

Abstract
Acute exposure to hypoxia decreases insulin sensitivity in healthy adult humans; the mechanism is unclear, but increased activation of the sympathetic nervous system may be involved. We have investigated the hypothesis that short-term sympathetic inhibition attenuates hypoxia induced insulin resistance. Insulin sensitivity (via the hyperinsulinaemic euglycaemic clamp) was determined in 10 healthy men (age 23 ± 1 years, body mass index 24.2 ± 0.8 kg m−2 (means ± SEM)), in a random order, during normoxia (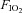 = 0.21), hypoxia (
= 0.21), hypoxia (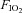 = 0.11), normoxia and sympathetic inhibition (via 48 h transdermal administration of the centrally acting α2-adrenergic receptor agonist, clonidine), and hypoxia and sympathetic inhibition. Oxyhaemoglobin saturation (pulse oximetry) was decreased (P < 0.001) with hypoxia (63 ± 2%) compared with normoxia (96 ± 0%), and was unaffected by sympathetic inhibition (P > 0.25). The area under the noradrenaline curve (relative to the normoxia response) was increased with hypoxia (137 ± 13%; P = 0.02); clonidine prevented the hypoxia induced increase (94 ± 14%; P = 0.43). The glucose infusion rate (adjusted for fat free mass and circulating insulin concentration) required to maintain blood glucose concentration at 5 mmol l−1 during administration of insulin was decreased in hypoxia compared with normoxia (225 ± 23 vs. 128 ± 30 nmol (kg fat free mass)−1 pmol l−1 min−1; P = 0.03), and unchanged during normoxia and sympathetic inhibition (219 ± 19; P = 0.86) and hypoxia and sympathetic inhibition (169 ± 23; P = 0.23). We conclude that short-term sympathetic inhibition attenuates hypoxia induced insulin resistance.
= 0.11), normoxia and sympathetic inhibition (via 48 h transdermal administration of the centrally acting α2-adrenergic receptor agonist, clonidine), and hypoxia and sympathetic inhibition. Oxyhaemoglobin saturation (pulse oximetry) was decreased (P < 0.001) with hypoxia (63 ± 2%) compared with normoxia (96 ± 0%), and was unaffected by sympathetic inhibition (P > 0.25). The area under the noradrenaline curve (relative to the normoxia response) was increased with hypoxia (137 ± 13%; P = 0.02); clonidine prevented the hypoxia induced increase (94 ± 14%; P = 0.43). The glucose infusion rate (adjusted for fat free mass and circulating insulin concentration) required to maintain blood glucose concentration at 5 mmol l−1 during administration of insulin was decreased in hypoxia compared with normoxia (225 ± 23 vs. 128 ± 30 nmol (kg fat free mass)−1 pmol l−1 min−1; P = 0.03), and unchanged during normoxia and sympathetic inhibition (219 ± 19; P = 0.86) and hypoxia and sympathetic inhibition (169 ± 23; P = 0.23). We conclude that short-term sympathetic inhibition attenuates hypoxia induced insulin resistance.
Key points
In low-oxygen environments, such as high-altitude, control of blood sugar is disrupted. Further, the activity of the sympathetic nervous system is known to increase when the availability of oxygen is decreased.
We have investigated the possibility that the increase in sympathetic activity is partially responsible for the disruption in blood sugar control.
Using gasbags filled with low-oxygen gas, together with a commonly used blood pressure medication (clonidine) that inhibits the sympathetic nervous system, we have shown that breathing low oxygen disrupts blood sugar control, and that this disruption is prevented when the nervous system is inhibited.
This finding has important implications for people travelling to high altitudes, and for people who suffer from conditions characterized by low oxygen, such as sleep apnoea and lung diseases.
Introduction
In healthy adult humans and laboratory animals, insulin sensitivity is decreased during short-term exposure to conditions of low oxygen, such as high altitude (Larsen et al. 1997), hypoxic gas breathing (Oltmanns et al. 2004), and/or hypobaria (Braun et al. 2001). The mechanism is unclear, but several studies have provided evidence for a role of increased sympathetic activation (Oltmanns et al. 2004). Further, in humans, hyperoxic gas breathing (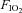 = 1.00) increases insulin sensitivity while decreasing circulating concentrations of plasma catecholamines (Wehrwein et al. 2010). Several clinical conditions, such as chronic obstructive pulmonary disease and sleep apnoea, are characterized by hypoxia, and the prevalence of insulin resistance and metabolic syndrome is high in these populations (Laaban et al. 2009; Gay, 2010; Schober et al. 2011), as is basal sympathetic activity (Narkiewicz et al. 1998; Heindl et al. 2001; Calbet, 2003). Identification of a physiological mechanism for hypoxia induced insulin resistance (e.g. sympathetic activation) may be of relevance for adults suffering from these hypoxia-related disorders. In light of these observations and important clinical implications, we have investigated the hypothesis that inhibition of the sympathetic nervous system will attenuate hypoxia induced insulin resistance in healthy adult humans.
= 1.00) increases insulin sensitivity while decreasing circulating concentrations of plasma catecholamines (Wehrwein et al. 2010). Several clinical conditions, such as chronic obstructive pulmonary disease and sleep apnoea, are characterized by hypoxia, and the prevalence of insulin resistance and metabolic syndrome is high in these populations (Laaban et al. 2009; Gay, 2010; Schober et al. 2011), as is basal sympathetic activity (Narkiewicz et al. 1998; Heindl et al. 2001; Calbet, 2003). Identification of a physiological mechanism for hypoxia induced insulin resistance (e.g. sympathetic activation) may be of relevance for adults suffering from these hypoxia-related disorders. In light of these observations and important clinical implications, we have investigated the hypothesis that inhibition of the sympathetic nervous system will attenuate hypoxia induced insulin resistance in healthy adult humans.
