

Abstract
Inadequate cerebral O2 availability has been proposed to be an important contributing factor to the development of central fatigue during strenuous exercise. Here we tested the hypothesis that supraspinal processes of fatigue would be increased after locomotor exercise in acute hypoxia compared to normoxia, and that such change would be related to reductions in cerebral O2 delivery and tissue oxygenation. Nine endurance-trained cyclists completed three constant-load cycling exercise trials at ∼80% of maximal work rate: (1) to the limit of tolerance in acute hypoxia; (2) for the same duration but in normoxia (control); and (3) to the limit of tolerance in normoxia. Throughout each trial, prefrontal cortex tissue oxygenation and middle cerebral artery blood velocity (MCAV) were assessed using near-infrared spectroscopy and transcranial Doppler sonography, respectively. Cerebral O2 delivery was calculated as the product of arterial O2 content and MCAV. Before and immediately after each trial, twitch responses to supramaximal femoral nerve stimulation and transcranial magnetic stimulation were obtained to assess neuromuscular and cortical function, respectively. Exercise time was reduced by 54% in hypoxia compared to normoxia (3.6 ± 1.3 vs. 8.1 ± 2.9 min; P < 0.001). Cerebral O2 delivery, cerebral oxygenation and maximum O2 uptake were reduced whereas muscle electromyographic activity was increased in hypoxia compared to control (P < 0.05). Maximum voluntary force and potentiated quadriceps twitch force were decreased below baseline after exercise in each trial; the decreases were greater in hypoxia compared to control (P < 0.001), but were not different in the exhaustive trials (P > 0.05). Cortical voluntary activation was also decreased after exercise in all trials, but the decline in hypoxia (Δ18%) was greater than in the normoxic trials (Δ5–9%) (P < 0.05). The reductions in cortical voluntary activation were paralleled by reductions in cerebral O2 delivery. The results suggest that curtailment of exercise performance in acute severe hypoxia is due, in part, to failure of drive from the motor cortex, possibly as a consequence of diminished O2 availability in the brain.
Key points
Processes leading to fatigue occur within the exercising muscle (peripheral fatigue) and the nervous system (central fatigue).
We asked whether central processes of fatigue would be increased after strenuous exercise in environments where oxygen availability is reduced (hypoxia) compared to the same absolute exercise intensity at sea-level.
Our main finding was that the contribution of central processes to fatigue was increased after exercise in hypoxia (equivalent to ∼3800 m above sea-level).
The greater amount of central fatigue in hypoxia was due to suboptimal neural output from the brain and was associated with reductions in oxygen availability.
The findings provide a plausible mechanism for why exercise performance is impaired at high altitude, and might help our understanding of exercise limitation in patients with reduced oxygen delivery to the brain.
Introduction
Muscle fatigue is characterised as an exercise-induced decrease in maximal force production or an inability to sustain further exercise at a required force (Gandevia, 2001). The exact mechanism of fatigue is not known, but presumably involves a complex interaction of peripheral and central processes (Gandevia, 2001; Allen et al. 2008). Peripheral fatigue is defined as a decrease in the force-generating capacity of skeletal muscle due to processes at, or distal to, the neuromuscular junction. Central fatigue is a progressive exercise-induced reduction in voluntary activation or neural drive to the muscle due to failure of the central nervous system to excite or drive motoneurones adequately. When high-intensity locomotor exercise is performed in hypoxia, the rate of development of peripheral fatigue is increased as demonstrated by a decline in the force response to motor nerve stimulation and an increased rate of rise in electromyogram (EMG) signals during the exercise (Amann et al. 2006a,b; Romer et al. 2006, 2007). Due to methodological limitations, however, there is not yet unequivocal evidence that central neural drive declines during exercise in normoxia or that the contribution of central processes to fatigue is increased in hypoxia (Amann & Calbet, 2008; Amann & Kayser, 2009).
Central fatigue has traditionally been identified by delivering a supramaximal stimulus to the peripheral motor nerve during an isometric maximal voluntary contraction (MVC; Merton, 1954). An increase in this ‘superimposed’ twitch after exercise implies a reduction in voluntary activation and hence central fatigue. A limitation of this conventional method, however, is that impairment in voluntary activation can be mediated at any site proximal to the motor axons such that the exact site of central fatigue cannot be determined (Taylor, 2009). Transcranial magnetic stimulation (TMS) has been used to further localise the site of central fatigue (Todd et al. 2003, 2004a). When TMS is delivered over the motor cortex during an MVC, it is common for a small twitch-like increment in force to occur in the contracting muscle. That is, despite maximal effort, motor cortical output at the time of stimulation is insufficient to drive the motoneurones optimally. An increase in this increment after exercise provides evidence of a reduced cortical voluntary activation, thereby indicating the presence of supraspinal fatigue (Gandevia et al. 1996; Todd et al. 2003). TMS has demonstrated reductions in cortical voluntary activation of ankle dorsiflexors after simulated marathon running (Ross et al. 2007) and of knee extensor muscles after intermittent, high-intensity cycling (Sidhu et al. 2009b), but has not been used to evaluate voluntary drive of involved muscles after sustained, high-intensity locomotor exercise in normoxia or after locomotor exercise of any duration in hypoxia.
Recent evidence suggests that reduced O2 availability in the brain might constitute a signal leading to central fatigue (Amann et al. 2007; Subudhi et al. 2007, 2008, 2009a; Rasmussen et al. 2010; Vogiatzis et al. 2011). We recently found that declines in cortical voluntary activation in response to single-limb knee-extensor contractions were greater in acute severe hypoxia compared to normoxia, and that the increased contribution of supraspinal fatigue to the loss of voluntary force occurred in line with the greatest cerebral deoxygenation (Goodall et al. 2010). Furthermore, the reduction in cerebral oxygenation accompanying strenuous locomotor exercise in hypoxia has been associated with a decrease in cortical voluntary activation of a muscle not directly involved in the exercise (Rasmussen et al. 2010). It is not yet known whether curtailment of locomotor exercise in hypoxia is attributable to an increased supraspinal contribution to fatigue consequent to a reduced O2 availability in the brain.
The aim of the present study, therefore, was to evaluate (using TMS) the contribution of central processes to fatigue of the human knee-extensor muscles in response to sustained high-intensity locomotor exercise in normoxia and acute severe hypoxia. A further aim was to determine whether central fatigue in these conditions is associated with restrictions in the provision of O2 to the brain. We hypothesised that the contribution of supraspinal fatigue would be increased after exercise in hypoxia compared to normoxia, and that such change would be related to reductions in cerebral O2 delivery and oxygenation.
Methods
Ethical approval
Written informed consent was obtained and the study conformed to the latest revision of the Declaration of Helsinki. All procedures were approved by Brunel University Research Ethics Committee.
Participants
Nine male endurance-trained cyclists volunteered to participate in the study (mean ± SD age, 28.1 ± 5.5 years; stature, 1.76 ± 0.07 m; body mass, 71.6 ± 8.8 kg; maximum O2 uptake (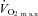 ), 61.1 ± 4.6 ml kg−1 min−1). The participants were non-smokers and free from cardiorespiratory disease. The participants were instructed to arrive at the laboratory in a rested and fully hydrated state, at least 3 h postprandial, and to avoid strenuous exercise in the 48 h preceding each testing session. They were also asked to refrain from caffeine and alcohol for 12 and 24 h before each test, respectively.
), 61.1 ± 4.6 ml kg−1 min−1). The participants were non-smokers and free from cardiorespiratory disease. The participants were instructed to arrive at the laboratory in a rested and fully hydrated state, at least 3 h postprandial, and to avoid strenuous exercise in the 48 h preceding each testing session. They were also asked to refrain from caffeine and alcohol for 12 and 24 h before each test, respectively.
Experimental design
Participants completed two sessions (preliminary and experimental) separated by at least 48 h during a 2 week period. During the preliminary session, the participants were thoroughly familiarised with the methods used to assess neuromuscular function. In addition, they performed a maximal incremental exercise test while breathing room air for the determination of gas-exchange threshold and 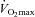 . During the experimental session, the participants performed three constant-load exercise trials, each separated by at least 1 h of rest, at a work rate calculated to require 60% of the difference between the gas-exchange threshold and
. During the experimental session, the participants performed three constant-load exercise trials, each separated by at least 1 h of rest, at a work rate calculated to require 60% of the difference between the gas-exchange threshold and 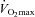 . This intensity was chosen in order that participants would be exercising above critical power (i.e. severe intensity), thereby ensuring attainment of
. This intensity was chosen in order that participants would be exercising above critical power (i.e. severe intensity), thereby ensuring attainment of 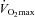 and maximising the tolerable duration of exercise in this domain. The order of exercise trials was: (1) to the limit of tolerance in hypoxia; (2) for the same duration but in normoxia (control); and (3) to the limit of tolerance in normoxia. The order of trials was not randomised as our study focused on the central mechanisms of fatigue, not on the well-established negative performance effects of hypoxia. For the hypoxia trial the participants breathed a humidified gas mixture (inspired O2 fraction (
and maximising the tolerable duration of exercise in this domain. The order of exercise trials was: (1) to the limit of tolerance in hypoxia; (2) for the same duration but in normoxia (control); and (3) to the limit of tolerance in normoxia. The order of trials was not randomised as our study focused on the central mechanisms of fatigue, not on the well-established negative performance effects of hypoxia. For the hypoxia trial the participants breathed a humidified gas mixture (inspired O2 fraction (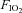 ), 0.13; inspired O2 tension, 92.7 ± 0.6 mmHg; altitude equivalent ∼3800 m above sea level), whereas for the control and normoxic trials the participants breathed room air (
), 0.13; inspired O2 tension, 92.7 ± 0.6 mmHg; altitude equivalent ∼3800 m above sea level), whereas for the control and normoxic trials the participants breathed room air (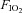 , 0.21). Whilst the conditions were not blinded, the participants were naive to the purpose of the study and unaware of the experimental hypotheses. Before and within 2.5 min after each exercise trial, twitch responses to supramaximal femoral nerve stimulation and TMS were obtained to assess neuromuscular function and voluntary activation. For the hypoxia trial, the post-exercise measurements were made while participants continued to breathe the test gas. Arterial blood, cerebrovascular and cardiorespiratory responses, as well as electromyographic (EMG) activity of the vastus lateralis, were assessed throughout each trial.
