Fouad-Tarazi FM, Estafanous FG. Effects of propranolol on myocardial performance during acute normovolemic hemodilution. J Cardiothorac Vasc Anesth. 1991;5:15–22. doi: 10.1016/1053-0770(91)90087-a. [DOI] [PubMed] [Google Scholar]
- Sielenkamper AW, Meyer J, Kloppenburg H, Eicker K, Van Aken H. The effects of sepsis on gut mucosal blood flow in rats. Eur J Anaesthesiol. 2001;18:673–678. doi: 10.1046/j.1365-2346.2001.00905.x. [DOI] [PubMed] [Google Scholar]
- Spindler V, Schlegel N, Waschke J. Role of GTPases in control of microvascular permeability. Cardiovasc Res. 2010;87:243–253. doi: 10.1093/cvr/cvq086. [DOI] [PubMed] [Google Scholar]
- Tofovic SP, Zacharia LC, Carcillo JA, Jackson EK. Inhibition of cytokine release by and cardiac effects of type IV phosphodiesterase inhibition in early, profound endotoxaemia in vivo. Clin Exp Pharmacol Physiol. 2000;27:787–792. doi: 10.1046/j.1440-1681.2000.03332.x. [DOI] [PubMed] [Google Scholar]
- Trzeciak S, Dellinger RP, Chansky ME, Arnold RC, Schorr C, Milcarek B, Hollenberg SM, Parrillo JE. Serum lactate as a predictor of mortality in patients with infection. Intensive Care Med. 2007;33:970–977. doi: 10.1007/s00134-007-0563-9. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci. 2008;1123:134–145. doi: 10.1196/annals.1420.016. [DOI] [PubMed] [Google Scholar]
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- Waschke J, Baumgartner W, Adamson RH, Zeng M, Aktories K, Barth H, Wilde C, Curry FE, Drenckhahn D. Requirement of Rac activity for maintenance of capillary endothelial barrier properties. Am J Physiol Heart Circ Physiol. 2004a;286:H394–H401. doi: 10.1152/ajpheart.00221.2003. [DOI] [PubMed] [Google Scholar]
- Waschke J, Drenckhahn D, Adamson RH, Barth H, Curry FE. cAMP protects endothelial barrier functions by preventing Rac-1 inhibition. Am J Physiol Heart Circ Physiol. 2004b;287:H2427–H2433. doi: 10.1152/ajpheart.00556.2004. [DOI] [PubMed] [Google Scholar]
- Waschke J, Drenckhahn D, Adamson RH, Curry FE. Role of adhesion and contraction in Rac 1-regulated endothelial barrier function in vivo and in vitro. Am J Physiol Heart Circ Physiol. 2004c;287:H704–H711. doi: 10.1152/ajpheart.01076.2003. [DOI] [PubMed] [Google Scholar]
- Witzenrath M, Gutbier B, Schmeck B, Tenor H, Seybold J, Kuelzer R, Grentzmann G, Hatzelmann A, van Laak V, Tschernig T, Mitchell TJ, Schudt C, Rosseau S, Suttorp N, Schutte H. Phosphodiesterase 2 inhibition diminished acute lung injury in murine pneumococcal pneumonia. Crit Care Med. 2009;37:584–590. doi: 10.1097/CCM.0b013e3181959814. [DOI] [PubMed] [Google Scholar]
- Wunder C, Brock RW, McCarter SD, Bihari A, Harris K, Eichelbronner O, Potter RF. Inhibition of haem oxygenase activity increases leukocyte accumulation in the liver following limb ischaemia-reperfusion in mice. J Physiol. 2002;540:1013–1021. doi: 10.1113/jphysiol.2001.015446. [DOI] [PMC free article] [PubMed] [Google Scholar]