It is unlikely that increased sympathetic activation accounts entirely for hypoxia induced insulin resistance, and thus we have also measured several circulating factors previously shown to be regulated by hypoxia that also possess insulin-(de-)sensitizing properties. Specifically, we determined circulating concentrations of the insulin sensitizing adipokine, adiponectin (Yamauchi et al. 2001), and several factors associated with insulin resistance, including pigment epithelium derived factor (Bell, 2011), oxidized low density lipoproteins (Park et al. 2011), tumour necrosis factor-α (Stanley et al. 2011) and non-esterified fatty acids (Chai et al. 2011).
Methods
Subjects
Twelve healthy adult males were recruited. Inclusion criteria included normal fasting blood glucose concentration (<5.5 mmol l−1 (<100 mg dl−1)), and normal blood pressure (<140/90 mmHg). Exclusion criteria included regular use of tobacco products or medications that might confound the interpretation of data, and contraindications to hypoxic gas breathing (e.g. asthma, pulmonary disorders). The experimental protocol conformed to the standards set by the Declaration of Helsinki of 1975, as revised in 1983, and was approved by the Institutional Review Board at Colorado State University. The nature, purpose and risks of the study were explained to each subject before written informed consent was obtained.
Overall experimental design
Following screening procedures, insulin sensitivity was determined, in a random order, during four conditions: (1) normoxic gas breathing (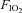 = 0.21); (2) hypoxic gas breathing (
= 0.21); (2) hypoxic gas breathing (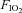 = 0.11); (3) normoxia during sympathetic inhibition (48 h transdermal administration of clonidine); and (4) hypoxia during sympathetic inhibition. Conditions were separated by a minimum of 7 days. All testing was performed early in the morning (beginning between 06.00 and 08.00 h) after a 12 h fast and 48 h abstention from exercise.
= 0.11); (3) normoxia during sympathetic inhibition (48 h transdermal administration of clonidine); and (4) hypoxia during sympathetic inhibition. Conditions were separated by a minimum of 7 days. All testing was performed early in the morning (beginning between 06.00 and 08.00 h) after a 12 h fast and 48 h abstention from exercise.
Screening procedures
Fat mass and fat free mass were measured using dual-energy x-ray absorptiometry (DXA-IQ; Lunar Radiation Corp., Madison, WI, USA, software v. 4.1). Maximal oxygen uptake was determined with a metabolic cart (Parvo Medics, Sandy, UT, USA) during incremental cycle ergometer or treadmill exercise to volitional fatigue, as previously described (Richards et al. 2010b). Prior to, during, and immediately following exercise, heart rate was determined via 12-lead electrocardiogram.
Insulin sensitivity
Insulin sensitivity was determined using the hyperinsulinaemic euglycaemic clamp technique (DeFronzo et al. 1979; Rattarasarn et al. 2004), as previously performed in our laboratory (Richards et al. 2010a); this is considered by most investigators to be the gold-standard measurement of insulin sensitivity (Bloomgarden, 2006). Briefly, intravenous catheters were inserted into an antecubital vein for infusion of glucose and insulin, and into a contralateral dorsal hand vein, warmed using a heated blanket for arterialized-venous blood sampling. A descending dose (127–40 mU m−2 body surface area min−1) of regular insulin (Humulin, Eli Lilly and Co., Indianapolis, IA, USA) was administered over the first 10 min, followed by a continuous infusion (40 mU m−2 body surface area min−1) from 10 to 180 min. Administration of a 20% dextrose solution was initiated at 4 min (2 mg kg−1 min−1) and adjusted as necessary to maintain blood glucose at a concentration of 5 mmol l−1 (90 mg dl−1) throughout the clamp period. Arterialized-venous blood samples (∼1 ml) were obtained every 5 min and blood glucose concentration was determined immediately using an automated device (2300 STAT Plus Glucose Lactate Analyser, YSI Inc., Yellow Springs, OH, USA). Insulin sensitivity was calculated from the mean rate of glucose infusion during the last 30 min of the clamp and expressed as units of glucose per kg fat free mass per unit of circulating insulin per min.
Normoxic/hypoxic gas breathing
Subjects wore an airtight facemask (7450 Series; Hans Rudolph, Inc., Shawnee, KS, USA) attached to a three-way, non-rebreathing valve (2730 Series; Han Rudolph, Inc.), connected via a hose to a 100 litre non-diffusing gasbag (6000 Series; Hans Rudolph, Inc.), which was filled with precision mixed gases (Airgas, Denver, CO, USA). Following basal measures of heart rate (3-lead electrocardiogram), blood pressure (automated physiological monitor; Cardiocap 5, GE Datex-Ohmeda, Madison, WI, USA), oxyhaemoglobin saturation (pulse oximeter; Cardiocap 5) and basal blood sampling, subjects began breathing from the gasbag. After 15 min the measurement of insulin sensitivity commenced; subjects continued to breathe from the gasbag throughout the remainder of the 180 min protocol. Heart rate and oxyhaemoglobin saturation were monitored continuously and recorded every 15 min. Blood pressure was measured and recorded every 15 min.