, 0.21). Whilst the conditions were not blinded, the participants were naive to the purpose of the study and unaware of the experimental hypotheses. Before and within 2.5 min after each exercise trial, twitch responses to supramaximal femoral nerve stimulation and TMS were obtained to assess neuromuscular function and voluntary activation. For the hypoxia trial, the post-exercise measurements were made while participants continued to breathe the test gas. Arterial blood, cerebrovascular and cardiorespiratory responses, as well as electromyographic (EMG) activity of the vastus lateralis, were assessed throughout each trial.
Preliminary session
Participants were thoroughly familiarised with receiving femoral nerve and cortical stimulation during brief, isometric contractions of the knee-extensors. In addition, the participants performed a maximal incremental exercise test on an electromagnetically braked cycle ergometer (Excalibur Sport, Lode, Groningen, The Netherlands). The test consisted of a 3 min rest period, followed by 3 min of ‘unloaded’ cycling (20 W) and a continuous ramped increase in work rate of 30 W min−1 to the limit of tolerance (20 rpm below self-selected cadence). Respiratory indices were assessed breath-by-breath using an online system (Quark b2, Cosmed, Rome, Italy) and averaged over consecutive 10 s epochs. The gas-exchange threshold was determined online using multiple parallel methods (Wasserman, 1984; Beaver et al. 1986). The 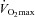 was taken as the highest 30 s mean value attained prior to the participant's volitional exhaustion, and verified using the appended-step method (Rossiter et al. 2006).
was taken as the highest 30 s mean value attained prior to the participant's volitional exhaustion, and verified using the appended-step method (Rossiter et al. 2006).
Force and EMG recordings
Knee-extensor force during voluntary and evoked contractions was measured using a calibrated load cell (ABA Ergo Meter, Globus Italia, Codogne, Italy). The load cell was fixed to a custom-built chair and connected to a non-compliant cuff attached around the participant's right leg just superior to the ankle malleoli. The load cell was adjusted to a height that was in the direct line of applied force. Participants sat upright in the chair with the hips and knees at 1.57 rad (90 deg) of flexion. EMG activity of the knee-extensors was recorded from the vastus lateralis. EMG activity of the knee-flexors was recorded from the lateral head of the biceps femoris. After the skin was shaved and cleaned, surface electrodes (Kendall H59P, Tyco Healthcare Group, Mansfield, MA, USA) were placed 2 cm apart over the muscle bellies. A reference electrode was placed over the patella. The electrodes were used to record the compound muscle action potential (M-wave) elicited by electrical stimulation of the femoral nerve, the motor evoked potential (MEP) elicited by TMS, the root-mean-square amplitude for maximal voluntary contractions (MVCRMS), and the integrated EMG activity of the vastus lateralis during exercise. Signals were amplified (gain 1000; 1902, Cambridge Electronic Design, Cambridge, UK), band-pass filtered (EMG only: 20–2000 Hz), digitised (4 kHz; micro 1401, Cambridge Electronic Design), then acquired and later analysed (Spike2 version 7.01, Cambridge Electronic Design).
Neuromuscular function
Force and EMG variables were assessed before and immediately after each trial. MVC force was determined from three maximal contractions. Femoral nerve stimulation was delivered during each of the contractions and an additional stimulus was delivered at rest, ∼2 s after the superimposed stimulus, to determine potentiated quadriceps twitch force (Qtw,pot) and peripheral voluntary activation (see ‘Data analysis’ below). TMS was delivered during brief (∼5 s) maximal and submaximal voluntary contractions for the determination of cortical voluntary activation. A set of contractions comprised 100, 75 and 50% MVC efforts separated by ∼5 s of rest. Contraction sets were repeated three times, with 15 s between each set. Visual feedback of the target force was provided via a computer monitor.
Femoral nerve stimulation
Single electrical stimuli of 200 μs pulse width were delivered to the right femoral nerve via surface electrode (CF3200, Nidd Valley Medical Ltd, Harrogate, UK) and constant-current stimulator (DS7AH, Digitimer Ltd, Welwyn Garden City, UK). The cathode was positioned over the nerve high in the femoral triangle; the anode was placed midway between the greater trochanter and the iliac crest (Sidhu et al. 2009b; Goodall et al. 2010). The site of stimulation that produced the largest resting twitch amplitude and M-wave (Mmax) was located. Single stimuli were delivered beginning at 100 mA and increasing by 20 mA until plateaus occurred in twitch amplitude and Mmax. Supramaximal stimulation was ensured by increasing the final intensity by 30% (mean current, 278 ± 49 mA). Muscle contractility was assessed for each peripherally derived resting twitch as twitch amplitude (Qtw: peak minus onset force), maximum rate of force development (MRFD), contraction time (CT), maximum relaxation rate (MRR), and one-half relaxation time (RT0.5). Membrane excitability was inferred from the peak-to-peak amplitude and area of the electrically evoked Mmax (Fowles et al. 2002).
Transcranial magnetic stimulation
TMS was delivered via a concave double cone coil (110 mm diameter; maximum output 1.4 T) powered by a mono-pulse magnetic stimulator (Magstim 200, The Magstim Company Ltd, Whitland, UK). The coil was held over the vertex to preferentially stimulate the left hemisphere (postero-anterior intracranial current flow), and was placed in an optimal position to elicit a large MEP in the vastus lateralis and a small MEP in the antagonist (biceps femoris). The optimal coil position (1.4 ± 0.5 cm lateral to the vertex) was marked on the scalp with indelible ink to ensure reproducibility of the stimulation. A stimulus–response curve was constructed to determine resting motor threshold for the knee extensors. Stimulator output was decreased by 5% increments from 80% until the MEP response was below 0.05 mV in more than half of eight stimuli (Sharshar et al. 2004). Resting motor threshold occurred at 52 ± 7% of maximum stimulator output, and during each of the experimental trials TMS was delivered at 130% of resting motor threshold (67 ± 9% maximum stimulator output). This stimulation intensity elicited a large MEP in the vastus lateralis (area between 60 and 100% of Mmax during knee-extensor contractions ≥50% MVC) while causing only a small MEP in the biceps femoris (amplitude ∼20% of MEP during knee-extensor contractions) before and after each trial.
Constant-load exercise
Participants sat on the cycle ergometer (Excalibur Sport) while baseline data were collected for 5 min. In the hypoxic condition the participants were switched to the test gas for 7 min. Inspired air was directed to the participants through 1.8 m of plastic tubing and valve arrangement that delivered compressed, medical-grade, dry gas (BOC, Guilford, UK) via a 500 litre Douglas bag. The gas was humidified by heating water in the bottom of the Douglas bag using a ceramic hotplate (Bibby HB500, Wolf Laboratories Ltd, York, UK). Next, the participants pedalled for 3 min at 20 W before a ‘step’ increase in work rate equivalent to 60% of the difference between gas-exchange threshold and 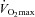 . The work rate was adjusted to accommodate the mean lag time of
. The work rate was adjusted to accommodate the mean lag time of 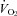 during ramp exercise, assumed to approximate two-thirds of the ramp rate (i.e. 20 W; Whipp et al. 1981). The work rate in all three trials was 292 ± 58 W (77 ± 5% of maximal work rate). The participants remained seated throughout exercise and maintained a target pedal cadence equivalent to that chosen during the incremental exercise test. Task-failure was defined as a 20 rpm drop in the target cadence.
during ramp exercise, assumed to approximate two-thirds of the ramp rate (i.e. 20 W; Whipp et al. 1981). The work rate in all three trials was 292 ± 58 W (77 ± 5% of maximal work rate). The participants remained seated throughout exercise and maintained a target pedal cadence equivalent to that chosen during the incremental exercise test. Task-failure was defined as a 20 rpm drop in the target cadence.
Arterial blood sampling
In those participants who could be successfully catheterised (n = 6), blood was sampled from a radial artery at pre-exercise baseline, after wash-in of the test gas (hypoxia only), after 1 min of severe exercise, every 2 min thereafter and at end-exercise The samples were assessed within 30 min for pH, base excess, O2 tension (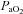 ), CO2 tension (
), CO2 tension ( ), haemoglobin concentration ([Hb]), O2 saturation (
), haemoglobin concentration ([Hb]), O2 saturation (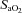 ) and lactate concentration ([Lac]a) using an automated analyser (ABL800 FLEX, Radiometer, Copenhagen, Denmark). Arterial O2 content (
) and lactate concentration ([Lac]a) using an automated analyser (ABL800 FLEX, Radiometer, Copenhagen, Denmark). Arterial O2 content (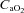 ) was calculated using the equation: ([Hb]× 1.39 ×
) was calculated using the equation: ([Hb]× 1.39 ×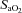 /100) + (
/100) + (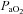 × 0.003).
× 0.003).
Cerebrovascular responses
Cerebral oxygenation was assessed using NIRS (INVOS 5100C, Somanetics, Troy, MI, USA). Two near-infrared sensors (SomaSensor, Somanetics) were placed on the skin over the right and left prefrontal cortex region of the forehead, and the signals were averaged to determine cerebral oxygenation. The sensors were secured to the skin using double-sided adhesive tape and shielded from ambient light using an elastic bandage. The sensors alternately emit near-infrared light at wavelengths of 730 and 810 nm. Each sensor contains two detectors located at 3 and 4 cm from the emitting source that detect oxygenated and deoxygenated states of Hb to estimate regional O2 saturation based on internal algorithms (Rasmussen et al. 2007). The NIRS data were acquired continuously and output every 5 s.
Blood velocity in the middle cerebral artery (MCAV) was determined using transcranial Doppler sonography (Doppler-Box, Compumedics DWL, Singen, Germany). A 2 MHz Doppler probe was positioned over the right middle cerebral artery using previously described search techniques (Aaslid et al. 1982) and was secured with an adjustable headset (DiaMon, Compumedics DWL). The mean depth for Doppler signals was 51 ± 3 mm. Arterial blood pressure was recorded with a transducer located at heart level (TruWave, Edwards Lifesciences, Nyon, Switzerland). Data were sampled at 200 Hz (PowerLab 16/30, ADInstruments Ltd, Oxford, UK) and processed offline (LabChart version 5.4.2, ADInstruments Ltd) for the continuous determination of MCAV and blood pressure. Cerebrovascular conductance (CVC) was calculated by dividing MCAV by blood pressure. Cerebral O2 delivery was calculated as the product of MCAV and 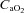 . Changes in MCAV should reflect changes in cerebral blood flow based on evidence that the middle cerebral artery changes minimally in response to hypoxia and hypocapnia (Poulin & Robbins, 1996; Serrador et al. 2000).
. Changes in MCAV should reflect changes in cerebral blood flow based on evidence that the middle cerebral artery changes minimally in response to hypoxia and hypocapnia (Poulin & Robbins, 1996; Serrador et al. 2000).
Cardiorespiratory and perceptual responses
Ventilatory and pulmonary gas exchange indices were assessed breath-by-breath using an online system (Quark b2, Cosmed). Heart rate was assessed beat-by-beat using a short-range telemetry system (Polar Electro Oy, Kempele, Finland) interfaced with the online system. Ratings of perceived exertion for dyspnoea and limb discomfort were obtained at baseline and end-exercise using Borg's modified CR10 scale (Borg, 1998).
Data analysis
Peripheral voluntary activation was quantified using twitch interpolation (Merton, 1954). Briefly, the force produced during a superimposed twitch (SIT) delivered within 0.5 s of attaining peak force during the MVC was compared with the force produced by a single twitch delivered during relaxation ∼2 s after the MVC: voluntary activation (%) = [1 – (SIT/Qtw,pot)]× 100. Cortical voluntary activation was assessed by measuring the force responses to motor-cortex stimulation during submaximal and maximal contractions. Because corticospinal excitability increases during voluntary contraction (Rothwell et al. 1991) it was necessary to estimate, rather than measure directly, the amplitude of the resting twitch evoked by motor-cortex stimulation. Briefly, the y-intercept of the linear regression between the mean amplitude of the three superimposed twitches evoked by TMS and voluntary force recorded during each 100, 75 and 50% MVC was taken as the estimated amplitude of the resting twitch (ERT: Todd et al. 2003, 2004b; Goodall et al. 2009). Cortical voluntary activation (%) was subsequently quantified using the equation: [1 – (SIT/ERT)]× 100. The reliability of TMS for the assessment of cortical voluntary activation and ERT for the knee extensors has been established in our laboratory (Goodall et al. 2009) and elsewhere (Sidhu et al. 2009a).
The peak-to-peak amplitude and area of evoked MEP and Mmax were measured offline. To ensure the motor cortex stimulus was activating a high proportion of the knee-extensor motor units, the area of vastus lateralis MEP was normalised to that of Mmax elicited during the MVC at the beginning of each trial (Taylor et al. 1999). The duration of the cortical silent period evoked by TMS during MVC was quantified as the duration from the point of cortical stimulation to the point when post-stimulus EMG exceeded ±2 SD of pre-stimulus EMG for >100 ms (Goodall et al. 2010). Vastus lateralis EMG signals during exercise were rectified and smoothed (15 ms), then quantified as the mean integrated area during each cycle revolution averaged over each 10 s period of exercise. A computer algorithm identified the onset and offset of activity where the rectified EMG signal deviated >2 SD from baseline for >100 ms.
Statistical analysis
Using a predicted effect size of 1.35 for changes in cortical voluntary activation (Goodall et al. 2010), an a priori power analysis (G*POWER 3 Software) revealed that nine participants would provide 75% power to detect differences at an α-level of 0.05. Two-way repeated measures ANOVA on trial and time was used to test for within-group differences. When ANOVA revealed significant interactions, pairwise contrasts were made using the Bonferroni method. Student's paired t test was used to assess baseline to post-exercise differences for each trial. Multiple regression analysis was used to assess correlations within participants (Bland & Altman, 1995), whereby cortical voluntary activation was treated as the dependent variable, cerebral oxygenation and cerebral O2 delivery as the independent variables, and participant as the categorical variable. Data are reported as means ± SD within the text and as means ± SEM in figures 1–5 and 7. Analyses were performed using SPSS (v. 15.1; SPSS Inc., Chicago, IL, USA). Statistical significance was set at P < 0.05.
Figure 1. Arterial blood parameters.
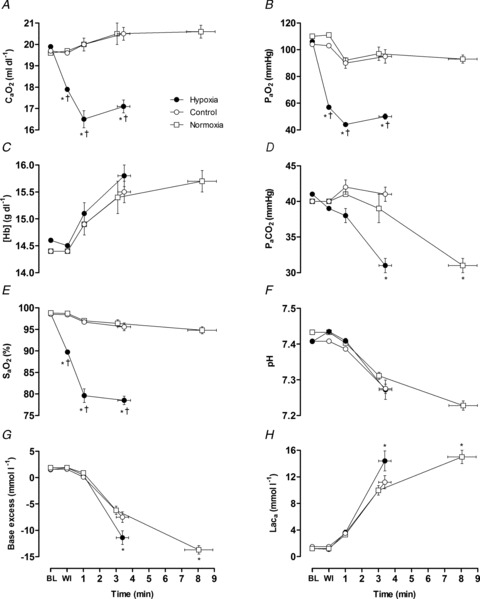
Arterial O2 content (A), O2 tension (B), haemoglobin concentration (C), CO2 tension (D), O2 saturation (E), pH (F), base excess (G) and lactate concentration (H). Values are plotted for pre-exercise baseline (BL), test gas wash-in (WI), for the duration of the shortest trial in that condition and extrapolated to the group mean exercise time. Data are means ± SEM for 6 participants. *P < 0.05 vs. control at the same time point; †P < 0.05 vs. normoxia at end-exercise.
Figure 5. Neuromuscular function.
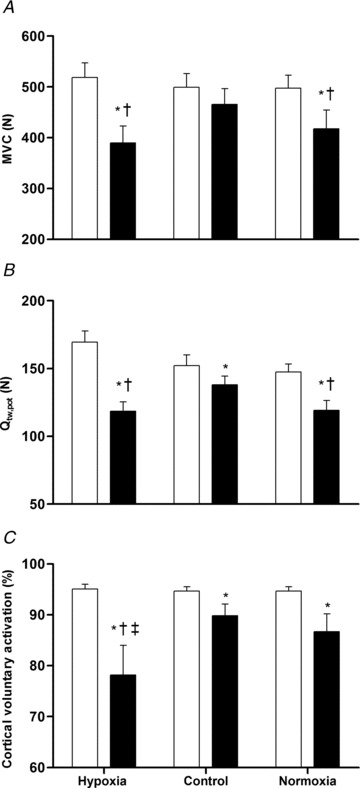
Maximum voluntary contraction (A), potentiated quadriceps twitch force (B) and cortical voluntary activation (C) at pre-exercise baseline (open bars) and immediately after exercise (filled bars). Data are means ± SEM for 9 participants. *P < 0.05 vs. respective baseline value; †P < 0.05 vs. control at the same time point (post-exercise); ‡P < 0.05 vs. normoxia at the same time point (post-exercise).
Figure 7. Relationships between cortical voluntary activation and cerebral haemodynamics.
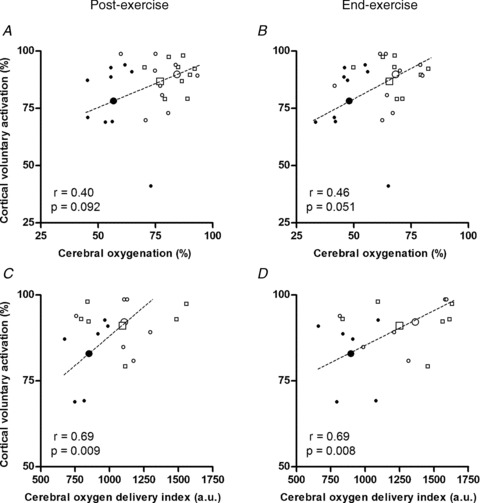
Cortical voluntary activation vs. cerebral oxygenation (A and B) and middle cerebral artery O2 delivery index (C and D). Cerebrovascular responses are shown for the period immediately after (<2.5 min) exercise when motor cortex stimulation was applied (A and C) and for the final minute of exercise (B and D). Data are individual (small symbols) and mean (large symbols) for 9 (A and B) and 6 (C and D) participants in hypoxia (filled circles), control (open circles) and normoxia (open squares). The regression lines are for group mean data. The correlation coefficients (r) and associated P-values are for repeated observations within participants (Bland & Altman, 1995).
Results
Exercise responses
Exercise performance was significantly reduced in hypoxia compared to normoxia (3.6 ± 1.3 vs. 8.1 ± 2.9 min, P < 0.001; −54%). No parameter differed across trials at resting baseline. 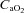 was significantly lower during hypoxic exercise compared to both normoxic trials, in association with a lower
was significantly lower during hypoxic exercise compared to both normoxic trials, in association with a lower 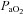 and
and 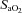 (Fig. 1). Cerebral oxygenation was significantly lower at task failure in hypoxia compared to control (48 ± 9 vs. 68 ± 10%; P < 0.001) and normoxia (48 ± 9 vs. 66 ± 11%; P < 0.001) (Fig. 2). In all participants, MCAV declined at a faster rate from peak values attained during the first 2 min of exercise in hypoxia compared to control (55.1 ± 11.6 vs. 65.4 ± 12.7 cm s−1; P = 0.13) and normoxia (55.1 ± 11.6 vs. 58.5 ± 14.2 cm s−1; P = 0.23) (Fig. 2). Consequently, cerebral O2 delivery was significantly lower during hypoxic exercise compared to the normoxic trials (Fig. 3). Mean arterial pressure and CVC did not differ across trials at baseline or during hypoxic wash-in. Mean arterial pressure rose throughout exercise by ∼30% (pooled mean = 136 ± 10 mmHg), but did not differ across trials at any time point. CVC followed similar kinetic responses to MCAV, increasing during the first 2 min of exercise before declining below baseline in hypoxia and normoxia.