Inhibition of the sympathetic nervous system
Forty-eight hours prior to two of the laboratory visits, transdermal clonidine administration (Catapres-TTS; 0.2 mg day−1) was initiated and continued until the end of the hyperinsulinaemic euglycaemic clamp. Clonidine is a blood pressure medication. Its mechanism of action is via pre-junctional stimulation of α2-adrenergic receptors, including those located in the locus coeruleus, resulting in centrally mediated peripheral sympathetic inhibition, as reflected by decreased plasma noradrenaline concentration (Newsom et al. 2010), decreased noradrenaline release (Schwartz et al. 1990a,b) and attenuated skeletal muscle sympathetic nerve activity (Muzi et al. 1992; Furlan et al. 2006; Newsom et al. 2010).
Blood collection and analysis
During each laboratory visit, arterialized-venous blood was sampled immediately prior to normoxic/hypoxic gas breathing, after 15 min of gas breathing, and during the final 5 min of the hyperinsulinaemic euglycaemic clamp (time: −15, 0 and 180 min, respectively). Blood (∼20 ml preserved with K3-ethylenediaminetetraacetic acid, plus ∼5 ml preserved with ethylene glycol tetraacetic acid/glutathione) was collected in chilled tubes, placed immediately on ice and centrifuged within 60 min of collection to isolate plasma. Plasma samples were stored at –80°C until analysis. Enzyme-linked immunosorbent assays (ELISA) were used to measure, in duplicate, plasma concentrations of C-peptide, adiponectin, and pigment epithelium derived factor (all Millipore Corp., Billerica, MA, USA), insulin and oxidized low density lipoproteins (both ALPCO Diagnostics, Salem, NH, USA), tumour necrosis factor-α (R&D Systems, Inc., Minneapolis, MN, USA), catecholamines (Rocky Mountain Diagnostics, Colorado Springs, CO, USA), and non-esterified fatty acids (Wako Diagnostics, Richmond, VA, USA).
Statistical analysis
This was a randomized, repeated measures study. Accordingly, we examined potential differences in insulin sensitivity due to main effects of 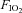 and presence/absence of sympathetic inhibition using repeated measures analysis of variance (ANOVA). Multiple comparisons of factor means were performed using the Newman–Keuls test. Plasma catecholamines were analysed by calculating an area under the curve (trapezoidal rule) above baseline, and comparing these values using repeated measures ANOVA. Other circulating factors were analysed with repeated measures ANOVA with time (−15, 0 and 180 min) as an additional factor. The level of statistical significance was set at P < 0.05. Data are expressed as means ± SEM.
and presence/absence of sympathetic inhibition using repeated measures analysis of variance (ANOVA). Multiple comparisons of factor means were performed using the Newman–Keuls test. Plasma catecholamines were analysed by calculating an area under the curve (trapezoidal rule) above baseline, and comparing these values using repeated measures ANOVA. Other circulating factors were analysed with repeated measures ANOVA with time (−15, 0 and 180 min) as an additional factor. The level of statistical significance was set at P < 0.05. Data are expressed as means ± SEM.
Results
Subject characteristics
Two of the 12 subjects recruited for participation were unable to complete the study due to intolerance of hypoxia. These subjects developed symptoms characteristic of acute mountain sickness that included nausea and headache, but recovered quickly (<10 min) after cessation of hypoxic gas breathing. Selected physiological characteristics of the remaining 10 subjects are presented in Table 1. As a group, the subjects were normal weight, normotensive, had normal fasting blood glucose values, and were of average aerobic fitness.
Table 1.
Physiological characteristics of the research participants (n = 10 men)
| Variable | Mean ± SEM |
|---|---|
| Age (years) | 23 ± 1 |
| Height (m) | 1.80 ± 0.01 |
| Body mass (kg) | 78.6 ± 2.6 |
| Body mass index (kg m−2) | 24.2 ± 0.8 |
| Fat mass (kg) | 15.2 ± 0.9 |
| % Body fat | 18.5 ± 1.2 |
| Fat free mass (kg) | 59.9 ± 1.7 |
| Maximal oxygen uptake (ml kg−1 min−1) | 46.8 ± 2.5 |
| Resting heart rate (beats min−1) | 59 ± 3 |
| Resting blood pressure (mmHg) | 120/72 ± 3/2 |
| Fasting blood glucose concentration (mmol l−1) | 4.51 ± 0.07 |
Data are means ± SEM.
Responses to hypoxia and clonidine
During hypoxic gas breathing, oxyhaemoglobin saturation (Fig. 1) was appreciably decreased (P < 0.01) compared with normoxia, but was unaffected by clonidine (P > 0.25). Plasma catecholamine concentrations prior to and throughout normoxia and hypoxia, with and without clonidine, are presented in Fig. 2A and B. Prior to normoxic/hypoxic gas breathing (that is, in the basal state) clonidine decreased noradrenaline (P = 0.05); the decrease in adrenaline did not attain statistical significance (P = 0.16). The area under the noradrenaline curve (relative to the normoxia response) was increased with hypoxia (137 ± 13%; P = 0.02); clonidine prevented the hypoxia induced increase (94 ± 14%; P = 0.43). The area under the adrenaline curve (relative to the normoxia response) was appreciably increased in hypoxia (371 ± 40%; P < 0.001). In contrast to noradrenaline, clonidine did not affect the hypoxia induced increase in adrenaline (346 ± 82%; P < 0.001).