(Fig. 1). Cerebral oxygenation was significantly lower at task failure in hypoxia compared to control (48 ± 9 vs. 68 ± 10%; P < 0.001) and normoxia (48 ± 9 vs. 66 ± 11%; P < 0.001) (Fig. 2). In all participants, MCAV declined at a faster rate from peak values attained during the first 2 min of exercise in hypoxia compared to control (55.1 ± 11.6 vs. 65.4 ± 12.7 cm s−1; P = 0.13) and normoxia (55.1 ± 11.6 vs. 58.5 ± 14.2 cm s−1; P = 0.23) (Fig. 2). Consequently, cerebral O2 delivery was significantly lower during hypoxic exercise compared to the normoxic trials (Fig. 3). Mean arterial pressure and CVC did not differ across trials at baseline or during hypoxic wash-in. Mean arterial pressure rose throughout exercise by ∼30% (pooled mean = 136 ± 10 mmHg), but did not differ across trials at any time point. CVC followed similar kinetic responses to MCAV, increasing during the first 2 min of exercise before declining below baseline in hypoxia and normoxia.
Figure 2. Cerebrovascular responses.
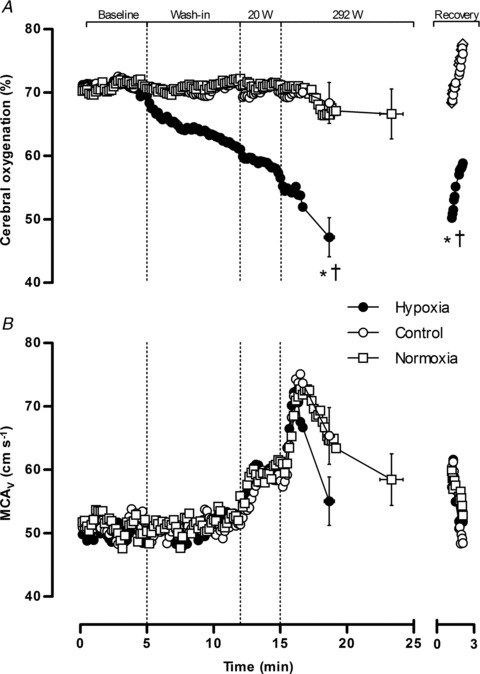
Cerebral oxygenation (A) and middle cerebral artery blood velocity (B). Data are averaged over 10 s epochs for pre-exercise baseline, test gas wash-in, unloaded cycling (20 W), severe constant-load exercise (292 W) and passive recovery. Values are plotted for the duration of the shortest trial in that condition and extrapolated to the group mean exercise time. Data are means ± SEM for 9 participants. *P < 0.05 vs. control at end-exercise or recovery; †P < 0.05 vs. normoxia at end-exercise or recovery.
Figure 3. Cerebral O2 delivery.
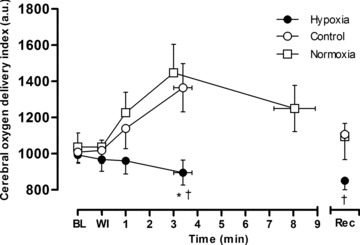
Middle cerebral artery O2 delivery index (MCAV×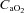 ) for pre-exercise baseline (BL), test gas wash-in (WI), severe constant-load exercise (292 W) and <2.5 min after exercise (Rec). Values are plotted for the duration of the shortest trial in that condition and extrapolated to the group mean exercise time. Data are means ± SEM for 6 participants. *P < 0.05 vs. control at end-exercise; †P < 0.05 vs. normoxia at end-exercise or recovery.
) for pre-exercise baseline (BL), test gas wash-in (WI), severe constant-load exercise (292 W) and <2.5 min after exercise (Rec). Values are plotted for the duration of the shortest trial in that condition and extrapolated to the group mean exercise time. Data are means ± SEM for 6 participants. *P < 0.05 vs. control at end-exercise; †P < 0.05 vs. normoxia at end-exercise or recovery.
Whole body 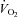 was not different across the three trials during unloaded cycling and the first 2 min of strenuous exercise, but was significantly reduced at end-exercise in hypoxia compared to control (Δ0.46 ± 0.12 L min−1, P = 0.002; −13%) and normoxia (Δ0.90 ± 0.18 L min−1, P = 0.001; −29%) (Fig. 4). Conversely, iEMG at end-exercise was significantly higher in hypoxia compared to control (P < 0.001; 80%) and tended to be higher compared to normoxia (P = 0.059; 16%). Dyspnoea and limb discomfort at end-exercise were generally higher in hypoxia compared to control (P = 0.066 and 0.030, respectively), but were not different compared to normoxia (Table 1).
was not different across the three trials during unloaded cycling and the first 2 min of strenuous exercise, but was significantly reduced at end-exercise in hypoxia compared to control (Δ0.46 ± 0.12 L min−1, P = 0.002; −13%) and normoxia (Δ0.90 ± 0.18 L min−1, P = 0.001; −29%) (Fig. 4). Conversely, iEMG at end-exercise was significantly higher in hypoxia compared to control (P < 0.001; 80%) and tended to be higher compared to normoxia (P = 0.059; 16%). Dyspnoea and limb discomfort at end-exercise were generally higher in hypoxia compared to control (P = 0.066 and 0.030, respectively), but were not different compared to normoxia (Table 1).
Figure 4. Whole-body O2 uptake and vastus lateralis integrated electromyographic activity.
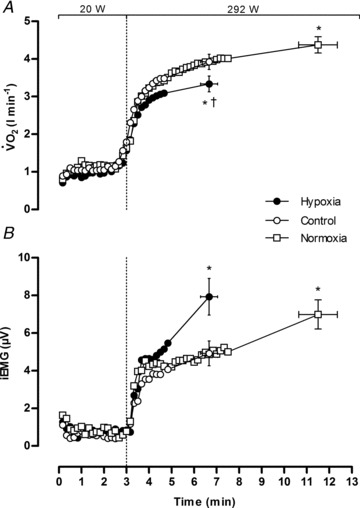
Data are averaged over 10 s epochs for unloaded cycling (20 W) and severe constant-load exercise (292 W). Values are plotted for the duration of the shortest trial in that condition and extrapolated to the group mean exercise time. Data are means ± SEM for 9 participants. *P < 0.05 vs. control at end-exercise; †P < 0.05 vs. normoxia at end-exercise.
Table 1.
Cardiorespiratory and perceptual responses at rest and during the final minute of constant-load exercise
| Hypoxia | Control | Normoxia | ||
|---|---|---|---|---|
| Exercise time (min) | 3.6 ± 1.3† | 3.6 ± 1.3† | 8.1 ± 2.9 | |
| HR (b min−1) | Rest | 72 ± 9*† | 65 ± 11 | 65 ± 10 |
| Final | 167 ± 14† | 165 ± 15† | 179 ± 13 | |
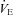 (l min−1) (l min−1) |
Rest | 15 ± 4 | 15 ± 5 | 14 ± 2 |
| Final | 158 ± 27* | 116 ± 16† | 160 ± 25 | |
| fR (b min−1) | Rest | 13.8 ± 2.5 | 15.2 ± 1.8 | 13.0 ± 3.4 |
| Final | 58.7 ± 13.8* | 39.5 ± 7.0† | 62.6 ± 1.8 | |
| VT (l) | Rest | 1.32 ± 0.56 | 1.03 ± 0.37 | 1.18 ± 0.58 |
| Final | 3.01 ± 0.60 | 3.20 ± 0.40 | 2.70 ± 0.50 | |
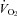 (l min−1) (l min−1) |
Rest | 0.47 ± 0.13 | 0.48 ± 0.87 | 0.46 ± 0.66 |
| Final | 4.50 ± 0.69 | 4.32 ± 0.45 | 4.44 ± 0.69 | |
| RPE, dyspnoea | Rest | 0.1 ± 0.1 | 0.0 ± 0.1 | 0.0 ± 0.1 |
| Final | 8.1 ± 2.4 | 6.2 ± 1.3† | 9.1 ± 0.7 | |
| RPE, limb | Rest | 0.0 ± 0.0 | 0.1 ± 0.3 | 0.2 ± 0.4 |
| Final | 8.7 ± 1.9* | 6.4 ± 1.7† | 9.2 ± 0.8 |
Values are means ± SD for 9 participants. HR, heart rate. 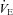 , minute ventilation; fR, respiratory frequency; VT, tidal volume;
, minute ventilation; fR, respiratory frequency; VT, tidal volume; 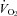 , O2 uptake;
, O2 uptake; 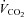 , CO2 output; RPE, ratings of perceived exertion. Note: resting values for hypoxia were measured during the final minute of the hypoxic wash-in; only HR increased from values attained before the wash-in (62 ± 12 beats min−1, P < 0.05).
, CO2 output; RPE, ratings of perceived exertion. Note: resting values for hypoxia were measured during the final minute of the hypoxic wash-in; only HR increased from values attained before the wash-in (62 ± 12 beats min−1, P < 0.05).
P < 0.05 vs. control at the same time point;
P < 0.05 vs. normoxia at the same time point.
Pre- and post-exercise responses
Peripheral responses
The effects of condition on neuromuscular responses to exercise are shown in Table 2. Baseline measures of neuromuscular function were not different across trials, except for Qtw,pot and ERT, which were significantly lower in the normoxic trials compared to hypoxia. After exercise, MVC force and Qtw,pot were reduced below baseline in all three trials; however, the reductions were greater in hypoxia and normoxia compared to control (MVC: −25 and −17%vs. −7%; Qtw,pot: −30 and −19%vs. −9%). ERT was also reduced below baseline after exercise in all three trials, and the decreases in hypoxia and normoxia exceeded those in the control trial. In line with the changes in Qtw,pot and ERT, there were alterations in several within-twitch measures of muscle contractility (MRFD, CT, MRR and RT0.5) that were generally greater in hypoxia compared to the normoxic trials. There was a trend for MVCRMS to be reduced after exercise in hypoxia (P = 0.071; −26%), but not in control (P = 0.24; −9%) or normoxia (P = 0.56; −6%). Conversely, there were no differences across trials in measures of membrane excitability (Mmax amplitude and area).