Figure 1. Oxyhaemoglobin saturation (via pulse oximetery) prior to (−15 min) and during (0–180 min) normoxia (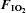 = 0.21) and hypoxia (
= 0.21) and hypoxia (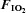 = 0.11), with and without sympathetic inhibition (48 h transdermal clonidine), during measurement of insulin sensitivity via the hyperinsulinaemic euglycaemic clamp.
= 0.11), with and without sympathetic inhibition (48 h transdermal clonidine), during measurement of insulin sensitivity via the hyperinsulinaemic euglycaemic clamp.
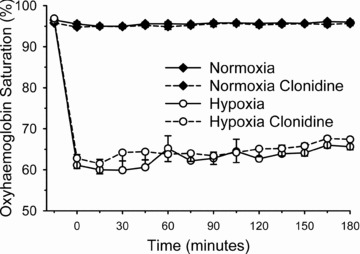
Hypoxic gas breathing appreciably decreased oxyhaemoglobin saturation (P < 0.01) compared with normoxia. Oxyhaemoglobin saturation was unaffected by clonidine (P > 0.25). Data are means ± SEM.
Figure 2. Plasma adrenaline (upper panel) and noradrenaline (lower panel) prior to (−15 min) and during (0–180 min) normoxia (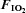 = 0.21) and hypoxia (
= 0.21) and hypoxia (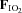 = 0.11), with and without sympathetic inhibition (48 h transdermal clonidine), during measurement of insulin sensitivity via the hyperinsulinaemic euglycaemic clamp.
= 0.11), with and without sympathetic inhibition (48 h transdermal clonidine), during measurement of insulin sensitivity via the hyperinsulinaemic euglycaemic clamp.
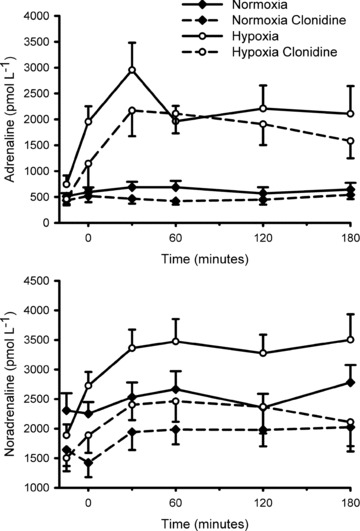
The area under the noradrenaline curve (relative to the normoxia response) was increased with hypoxia (137 ± 13%; P = 0.02); clonidine prevented the hypoxia induced increase (94 ± 14%; P = 0.43). The area under the adrenaline curve (relative to the normoxia response) was appreciably increased in hypoxia (371 ± 40%; P < 0.001). Clonidine did not affect the hypoxia induced increase in adrenaline (346 ± 82%; P < 0.001). Data are means ± SEM.
Heart rate and blood pressure, prior to and during normoxia and hypoxia, with and without clonidine, are presented in Table 2. Prior to normoxic or hypoxic gas breathing, clonidine decreased resting heart rate (P < 0.04) and resting systolic blood pressure (P < 0.05). The decrease in diastolic blood pressure with clonidine did not attain statistical significance (P < 0.08). During hypoxia and the hyperinsulinaemic euglycaemic clamp, heart rate was increased (P < 0.001); clonidine attenuated the hypoxia induced increase in heart rate (P = 0.006). Hypoxia did not affect diastolic blood pressure (P = 0.10) but there was a main effect (lowering) with clonidine (P < 0.001). Neither hypoxia nor clonidine influenced the average systolic blood pressure (P > 0.41) during the measurement of insulin sensitivity.
Table 2.
Heart rate and blood pressure, prior to and during normoxic and hypoxic gas breathing, with and without sympathetic inhibition (48 h transdermal clonidine)
Normoxia (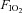 = 0.21) = 0.21) |
Hypoxia (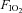 = 0.11) = 0.11) |
|||||||
|---|---|---|---|---|---|---|---|---|
| No clonidine | Clonidine | No clonidine | With clonidine | |||||
| Prior to | During | Prior to | During | Prior to | During | Prior to | During | |
| HR (b min−1) | 59 ± 3 | 68 ± 2 | 55 ± 4 | 61 ± 3 | 57 ± 2 | 84 ± 4 | 49 ± 2 | 76 ± 3 |
| SBP (mmHg) | 120 ± 3 | 124 ± 3 | 110 ± 3 | 114 ± 3 | 123 ± 3 | 122 ± 2 | 113 ± 3 | 125 ± 9 |
| DBP (mmHg) | 72 ± 2 | 71 ± 2 | 67 ± 3 | 64 ± 3 | 75 ± 3 | 68 ± 2 | 66 ± 2 | 63 ± 2 |
Data are means ± SEM. ‘Prior to’ data collected before breathing normoxic or hypoxic gas. ‘During’ data are averaged values collected throughout the entire hyperinsulinaemic euglycaemic clamp procedure (180 min). See text for details pertaining to statistical analysis. SBP: systolic blood pressure. DBP: diastolic blood pressure.
Collectively, these data suggest that hypoxia induced appreciable and predictable physiological effects (decreased oxyhaemoglobin saturation, and increased plasma catecholamines and heart rate). Clonidine decreased basal plasma noradrenaline concentration, heart rate and systolic blood pressure, and attenuated the noradrenaline and heart rate response to hypoxia, suggesting that clonidine inhibited the sympathetic nervous system.
Insulin sensitivity
The glucose infusion rates (adjusted for fat free mass and circulating insulin concentration) required to maintain blood glucose concentration at 5 mmol l−1 during administration of insulin, while breathing normoxic and hypoxic gases with and without sympathetic inhibition are presented in Fig. 3. Statistical analysis revealed an interaction between 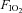 and sympathetic inhibition (P = 0.046), but no main effect of either the absence/presence of sympathetic inhibition or
and sympathetic inhibition (P = 0.046), but no main effect of either the absence/presence of sympathetic inhibition or 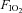 (both P > 0.06). Post hoc analysis showed that, compared with normoxic values, insulin sensitivity was decreased in hypoxia (P = 0.03), unchanged during normoxia and sympathetic inhibition (P = 0.86), and unchanged during hypoxia and sympathetic inhibition (P = 0.23). End-clamp blood glucose concentrations were not different between conditions (Normoxia: 5.00 ± 0.04; Normoxia-Clonidine: 5.02 ± 0.03; Hypoxia: 4.99 ± 0.06; Hypoxia-Clonidine: 5.04 ± 0.04 mmol l−1; P = 0.77). Plasma insulin concentrations were approximately 10-fold higher than pre-clamp concentrations (P < 0.001), but there was no interaction between time, hypoxia and sympathetic inhibition (Fig. 3; P = 0.22). The increased end-clamp plasma insulin concentrations were attributed to insulin administration as plasma C-peptide concentrations (reflective of insulin release from the pancreas) were similar between pre- and end-clamp measurements (Fig. 4; P > 0.41); nor was there any interaction between time, hypoxia and sympathetic inhibition (Fig. 4; P > 0.84).
(both P > 0.06). Post hoc analysis showed that, compared with normoxic values, insulin sensitivity was decreased in hypoxia (P = 0.03), unchanged during normoxia and sympathetic inhibition (P = 0.86), and unchanged during hypoxia and sympathetic inhibition (P = 0.23). End-clamp blood glucose concentrations were not different between conditions (Normoxia: 5.00 ± 0.04; Normoxia-Clonidine: 5.02 ± 0.03; Hypoxia: 4.99 ± 0.06; Hypoxia-Clonidine: 5.04 ± 0.04 mmol l−1; P = 0.77). Plasma insulin concentrations were approximately 10-fold higher than pre-clamp concentrations (P < 0.001), but there was no interaction between time, hypoxia and sympathetic inhibition (Fig. 3; P = 0.22). The increased end-clamp plasma insulin concentrations were attributed to insulin administration as plasma C-peptide concentrations (reflective of insulin release from the pancreas) were similar between pre- and end-clamp measurements (Fig. 4; P > 0.41); nor was there any interaction between time, hypoxia and sympathetic inhibition (Fig. 4; P > 0.84).
Figure 3. The rate of intravenous glucose administration (adjusted for fat free mass and circulating insulin concentration) required to maintain blood glucose concentration at 5 mmol l−1 during standardized intravenous insulin administration during normoxia (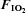 = 0.21) and hypoxia (
= 0.21) and hypoxia (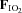 = 0.11), with and without sympathetic inhibition (48 h transdermal clonidine).
= 0.11), with and without sympathetic inhibition (48 h transdermal clonidine).
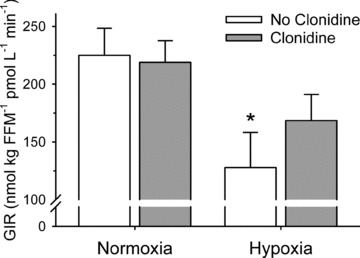
Data are means ± SEM. *Difference from normoxia without clonidine (P < 0.05). GIR: glucose infusion rate.
Figure 4. Plasma concentrations of factors known to influence insulin sensitivity that are partially regulated by hypoxia prior to (−15 min) and during (0 and 180 min) normoxia (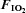 = 0.21) and hypoxia (
= 0.21) and hypoxia (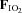 = 0.11), with and without sympathetic inhibition (48 h transdermal clonidine), during measurement of insulin sensitivity via the hyperinsulinaemic euglycaemic clamp.
= 0.11), with and without sympathetic inhibition (48 h transdermal clonidine), during measurement of insulin sensitivity via the hyperinsulinaemic euglycaemic clamp.
Data are means ± SEM. P values reflect interaction between time, 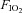 and clonidine. NEFA: non-esterified fatty acids. PEDF: pigment epithelium derived factor. LDL: low density lipoprotein. TNFα: tumour necrosis factor α.
and clonidine. NEFA: non-esterified fatty acids. PEDF: pigment epithelium derived factor. LDL: low density lipoprotein. TNFα: tumour necrosis factor α.
Additional blood parameters
Additional blood parameters thought to be regulated by hypoxia and able to exert an effect on insulin sensitivity are presented in Fig. 4. The interaction of time, hypoxia and sympathetic inhibition did not attain statistical significance (P = 0.058) for non-esterified fatty acids. Similarly, none of the other parameters were influenced by time, hypoxia and/or sympathetic inhibition (Fig. 4; all P≥ 0.30).
Discussion
The novel finding of this investigation is that short-term sympathetic inhibition attenuates hypoxia induced insulin resistance. This is based on the observation that the glucose infusion rate during a hyperinsulinaemic euglycaemic clamp, performed during hypoxia, was decreased by approximately 50% compared to the same test performed in normoxia; when the hypoxia test was repeated during sympathetic inhibition with clonidine, the difference between glucose infusion rates in normoxia and hypoxia was abrogated.