Table 2.
Neuromuscular function before and immediately after constant-load exercise
| Hypoxia | Control | Normoxia | ||
|---|---|---|---|---|
| MVC | Pre | 519 ± 87 | 499 ± 81 | 498 ± 76 |
| (N) | Post | 390 ± 97* | 466 ± 93† | 418 ± 110 |
| MVCRMS | Pre | 0.38 ± 0.19 | 0.34 ± 0.18 | 0.34 ± 0.13 |
| (mV) | Post | 0.28 ± 0.15 | 0.31 ± 0.17 | 0.32 ± 0.12 |
| Qtw,pot | Pre | 170 ± 25*† | 152 ± 23 | 148 ± 18 |
| (N) | Post | 119 ± 21* | 138 ± 21† | 119 ± 22 |
| ERT | Pre | 154 ± 42*† | 130 ± 34 | 128 ± 34 |
| (N) | Post | 82 ± 30* | 104 ± 28† | 82 ± 21 |
| MRFD | Pre | 5.0 ± 1.4 | 3.8 ± 0.9 | 3.6 ± 0.6 |
| (N ms−1) | Post | 2.7 ± 0.5*† | 3.4 ± 0.7 | 3.1 ± 0.5 |
| CT | Pre | 90 ± 6 | 89 ± 11 | 87 ± 13 |
| (ms) | Post | 92 ± 7 | 83 ± 12 | 85 ± 12 |
| MRR | Pre | −1.74 ± 0.43 | −1.55 ± 0.42 | −1.67 ± 0.57 |
| (N ms−1) | Post | −0.93 ± 0.24* | −1.21 ± 0.33 | −1.25 ± 0.57 |
| RT0.5 | Pre | 73 ± 9 | 71 ± 11 | 73 ± 12 |
| (ms) | Post | 98 ± 22*† | 84 ± 17 | 85 ± 12 |
| Mmax amplitude | Pre | 6.3 ± 2.5 | 6.2 ± 2.5 | 6.2 ± 2.3 |
| (mV) | Post | 6.8 ± 2.9 | 6.2 ± 3.2 | 6.5 ± 2.9 |
| Mmax area | Pre | 61 ± 29 | 57 ± 26 | 57 ± 23 |
| (μV s−1) | Post | 67 ± 26 | 60 ± 28 | 64 ± 25 |
| Cortical VA | Pre | 95.1 ± 2.7 | 94.7 ± 2.5 | 94.7 ± 2.5 |
| (%) | Post | 78.2 ± 17.4*† | 89.8 ± 7.0 | 86.7 ± 10.5 |
| Peripheral | Pre | 93.6 ± 3.0 | 93.5 ± 4.2 | 92.1 ± 4.5 |
| VA (%) | Post | 87.3 ± 7.5 | 89.0 ± 7.2 | 86.8 ± 7.6 |
| CSP | Pre | 244 ± 81 | 236 ± 68 | 235 ± 83 |
| (ms) | Post | 250 ± 70 | 233 ± 72 | 220 ± 62 |
Values are means ± SD for 9 participants. MVC, maximal voluntary contraction; MVCRMS, root-mean-square amplitude during MVC; Qtw,pot, potentiated quadriceps twitch; ERT, estimated resting twitch derived using cortical stimulation; MRFD, maximum rate of force development; CT, contraction time; MRR, maximum relaxation rate; RT0.5, one-half relaxation time; Mmax, maximum M-wave; VA, voluntary activation; CSP, cortical silent period.
P < 0.05 vs. control at the same time-point;
P < 0.05 vs. normoxia at the same time-point.
Central responses
Cortical voluntary activation after exercise was reduced below baseline in hypoxia (P = 0.016; Δ18%), control (P = 0.028; Δ5%) and normoxia (P = 0.030; Δ9%). In contrast to the aforementioned reductions in MVC, Qtw and ERT, the decreases in cortical voluntary activation were greater in hypoxia compared to both control (P = 0.024) and normoxia (P = 0.047) (Fig. 5). Voluntary activation at baseline estimated using motor nerve stimulation was comparable to that derived using TMS (∼93 vs. 95%). After exercise, these peripherally derived estimates of voluntary activation were reduced below baseline (P < 0.027), but did not differ across trials (P = 0.38; Table 2). Corticospinal excitability (MEP area) during each contraction intensity (100, 75 and 50% MVC) was increased by ∼20% after exercise in hypoxia (P < 0.025; Fig. 6), but was unchanged in the normoxic trials. Cortical inhibition (cortical silent period) during maximal contractions did not change in any of the trials (P = 0.64; Table 2).
Figure 6. Representative EMG recordings.
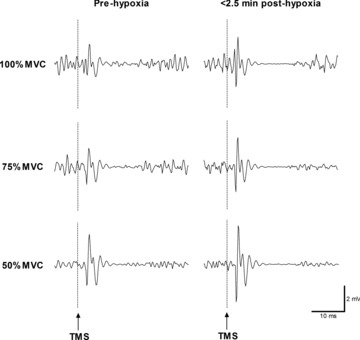
Data show motor evoked potentials (MEP) recorded from the vastus lateralis in response to TMS at 100, 75 and 50% MVC before and immediately after exercise in hypoxia in a single participant. Note the larger MEP elicited by TMS after hypoxia. MEP was not altered after the normoxic trials (data not shown).
Relationships between cortical voluntary activation and cerebral haemodynamics
There were positive relationships between cortical voluntary activation, cerebral oxygenation and O2 delivery measured after exercise (Fig. 7A and C). Similar relationships were apparent when cortical voluntary activation, assumed to be the same at end-exercise as after the exercise, was compared to the cerebral oxygenation and O2 delivery measured during the final minute of exercise (Fig. 7B and D).
Discussion
The main finding was that supraspinal fatigue contributed to the loss of voluntary force after sustained high-intensity locomotor exercise in normoxia and acute severe hypoxia. Cortical voluntary activation declined after exercise in both conditions, but the decline was more than twofold greater for exercise performed to the limit of tolerance in hypoxia compared to the same duration of exercise in normoxia, and onefold greater compared to exercise performed to the limit of tolerance in normoxia. Importantly, the declines in cortical voluntary activation occurred in parallel with reductions in cerebral O2 delivery and oxygenation. Thus, we confirm our hypothesis that acute severe hypoxia exaggerates the supraspinal component of central fatigue in healthy humans, possibly through mechanisms sensitive to reduced O2 availability in the brain.
Fatigue during normoxic and hypoxic exercise: central and peripheral components
A key aim of this study was to determine whether a depression in central neural output to exercising muscles contributes to fatigue in normoxic and hypoxic exercise. Motor cortex stimulation has been used to show that supraspinal fatigue of the knee-extensor muscles is present in normoxia after brief and sustained isometric contractions (Goodall et al. 2009; Sidhu et al. 2009a) as well as intermittent locomotor exercise (Sidhu et al. 2009b). We have also used this technique to show that supraspinal fatigue after intermittent isometric contractions is exacerbated in acute severe hypoxia (Goodall et al. 2010). The present study is the first to use motor cortex stimulation to explore whether there is a supraspinal contribution to fatigue of the knee extensors after sustained locomotor exercise. We found that exercise in normoxia elicited reductions in maximal voluntary force, force evoked by supramaximal stimulation of the femoral nerve, and voluntary activation determined using motor cortex stimulation; these reductions were greater in acute severe hypoxia compared to exercise of the same duration and absolute intensity in normoxia (Fig. 5). Thus, high-intensity, sustained locomotor exercise elicited peripheral as well as central (supraspinal) fatigue, and hypoxia accelerated the development of both components. The reduction in cortical voluntary activation was twofold greater after exercise in hypoxia compared to control. This further reduction in cortical voluntary activation indicates that strenuous exercise in severe hypoxia reduces the capacity of the motor cortex to drive knee-extensor muscles. Whilst several studies have suggested that central motor drive might be affected in severe hypoxia (Amann et al. 2007; Subudhi et al. 2007, 2008, 2009a; Vogiatzis et al. 2011), our study is the first to actually demonstrate an increased contribution of supraspinal processes to fatigue after locomotor exercise in this condition.
As expected, there was a significant reduction in exercise performance in hypoxia. This was accompanied by a post-exercise decrease in maximal voluntary force that was not different compared to normoxia (Fig. 5). Thus, there were near-identical levels of muscle fatigue in hypoxia and normoxia at the limit of exercise tolerance. After both of the exhaustive trials there was a further increment in knee-extensor force elicited by cortical stimulation during voluntary contractions, signifying that supraspinal fatigue contributed to the reduction in voluntary force production. Furthermore, there was a greater reduction in cortical voluntary activation in hypoxia compared to normoxia despite apparent similar levels of peripheral fatigue. The similar force output (peripheral twitch and MVC) with different levels of cortical voluntary activation suggest that the muscle may have been more effective during hypoxia. Relaxation and contraction rates of the twitch decreased with fatigue in all three conditions, but the changes were most pronounced in hypoxia (Table 2). Thus, the additional slowing of the muscle in hypoxia may have optimised force for a similar rate of motoneurone firing (Barry & Enoka, 2007).
Because the relationship between force output and cortical voluntary activation of knee-extensor muscles is linear between 50 and 100% of MVC (Sidhu et al. 2009b; Goodall et al. 2010), it was possible to determine the contribution of supraspinal fatigue to the total force loss. MVC force decreased to 75% of baseline after exercise in hypoxia and 83% in normoxia, whereas cortical voluntary activation dropped by 18 and 9%, respectively. In the absence of supraspinal fatigue, the MVC would have only dropped to 89% in hypoxia and 90% in normoxia. The remainder of the drop in voluntary force was due to a reduced cortical voluntary activation, such that supraspinal fatigue contributed 56% of the overall force loss in hypoxia and 41% of the overall force loss in normoxia. These findings indicate that exercise in hypoxia exacerbated the supraspinal component of central fatigue and increased the relative contribution of supraspinal fatigue to the total force loss.