Our observation of hypoxia induced insulin resistance is supported by numerous past observations of decreased insulin sensitivity in healthy adult humans and laboratory animals during short-term exposure to conditions of low oxygen, such as high altitude (Larsen et al. 1997), hypoxic gas breathing (Oltmanns et al. 2004), and hypobaria (Braun et al. 2001). Similarly, the increase in sympathetic activation in response to decreased oxygen availability has been well described (Hansen & Sander, 2003; Prabhakar & Kumar, 2010; Zoccal & Machado, 2011), and has been partially attributed to maintaining oxygen delivery in the face of decreased physiological 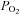 . Initially (acute exposure) this is achieved in part via increased cardiac output, and then eventually with haemo-concentration. Sympathetic activity is increased to support blood pressure following the plasma volume loss that accompanies haemo-concentration. While activation of the sympathetic nervous system might at first appear favourable, it may also have several adverse metabolic consequences, including disruptions in glucose regulation (Nonogaki, 2000; Vicini et al. 2002). These disruptions are realized via adrenaline mediated glycogenolysis (Vicini et al. 2002; Oltmanns et al. 2004), inhibition of hexokinase (Raz et al. 1991), and inhibition of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase (PI3-kinase) activity in skeletal muscle (Hunt & Ivy, 2002), and via noradrenaline stimulated lipolysis leading to increased circulating non-esterified fatty acids and impaired insulin-stimulated glucose transport (Karlsson, 1999; Nonogaki, 2000).
. Initially (acute exposure) this is achieved in part via increased cardiac output, and then eventually with haemo-concentration. Sympathetic activity is increased to support blood pressure following the plasma volume loss that accompanies haemo-concentration. While activation of the sympathetic nervous system might at first appear favourable, it may also have several adverse metabolic consequences, including disruptions in glucose regulation (Nonogaki, 2000; Vicini et al. 2002). These disruptions are realized via adrenaline mediated glycogenolysis (Vicini et al. 2002; Oltmanns et al. 2004), inhibition of hexokinase (Raz et al. 1991), and inhibition of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase (PI3-kinase) activity in skeletal muscle (Hunt & Ivy, 2002), and via noradrenaline stimulated lipolysis leading to increased circulating non-esterified fatty acids and impaired insulin-stimulated glucose transport (Karlsson, 1999; Nonogaki, 2000).
Several investigators have raised the possibility that hypoxia induced insulin resistance can be partially attributed to sympathetic activation (Oltmanns et al. 2004), although very few studies have directly addressed the issue and the data have been inconclusive. For example, in mice, the hypoxia induced decrease in insulin sensitivity was unaffected by the administration of the ganglionic antagonist, hexamethonium (Iiyori et al. 2007). However, the dose of hexamethonium may not have been sufficient to achieve full ganglionic blockade as the chronotropic response to an acute phenylephrine mediated increase in total peripheral resistance remained intact (Iiyori et al. 2007). In another study, insulin resistance (described using the homeostasis model assessment of insulin resistance; HOMA-IR) was increased in women during simulated altitude (hypobaric chamber); α1-adrenergic receptor antagonism (via oral prazosin) did not affect the hypobaria induced insulin resistance (Braun et al. 2001). Blockade of α1-adrenergic receptors is unlikely to influence sympathetic stimulation of glycogenolysis or sympathetic inhibition of insulin release on account that these are primarily β-adrenergic receptor and α2-adrenergic receptor mediated functions, respectively. In the current investigation, rather than block peripheral adrenergic receptors we chose to inhibit central sympathetic activation with the pre-junctional α2-adrenergic receptor agonist, clonidine. We, and others, have previously demonstrated that the dose of clonidine used in the current study decreases skeletal muscle sympathetic nerve activity (Newsom et al. 2010), noradrenaline release (Schwartz et al. 1990a,b) and plasma noradrenaline concentration (Newsom et al. 2010) in adult humans. Data from the current investigation are consistent with these previous reports.
Noteworthy, in the current study, although the noradrenaline response to hypoxia was inhibited, the influence of clonidine on adrenaline did not attain statistical significance. Further, inspection of the glucose infusion rate data (Fig. 3) reveals that although there was no statistical difference between insulin sensitivity determined in normoxia and in hypoxia during sympathetic inhibition (P = 0.23), the latter was still only ∼75% of the former (mean difference: –50.2 ± 14.5 nmol (kg fat free mass)−1 pmol l−1 min−1). This suggests that increased noradrenaline during hypoxia does not account entirely for the hypoxia induced insulin resistance. Given the previously described influence of adrenaline on glucose regulation (Vicini et al. 2002) and the absence of a significant effect of clonidine on the adrenaline response to hypoxia, adrenaline may have contributed to hypoxia induced insulin resistance. Alternatively, we considered several other circulating factors that are known to influence insulin sensitivity and may be regulated in part by hypoxia (Fig. 4). The interaction of time, hypoxia and sympathetic inhibition did not attain statistical significance (P = 0.058) for non-esterified fatty acids. It is plausible that as a result of sympathetically mediated lipolysis, non-esterified fatty acids may have contributed to decreased insulin sensitivity with hypoxia, but our experimental design was not adequately powered to support/refute this notion. Unexpectedly, none of the other circulating factors were appreciably affected by hypoxia (or sympathetic inhibition). It is possible that, independently, none of the factors decreased insulin sensitivity in hypoxia, but collectively, small changes in all of the factors acted symbiotically to disrupt glucose regulation. This possibility could be addressed in future hypoxia studies with simultaneous administration of antioxidant, anti-inflammatory and anti-lipolytic agents in addition to sympathetic inhibitors. Further, synthesis of adrenaline (and other catecholamines) could be inhibited with the tyrosine hydroxylase inhibitor, α-methyl-p-tyrosine.