Neither MEP area nor the ongoing period of EMG silence was altered as a consequence of exercise in normoxia. This implies that sustained, high-intensity locomotor exercise does not impair the responsiveness of neurones in the pathway from motor cortex to muscle output. As output from the motor cortex was insufficient for complete muscle activation after exercise, it is likely that the physiological signals which reduced voluntary drive acted upstream of the motor cortical outputs (see ‘Neural drive and O2 availability’). In contrast, hypoxia elicited a small but significant increase in the MEP response to cortical stimulation during each of the contraction intensities after exercise. Whilst transient increases in MEP and cortical silent period have been noted during maximal sustained contractions (Taylor et al. 1996), changes are generally not evident after fatiguing intermittent contractions (Goodall et al. 2010) or short-duration, high-intensity locomotor exercise (Sidhu et al. 2009b). The observed increase in MEP after exercise in hypoxia was unlikely to be due to alterations in muscle fibre action potential transmission as M-wave characteristics were not different across conditions. The increase might have been due to alterations in motoneurone firing. There was a 26% reduction in maximal EMG in hypoxia, whereas smaller reductions were noted in control and normoxia (9 and 6%, respectively). Since a reduction in maximal EMG is indicative of decreased motoneurone firing rates (Garland & McComas, 1990), the increased MEP after exercise in hypoxia might have been a compensation for reduced spinal excitability. Possible mechanisms include an enhanced descending excitatory output from the motor cortex and/or changes in the intrinsic properties of motoneurones (Taylor et al. 2006).
Neural drive and O2 availability
An important question arising from the effects of exercise on cortical function is whether these events were a consequence of metabolic disturbances in peripheral tissues and/or alterations in cerebral O2 availability. The aforementioned changes in stimulated force (i.e. peripheral fatigue) were accompanied by similar changes in quadriceps iEMG during exercise under comparable experimental conditions. Specifically, iEMG increased throughout exercise in both normoxia and hypoxia, but did not differ between conditions at the limit of exercise tolerance (Fig. 4B). That exercise time was significantly shorter in hypoxia for the same end-exercise iEMG implies that the accelerated rise in iEMG was the result of an increased motor unit recruitment associated with a shift upwards in the relative intensity of exercise, as evidenced by the 13–29% decrease in whole-body 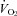 in this condition (Fig. 4A). The increased intensity of exercise in hypoxia would have been brought about by an increase in the percentage of type II fibres activated (Taylor et al. 1997). Heightened recruitment of type II fibres is known to accelerate the accumulation of muscle metabolites, confirmed in the present study by the quicker exercise-induced rise in blood lactate concentration in hypoxia (Fig. 1H). Firing of group III and IV muscle afferents is increased in hypoxia (Hill et al. 1992; Arbogast et al. 2000). In turn, there is evidence that these metabosensitive afferents act upstream of the motor cortex to inhibit voluntary descending drive (Gandevia, 1998; Martin et al. 2006) and influence the magnitude of central motor drive during high-intensity locomotor exercise (Amann et al. 2009, 2011). Collectively, these findings suggest that exercise-induced disturbances in the metabolic milieu of fatiguing muscles might have contributed, in part, to the reduction in cortical voluntary activation and exercise limitation in normoxia and to the additional supraspinal fatigue and curtailment of exercise performance in hypoxia.
in this condition (Fig. 4A). The increased intensity of exercise in hypoxia would have been brought about by an increase in the percentage of type II fibres activated (Taylor et al. 1997). Heightened recruitment of type II fibres is known to accelerate the accumulation of muscle metabolites, confirmed in the present study by the quicker exercise-induced rise in blood lactate concentration in hypoxia (Fig. 1H). Firing of group III and IV muscle afferents is increased in hypoxia (Hill et al. 1992; Arbogast et al. 2000). In turn, there is evidence that these metabosensitive afferents act upstream of the motor cortex to inhibit voluntary descending drive (Gandevia, 1998; Martin et al. 2006) and influence the magnitude of central motor drive during high-intensity locomotor exercise (Amann et al. 2009, 2011). Collectively, these findings suggest that exercise-induced disturbances in the metabolic milieu of fatiguing muscles might have contributed, in part, to the reduction in cortical voluntary activation and exercise limitation in normoxia and to the additional supraspinal fatigue and curtailment of exercise performance in hypoxia.
On the other hand, the greater reduction in cortical voluntary activation after exhaustive exercise in hypoxia versus normoxia was accompanied by comparable levels of metabolite accumulation and peripheral fatigue. It seems unlikely, therefore, that the increased contribution of supraspinal processes to fatigue in hypoxia was due to inhibitory neural feedback from metabolic disturbances in fatiguing muscles. Rather, we contend that the additional decrease in cortical motor drive in hypoxia was the result of reduced O2 availability in the brain. Cerebral blood flow velocity was unaffected by the hypoxic wash-in, increased early during exercise in both conditions, and then decreased towards baseline; in hypoxia, however, the decline occurred at a faster rate than during the normoxic trials (Fig. 2B), in agreement with previous findings during exhaustive heat stress exercise compared to control trials (Gonzalez-Alonso et al., 2004). Whilst some investigators have found a significant increase in cerebral blood flow velocity at rest and during moderate exercise (e.g. Rasmussen et al. 2010; Vogiatzis et al. 2011), this is not a universal finding (e.g. Imray et al. 2005; Rasmussen et al. 2007; Subudhi et al. 2008, 2009b). The reasons for the discrepancy are not entirely clear, but might include the severity of hypoxia, the method of assessment, and/or the mitigating effect of hyperventilation-induced hypocapnia when CO2 is uncontrolled. The faster fall in cerebral blood flow velocity during exercise in hypoxia occurred in parallel with a reduction in 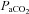 , a pattern that was not observed in control (Fig. 1D). It is well established that cerebral blood flow is highly sensitive to changes in
, a pattern that was not observed in control (Fig. 1D). It is well established that cerebral blood flow is highly sensitive to changes in 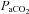 (Brugniaux et al. 2007). Furthermore, the reduction in
(Brugniaux et al. 2007). Furthermore, the reduction in 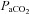 during hypoxia occurred in parallel with a reduction in cerebral vascular conductance, indicative of vasoconstriction (Claassen et al. 2007). No differences were evident in arterial pressure. Thus, the reduction in cerebral vascular conductance was primarily a result of the reduction in cerebral blood flow, which ultimately challenged cerebral O2 delivery (Fig. 3). The finding of lower cerebral O2 delivery towards the end of sustained high-intensity exercise in hypoxia compared to normoxia is in accordance with a recent report on frontal cerebral cortex O2 delivery during discontinuous incremental exercise (Vogiatzis et al. 2011).
during hypoxia occurred in parallel with a reduction in cerebral vascular conductance, indicative of vasoconstriction (Claassen et al. 2007). No differences were evident in arterial pressure. Thus, the reduction in cerebral vascular conductance was primarily a result of the reduction in cerebral blood flow, which ultimately challenged cerebral O2 delivery (Fig. 3). The finding of lower cerebral O2 delivery towards the end of sustained high-intensity exercise in hypoxia compared to normoxia is in accordance with a recent report on frontal cerebral cortex O2 delivery during discontinuous incremental exercise (Vogiatzis et al. 2011).
In contrast to the response for cerebral blood flow velocity, cerebral oxygenation decreased below baseline during wash-in of the hypoxic test gas and decreased further with exercise (Fig. 2A); the reductions were marginal during the normoxic trials. Importantly, the reduction in cerebral oxygenation (and O2 delivery) measured either immediately after exercise, when motor cortex stimulation was applied, or during the period immediately prior to exercise termination was associated with the aforementioned declines in cortical voluntary activation (Fig. 7). This finding suggests that the relative importance of inhibitory feedback on central motor output was diminished in the face of severe cerebral hypoxaemia. That hypoxia exacerbated the reduction in cerebral oxygenation during constant-load exercise is in agreement with studies that have used incremental exercise protocols (Subudhi et al. 2007, 2008, 2009a; Vogiatzis et al. 2011). Subudhi et al. (2009a), for example, found that overall patterns of cortical deoxygenation during strenuous exercise were similar in prefrontal, premotor and motor regions. Since deoxygenation occurs within motor regions during strenuous exercise in hypoxia, it seems plausible that hypoxia influences cortical motor output and limits exercise performance.
Our finding of a link between cerebral oxygenation and cortical motor output is in accordance with studies that have shown an increase in exercise capacity with reversal of arterial deoxygenation in acute severe hypoxia (Amann et al. 2007; Subudhi et al. 2008). A drawback of reoxygenation studies, however, is that increasing arterial oxygenation to normoxic resting levels affects not only the brain but the entire organism, such that other mechanisms might also be involved. More direct support for our proposed link between cerebral oxygenation and central motor drive stems from the recent finding that cortical voluntary activation of an uninvolved muscle is reduced during strenuous exercise in acute hypoxia in parallel with cerebral deoxygenation and alterations in cerebral metabolism (Rasmussen et al., 2010). The exact mechanism for an effect of cerebral O2 availability on cortical motor output is unknown, but alterations in the turnover of several brain neurotransmitters, including serotonin, dopamine and noradrenaline, have been suggested as significant determinants of central fatigue (Roelands & Meeusen, 2010). Whilst our data do not allow us to speak directly to these possibilities, the positive relationships between cortical voluntary activation, cerebral oxygenation and O2 delivery suggest that a link does exists between central motor drive and cerebral O2 availability.
Technical considerations
Motor nerve estimates of voluntary activation were reduced after exercise in hypoxia and normoxia, but were not different between conditions (Table 2). Whilst this is a common finding (Amann et al. 2006a,b; Romer et al. 2006, 2007), it is difficult to reconcile with our one- to twofold greater reduction in cortical voluntary activation (measured using TMS) after exercise in hypoxia compared to normoxia. This mismatch between falls in voluntary activation measured with motor nerve and motor cortical stimulation is consistent with previous studies that have applied these techniques to the knee extensors (Sidhu et al. 2009b; Goodall et al. 2010) and the elbow flexors (Todd et al. 2003). A problem with making direct quantitative comparisons of voluntary activation derived using these techniques is that both types of stimuli can activate different muscles and the shapes of the voluntary-force versus superimposed-twitch relationships differ (Todd et al. 2003; Taylor et al. 2006). A non-linear relationship is evident when using motor nerve stimulation at high contraction strengths, such that changes in voluntary force elicit minimal changes in superimposed twitch size (Allen et al. 1998; Todd et al. 2003). Additionally, impairments in voluntary activation identified using peripheral motor nerve stimulation might be mediated at any site proximal to the motor axons. Thus, the exact site of failure cannot be determined (see also Introduction). Moreover, the validity of this method for the assessment of voluntary activation, and hence central fatigue, has recently been questioned. Briefly, Taylor (2009) argued that whilst motor nerve estimates of voluntary activation do quantify how much of a muscle's total force is produced by volition, the method does not measure descending drive to the motoneurones or take into account the non-linear input–output relation of the motoneurone pool (see also above).
In line with other studies that have measured exercise-induced fatigue of the knee extensors (e.g. Amann et al. 2006a, 2009), our fatigue measurements were completed within 2.5 min after exercise termination. Corticospinal excitability has been shown to recover within 1 min after exercise (Taylor et al. 1996). Thus, the current experimental design might have been insufficient to capture all elements of central fatigue. However, the time taken to assess fatigue after exercise was kept constant for all three trials and even after >1 min of recovery there was still significant supraspinal fatigue, testifying to the robustness of the response.
A further consideration is the slight, albeit significant, reduction in baseline potentiated twitch force across trials. Due to the invasiveness of our procedures (an arterial catheter was in place for several hours), it was neither feasible nor ethical to increase the recovery between trials beyond 1 h. Importantly, comparisons were for the absolute values after exercise rather than the percentage changes pre- to post-exercise. Hence, the slight reduction in baseline twitch force (i.e. peripheral fatigue) did not affect our overall conclusion that cortical voluntary activation (i.e. central fatigue) was suppressed more after exercise in hypoxia versus normoxia.
Conclusions
The novel finding was that supraspinal fatigue was greater in acute severe hypoxia compared to normoxia. Cortical voluntary activation declined after exercise in both conditions, but the decline was one- to twofold greater in hypoxia compared to normoxia. In line with our hypothesis, the declines in cortical voluntary activation occurred in parallel with reductions in cerebral O2 delivery and oxygenation. Thus, the observed decrease in exercise performance in hypoxia might have been related, in part, to suboptimal output from the motor cortex, possibly as a consequence of reduced O2 availability in the brain. The findings provide a plausible mechanism for the curtailment of exercise in severe hypoxia and might help us to understand exercise limitation in patients with impaired cerebrovascular oxygenation.
Acknowledgments
We thank Dr Kameljit Kalsi and Mr Christopher Stock for assistance during data collection.
Glossary
Abbreviations
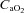
arterial O2 content
- CT
contraction time
- CVC
cerebrovascular conductance
- ERT
estimated resting twitch
- fR
respiratory frequency
- [Hb]
haemoglobin concentration
- iEMG
integrated electromyographic activity
- MCAV
middle cerebral artery blood velocity
- MVC
maximum voluntary contraction
- MEP
motor evoked potential
- MRFD
maximum rate of force development
- MRR
maximum relaxation rate
- Mmax
maximum M-wave
- Qtw,pot
potentiated quadriceps twitch force
- rms
root mean square
- RT0.5
one-half relaxation time
- SIT
superimposed twitch
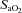
arterial O2 saturation
- TMS
transcranial magnetic stimulation
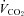
carbon dioxide output
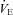
minute ventilation
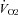
O2 uptake
- VT
tidal volume
Author contributions
All of the experiments were performed at the Centre for Sports Medicine and Human Performance, Brunel University. S.G. contributed to study design, data collection, data analysis, data interpretation and manuscript drafting. J.G.-A. contributed to study design, data collection, data interpretation and manuscript revision. L.A. performed invasive catheteristion procedures and provided medical support. E.R. contributed to conception and design of the experiments, data interpretation and manuscript revision. L.R. contributed to conception and design of the experiments, data collection, data interpretation, manuscript drafting and revision, and the editorial process. All authors approved the final version of the manuscript.
Author's present address
S. Goodall: School of Life Sciences, Northumbria University, Newcastle upon Tyne, UK.
References
- Aaslid R, Markwalder TM, Nornes H. Noninvasive transcranial Doppler ultrasound recording of flow velocity in basal cerebral arteries. J Neurosurg. 1982;57:769–774. doi: 10.3171/jns.1982.57.6.0769. [DOI] [PubMed] [Google Scholar]
- Allen DG, Lamb GD, Westerblad H. Skeletal muscle fatigue: cellular mechanisms. Physiol Rev. 2008;88:287–332. doi: 10.1152/physrev.00015.2007. [DOI] [PubMed] [Google Scholar]
- Allen GM, McKenzie DK, Gandevia SC. Twitch interpolation of the elbow flexor muscles at high forces. Muscle Nerve. 1998;21:318–328. doi: 10.1002/(sici)1097-4598(199803)21:3<318::aid-mus5>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Amann M, Blain GM, Proctor LT, Sebranek JJ, Pegelow DF, Dempsey JA. Implications of group III and IV muscle afferents for high-intensity endurance exercise performance in humans. J Physiol. 2011;589:5299–5309. doi: 10.1113/jphysiol.2011.213769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann M, Calbet JA. Convective oxygen transport and fatigue. J Appl Physiol. 2008;104:861–870. doi: 10.1152/japplphysiol.01008.2007. [DOI] [PubMed] [Google Scholar]
- Amann M, Eldridge MW, Lovering AT, Stickland MK, Pegelow DF, Dempsey JA. Arterial oxygenation influences central motor output and exercise performance via effects on peripheral locomotor muscle fatigue in humans. J Physiol. 2006a;575:937–952. doi: 10.1113/jphysiol.2006.113936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann M, Kayser B. Nervous system function during exercise in hypoxia. High Alt Med Biol. 2009;10:149–164. doi: 10.1089/ham.2008.1105. [DOI] [PubMed] [Google Scholar]
- Amann M, Proctor LT, Sebranek JJ, Pegelow DF, Dempsey JA. Opioid-mediated muscle afferents inhibit central motor drive and limit peripheral muscle fatigue development in humans. J Physiol. 2009;587:271–283. doi: 10.1113/jphysiol.2008.163303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann M, Romer LM, Pegelow DF, Jacques AJ, Hess CJ, Dempsey JA. Effects of arterial oxygen content on peripheral locomotor muscle fatigue. J Appl Physiol. 2006b;101:119–127. doi: 10.1152/japplphysiol.01596.2005. [DOI] [PubMed] [Google Scholar]
- Amann M, Romer LM, Subudhi AW, Pegelow DF, Dempsey JA. Severity of arterial hypoxaemia affects the relative contributions of peripheral muscle fatigue to exercise performance in healthy humans. J Physiol. 2007;581:389–403. doi: 10.1113/jphysiol.2007.129700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbogast S, Vassilakopoulos T, Darques JL, Duvauchelle JB, Jammes Y. Influence of oxygen supply on activation of group IV muscle afferents after low-frequency muscle stimulation. Muscle Nerve. 2000;23:1187–1193. doi: 10.1002/1097-4598(200008)23:8<1187::aid-mus5>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Barry BK, Enoka RM. The neurobiology of muscle fatigue: 15 years later. Integr Comp Biol. 2007;47:465–473. doi: 10.1093/icb/icm047. [DOI] [PubMed] [Google Scholar]
- Beaver WL, Wasserman K, Whipp BJ. A new method for detecting anaerobic threshold by gas exchange. J Appl Physiol. 1986;60:2020–2027. doi: 10.1152/jappl.1986.60.6.2020. [DOI] [PubMed] [Google Scholar]
- Bland JM, Altman DG. Calculating correlation coefficients with repeated observations: Part 1 – Correlation within subjects. BMJ. 1995;310:446. doi: 10.1136/bmj.310.6977.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg G. Borg's Perceived Exertion and Pain Scales. Champaign, IL: Human Kinetics; 1998. [Google Scholar]
- Brugniaux JV, Hodges AN, Hanly PJ, Poulin MJ. Cerebrovascular responses to altitude. Respir Physiol Neurobiol. 2007;158:212–223. doi: 10.1016/j.resp.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Claassen JA, Zhang R, Fu Q, Witkowski S, Levine BD. Transcranial Doppler estimation of cerebral blood flow and cerebrovascular conductance during modified rebreathing. J Appl Physiol. 2007;102:870–877. doi: 10.1152/japplphysiol.00906.2006. [DOI] [PubMed] [Google Scholar]