The current study has a number of important implications. Several clinical conditions, such as chronic obstructive pulmonary disease and sleep apnoea, are characterized by hypoxia, and the prevalence of insulin resistance and metabolic syndrome is high in adults with these conditions (Wolk & Somers, 2007; Laaban et al. 2009; Schober et al. 2011), as is basal sympathetic activity (Narkiewicz et al. 1998; Heindl et al. 2001). Our data reinforce previous suggestions that the heightened sympathetic activity may contribute to the development of insulin resistance (Landsberg, 1990; Lambert & Lambert, 2011) and support the potential use of clonidine in these populations to prevent the decrease in insulin sensitivity. Further, millions of adults recreate in high altitude environments. Our data provide a potential mechanism to explain previous observations of decreased insulin action at high altitude (Larsen et al. 1997; de Mol et al. 2011).
There are several limitations in the current study that warrant brief discussion. The hyperinsulinaemic euglycaemic clamp technique is considered by many to be the gold-standard measure of insulin sensitivity (Bloomgarden, 2006). Unfortunately it does not provide information specific to hepatic insulin sensitivity. It is plausible that adrenaline stimulated hepatic glucose production may have contributed to the hypoxia induced insulin resistance, although given the 10-fold increase in circulating insulin concentration during the clamp, this seems unlikely. This issue could be addressed with incorporation of labelled glucose and a two-step hyperinsulinaemic euglycaemic clamp.
Similar to the previous limitation, we have no data pertaining to mechanisms of hypoxia induced insulin resistance in peripheral tissues (e.g. skeletal muscle). This limitation could be addressed in future studies with the addition of tissue samples analysed for insulin signalling related parameters.
All of the research participants in the current study were men. This limits the implications of our data for women, although based on previous observations in women (Braun et al. 2001) there is no reason to expect the primary outcome would be any different.
Finally, the dosing regime for clonidine was based on an absolute dose and administered transdermally. Differences in body mass, body composition and transdermal absorption may have contributed to variability in the primary outcome variables, both within and between subjects.
In summary, short-term sympathetic inhibition with clonidine attenuates hypoxia induced insulin resistance in young healthy adults. These data may have important implications for glucose regulation at high altitude, and metabolic disease risk for patients with disorders characterized by hypoxia, such as sleep apnoea and chronic obstructive pulmonary disease.
Acknowledgments
It was supported by the Defense Advanced Research Projects Agency (DARPA) N66001-10-c-2134. We are grateful for the technical and administrative assistance provided by Scott E. Binns, Anna L. Klochak, Steve E. Szallar, and Lacey M. Wood.
Glossary
Abbreviations
- HOMA-IR
homeostasis model assessment of insulin resistance
- IRS-1
insulin receptor substrate-1
- PI3
phosphatidylinositol 3
Author contributions
This study was performed in the Human Performance/Clinical Research Laboratory at Colorado State University. Data were collected and analysed by G.L.P., R.L.S., M.L.S. and C.B. D.G.L. and G.J.L. provided medical oversight. All authors contributed to the conception and design of the experiment, and writing/editing of this manuscript.
References
- Bell C. Pigment epithelium-derived factor: a not so sympathetic regulator of insulin resistance? Exerc Sport Sci Rev. 2011;39:187–190. doi: 10.1097/JES.0b013e31822673f0. [DOI] [PubMed] [Google Scholar]
- Bloomgarden ZT. Measures of insulin sensitivity. Clin Lab Med. 2006;26:611–633. doi: 10.1016/j.cll.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Braun B, Rock PB, Zamudio S, Wolfel GE, Mazzeo RS, Muza SR, Fulco CS, Moore LG, Butterfield GE. Women at altitude: short-term exposure to hypoxia and/or α1-adrenergic blockade reduces insulin sensitivity. J Appl Physiol. 2001;91:623–631. doi: 10.1152/jappl.2001.91.2.623. [DOI] [PubMed] [Google Scholar]
- Calbet JA. Chronic hypoxia increases blood pressure and noradrenaline spillover in healthy humans. J Physiol. 2003;551:379–386. doi: 10.1113/jphysiol.2003.045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai W, Liu J, Jahn LA, Fowler DE, Barrett EJ, Liu Z. Salsalate attenuates free fatty acid-induced microvascular and metabolic insulin resistance in humans. Diabetes Care. 2011;34:1634–1638. doi: 10.2337/dc10-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mol P, de Vries ST, de Koning EJ, Gans RO, Tack CJ, Bilo HJ. Increased insulin requirements during exercise at very high altitude in type 1 diabetes. Diabetes Care. 2011;34:591–595. doi: 10.2337/dc10-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol Endocrinol Metab. 1979;237:E214–223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- Furlan R, Ardizzone S, Palazzolo L, Rimoldi A, Perego F, Barbic F, Bevilacqua M, Vago L, Bianchi Porro G, Malliani A. Sympathetic overactivity in active ulcerative colitis: effects of clonidine. Am J Physiol Regul Integr Comp Physiol. 2006;290:R224–232. doi: 10.1152/ajpregu.00442.2005. [DOI] [PubMed] [Google Scholar]