- Fowles JR, Green HJ, Tupling R, O’Brien S, Roy BD. Human neuromuscular fatigue is associated with altered Na+-K+-ATPase activity following isometric exercise. J Appl Physiol. 2002;92:1585–1593. doi: 10.1152/japplphysiol.00668.2001. [DOI] [PubMed] [Google Scholar]
- Gandevia SC. Neural control in human muscle fatigue: changes in muscle afferents, motoneurones and motor cortical drive. Acta Physiol Scand. 1998;162:275–283. doi: 10.1046/j.1365-201X.1998.0299f.x. [DOI] [PubMed] [Google Scholar]
- Gandevia SC. Spinal and supraspinal factors in human muscle fatigue. Physiol Rev. 2001;81:1725–1789. doi: 10.1152/physrev.2001.81.4.1725. [DOI] [PubMed] [Google Scholar]
- Gandevia SC, Allen GM, Butler JE, Taylor JL. Supraspinal factors in human muscle fatigue: evidence for suboptimal output from the motor cortex. J Physiol. 1996;490:529–536. doi: 10.1113/jphysiol.1996.sp021164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland SJ, McComas AJ. Reflex inhibition of human soleus muscle during fatigue. J Physiol. 1990;429:17–27. doi: 10.1113/jphysiol.1990.sp018241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Alonso J, Dalsgaard MK, Osada T, Volianitis S, Dawson EA, Yoshiga CC, Secher NH. Brain and central haemodynamics and oxygenation during maximal exercise in humans. J Physiol. 2004;557:331–342. doi: 10.1113/jphysiol.2004.060574. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodall S, Romer LM, Ross EZ. Voluntary activation of human knee extensors measured using transcranial magnetic stimulation. Exp Physiol. 2009;94:995–1004. doi: 10.1113/expphysiol.2009.047902. [DOI] [PubMed] [Google Scholar]
- Goodall S, Ross EZ, Romer LM. Effect of graded hypoxia on supraspinal contributions to fatigue with unilateral knee-extensor contractions. J Appl Physiol. 2010;109:1842–1851. doi: 10.1152/japplphysiol.00458.2010. [DOI] [PubMed] [Google Scholar]
- Hill JM, Pickar JG, Parrish MD, Kaufman MP. Effects of hypoxia on the discharge of group III and IV muscle afferents in cats. J Appl Physiol. 1992;73:2524–2529. doi: 10.1152/jappl.1992.73.6.2524. [DOI] [PubMed] [Google Scholar]
- Imray CH, Myers SD, Pattinson KT, Bradwell AR, Chan CW, Harris S, Collins P, Wright AD. Effect of exercise on cerebral perfusion in humans at high altitude. J Appl Physiol. 2005;99:699–706. doi: 10.1152/japplphysiol.00973.2004. [DOI] [PubMed] [Google Scholar]
- Martin PG, Smith JL, Butler JE, Gandevia SC, Taylor JL. Fatigue-sensitive afferents inhibit extensor but not flexor motoneurons in humans. J Neurosci. 2006;26:4796–4802. doi: 10.1523/JNEUROSCI.5487-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merton PA. Voluntary strength and fatigue. J Physiol. 1954;123:553–564. doi: 10.1113/jphysiol.1954.sp005070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin MJ, Robbins PA. Indexes of flow and cross-sectional area of the middle cerebral artery using doppler ultrasound during hypoxia and hypercapnia in humans. Stroke. 1996;27:2244–2250. doi: 10.1161/01.str.27.12.2244. [DOI] [PubMed] [Google Scholar]
- Rasmussen P, Dawson EA, Nybo L, van Lieshout JJ, Secher NH, Gjedde A. Capillary-oxygenation-level-dependent near-infrared spectrometry in frontal lobe of humans. J Cereb Blood Flow Metab. 2007;27:1082–1093. doi: 10.1038/sj.jcbfm.9600416. [DOI] [PubMed] [Google Scholar]
- Rasmussen P, Nielsen J, Overgaard M, Krogh-Madsen R, Gjedde A, Secher NH, Petersen NC. Reduced muscle activation during exercise related to brain oxygenation and metabolism in humans. J Physiol. 2010;588:1985–1995. doi: 10.1113/jphysiol.2009.186767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelands B, Meeusen R. Alterations in central fatigue by pharmacological manipulations of neurotransmitters in normal and high ambient temperature. Sports Med. 2010;40:229–246. doi: 10.2165/11533670-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Romer LM, Haverkamp HC, Amann M, Lovering AT, Pegelow DF, Dempsey JA. Effect of acute severe hypoxia on peripheral fatigue and endurance capacity in healthy humans. Am J Physiol Regul Integr Comp Physiol. 2007;292:R598–606. doi: 10.1152/ajpregu.00269.2006. [DOI] [PubMed] [Google Scholar]
- Romer LM, Haverkamp HC, Lovering AT, Pegelow DF, Dempsey JA. Effect of exercise-induced arterial hypoxemia on quadriceps muscle fatigue in healthy humans. Am J Physiol Regul Integr Comp Physiol. 2006;290:R365–375. doi: 10.1152/ajpregu.00332.2005. [DOI] [PubMed] [Google Scholar]
- Ross EZ, Middleton N, Shave R, George K, Nowicky A. Corticomotor excitability contributes to neuromuscular fatigue following marathon running in man. Exp Physiol. 2007;92:417–426. doi: 10.1113/expphysiol.2006.035972. [DOI] [PubMed] [Google Scholar]
- Rossiter HB, Kowalchuk JM, Whipp BJ. A test to establish maximum O2 uptake despite no plateau in the O2 uptake response to ramp incremental exercise. J Appl Physiol. 2006;100:764–770. doi: 10.1152/japplphysiol.00932.2005. [DOI] [PubMed] [Google Scholar]
- Rothwell JC, Thompson PD, Day BL, Boyd S, Marsden CD. Stimulation of the human motor cortex through the scalp. Exp Physiol. 1991;76:159–200. doi: 10.1113/expphysiol.1991.sp003485. [DOI] [PubMed] [Google Scholar]
- Serrador JM, Picot PA, Rutt BK, Shoemaker JK, Bondar RL. MRI measures of middle cerebral artery diameter in conscious humans during simulated orthostasis. Stroke. 2000;31:1672–1678. doi: 10.1161/01.str.31.7.1672. [DOI] [PubMed] [Google Scholar]
- Sharshar T, Hopkinson NS, Jonville S, Prigent H, Carlier R, Dayer MJ, Swallow EB, Lofaso F, Moxham J, Polkey MI. Demonstration of a second rapidly conducting cortico-diaphragmatic pathway in humans. J Physiol. 2004;560:897–908. doi: 10.1113/jphysiol.2004.061150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu SK, Bentley DJ, Carroll TJ. Cortical voluntary activation of the human knee extensors can be reliably estimated using transcranial magnetic stimulation. Muscle Nerve. 2009a;39:186–196. doi: 10.1002/mus.21064. [DOI] [PubMed] [Google Scholar]
- Sidhu SK, Bentley DJ, Carroll TJ. Locomotor exercise induces long-lasting impairments in the capacity of the human motor cortex to voluntarily activate knee extensor muscles. J Appl Physiol. 2009b;106:556–565. doi: 10.1152/japplphysiol.90911.2008. [DOI] [PubMed] [Google Scholar]
- Subudhi AW, Dimmen AC, Roach RC. Effects of acute hypoxia on cerebral and muscle oxygenation during incremental exercise. J Appl Physiol. 2007;103:177–183. doi: 10.1152/japplphysiol.01460.2006. [DOI] [PubMed] [Google Scholar]
- Subudhi AW, Lorenz MC, Fulco CS, Roach RC. Cerebrovascular responses to incremental exercise during hypobaric hypoxia: effect of oxygenation on maximal performance. Am J Physiol Heart Circ Physiol. 2008;294:H164–171. doi: 10.1152/ajpheart.01104.2007. [DOI] [PubMed] [Google Scholar]
- Subudhi AW, Miramon BR, Granger ME, Roach RC. Frontal and motor cortex oxygenation during maximal exercise in normoxia and hypoxia. J Appl Physiol. 2009a;106:1153–1158. doi: 10.1152/japplphysiol.91475.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subudhi AW, Panerai RB, Roach RC. Acute hypoxia impairs dynamic cerebral autoregulation: results from two independent techniques. J Appl Physiol. 2009b;107:1165–1171. doi: 10.1152/japplphysiol.00498.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AD, Bronks R, Smith P, Humphries B. Myoelectric evidence of peripheral muscle fatigue during exercise in severe hypoxia: some references to m. vastus lateralis myosin heavy chain composition. Eur J Appl Physiol. 1997;75:151–159. doi: 10.1007/s004210050140. [DOI] [PubMed] [Google Scholar]
- Taylor JL. Point: the interpolated twitch does/does not provide a valid measure of the voluntary activation of muscle. J Appl Physiol. 2009;107:354–355. doi: 10.1152/japplphysiol.91220.2008. [DOI] [PubMed] [Google Scholar]
- Taylor JL, Butler JE, Allen GM, Gandevia SC. Changes in motor cortical excitability during human muscle fatigue. J Physiol. 1996;490:519–528. doi: 10.1113/jphysiol.1996.sp021163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JL, Butler JE, Gandevia SC. Altered responses of human elbow flexors to peripheral-nerve and cortical stimulation during a sustained maximal voluntary contraction. Exp Brain Res. 1999;127:108–115. doi: 10.1007/s002210050779. [DOI] [PubMed] [Google Scholar]
- Taylor JL, Todd G, Gandevia SC. Evidence for a supraspinal contribution to human muscle fatigue. Clin Exp Pharmacol Physiol. 2006;33:400–405. doi: 10.1111/j.1440-1681.2006.04363.x. [DOI] [PubMed] [Google Scholar]
- Todd G, Gorman RB, Gandevia SC. Measurement and reproducibility of strength and voluntary activation of lower-limb muscles. Muscle Nerve. 2004a;29:834–842. doi: 10.1002/mus.20027. [DOI] [PubMed] [Google Scholar]
- Todd G, Taylor JL, Gandevia SC. Measurement of voluntary activation of fresh and fatigued human muscles using transcranial magnetic stimulation. J Physiol. 2003;551:661–671. doi: 10.1113/jphysiol.2003.044099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd G, Taylor JL, Gandevia SC. Reproducible measurement of voluntary activation of human elbow flexors with motor cortical stimulation. J Appl Physiol. 2004b;97:236–242. doi: 10.1152/japplphysiol.01336.2003. [DOI] [PubMed] [Google Scholar]
- Vogiatzis I, Louvaris Z, Habazettl H, Athanasopoulos D, Andrianopoulos V, Cherouveim E, Wagner H, Roussos C, Wagner PD, Zakynthinos S. Frontal cerebral cortex blood flow, oxygen delivery and oxygenation during normoxic and hypoxic exercise in athletes. J Physiol. 2011;589:4027–4039. doi: 10.1113/jphysiol.2011.210880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman K. The anaerobic threshold measurement to evaluate exercise performance. Am Rev Respir Dis. 1984;129:S35–40. doi: 10.1164/arrd.1984.129.2P2.S35. [DOI] [PubMed] [Google Scholar]
- Whipp BJ, Davis JA, Torres F, Wasserman K. A test to determine parameters of aerobic function during exercise. J Appl Physiol. 1981;50:217–221. doi: 10.1152/jappl.1981.50.1.217. [DOI] [PubMed] [Google Scholar]