- Gay PC. Sleep and sleep-disordered breathing in the hospitalized patient. Respir Care. 2010;55:1240–1254. [PubMed] [Google Scholar]
- Hansen J, Sander M. Sympathetic neural overactivity in healthy humans after prolonged exposure to hypobaric hypoxia. J Physiol. 2003;546:921–929. doi: 10.1113/jphysiol.2002.031765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindl S, Lehnert M, Criee CP, Hasenfuss G, Andreas S. Marked sympathetic activation in patients with chronic respiratory failure. Am J Respir Crit Care Med. 2001;164:597–601. doi: 10.1164/ajrccm.164.4.2007085. [DOI] [PubMed] [Google Scholar]
- Hunt DG, Ivy JL. Epinephrine inhibits insulin-stimulated muscle glucose transport. J Appl Physiol. 2002;93:1638–1643. doi: 10.1152/japplphysiol.00445.2002. [DOI] [PubMed] [Google Scholar]
- Iiyori N, Alonso LC, Li J, Sanders MH, Garcia-Ocana A, O’Doherty RM, Polotsky VY, O’Donnell CP. Intermittent hypoxia causes insulin resistance in lean mice independent of autonomic activity. Am J Respir Crit Care Med. 2007;175:851–857. doi: 10.1164/rccm.200610-1527OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson AK. Insulin resistance and sympathetic function in high spinal cord injury. Spinal Cord. 1999;37:494–500. doi: 10.1038/sj.sc.3100844. [DOI] [PubMed] [Google Scholar]
- Laaban JP, Daenen S, Leger D, Pascal S, Bayon V, Slama G, Elgrably F. Prevalence and predictive factors of sleep apnoea syndrome in type 2 diabetic patients. Diabetes Metab. 2009;35:372–377. doi: 10.1016/j.diabet.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Lambert EA, Lambert GW. Stress and its role in sympathetic nervous system activation in hypertension and the metabolic syndrome. Curr Hypertens Rep. 2011;13:244–248. doi: 10.1007/s11906-011-0186-y. [DOI] [PubMed] [Google Scholar]
- Landsberg L. Insulin resistance, energy balance and sympathetic nervous system activity. Clin Exp Hypertens A. 1990;12:817–830. doi: 10.3109/10641969009073502. [DOI] [PubMed] [Google Scholar]
- Larsen JJ, Hansen JM, Olsen NV, Galbo H, Dela F. The effect of altitude hypoxia on glucose homeostasis in men. J Physiol. 1997;504:241–249. doi: 10.1111/j.1469-7793.1997.241bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzi M, Goff DR, Kampine JP, Roerig DL, Ebert TJ. Clonidine reduces sympathetic activity but maintains baroreflex responses in normotensive humans. Anesthesiology. 1992;77:864–871. doi: 10.1097/00000542-199211000-00005. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJ, Cooley RL, Dyken ME, Somers VK. Sympathetic activity in obese subjects with and without obstructive sleep apnea. Circulation. 1998;98:772–776. doi: 10.1161/01.cir.98.8.772. [DOI] [PubMed] [Google Scholar]
- Newsom SA, Richards JC, Johnson TK, Kuzma JN, Lonac MC, Paxton RJ, Rynn GM, Voyles WF, Bell C. Short-term sympathoadrenal inhibition augments the thermogenic response to β-adrenergic receptor stimulation. J Endocrinol. 2010;206:307–315. doi: 10.1677/JOE-10-0152. [DOI] [PubMed] [Google Scholar]
- Nonogaki K. New insights into sympathetic regulation of glucose and fat metabolism. Diabetologia. 2000;43:533–549. doi: 10.1007/s001250051341. [DOI] [PubMed] [Google Scholar]
- Oltmanns KM, Gehring H, Rudolf S, Schultes B, Rook S, Schweiger U, Born J, Fehm HL, Peters A. Hypoxia causes glucose intolerance in humans. Am J Respir Crit Care Med. 2004;169:1231–1237. doi: 10.1164/rccm.200308-1200OC. [DOI] [PubMed] [Google Scholar]
- Park SH, Kim JY, Lee JH, Park HY. Elevated oxidized low-density lipoprotein concentrations in postmenopausal women with the metabolic syndrome. Clin Chim Acta. 2011;412:435–440. doi: 10.1016/j.cca.2010.11.017. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Kumar GK. Mechanisms of sympathetic activation and blood pressure elevation by intermittent hypoxia. Respir Physiol Neurobiol. 2010;174:156–161. doi: 10.1016/j.resp.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattarasarn C, Leelawattana R, Soonthornpun S, Setasuban W, Thamprasit A. Gender differences of regional abdominal fat distribution and their relationships with insulin sensitivity in healthy and glucose-intolerant Thais. J Clin Endocrinol Metab. 2004;89:6266–6270. doi: 10.1210/jc.2004-0209. [DOI] [PubMed] [Google Scholar]
- Raz I, Katz A, Spencer MK. Epinephrine inhibits insulin-mediated glycogenesis but enhances glycolysis in human skeletal muscle. Am J Physiol Endocrinol Metab. 1991;260:E430–435. doi: 10.1152/ajpendo.1991.260.3.E430. [DOI] [PubMed] [Google Scholar]
- Richards JC, Johnson TK, Kuzma JN, Lonac MC, Schweder MM, Voyles WF, Bell C. Short-term sprint interval training increases insulin sensitivity in healthy adults but does not affect the thermogenic response to β-adrenergic stimulation. J Physiol. 2010a;588:2961–2972. doi: 10.1113/jphysiol.2010.189886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JC, Lonac MC, Johnson TK, Schweder MM, Bell C. Epigallocatechin-3-gallate increases maximal oxygen uptake in adult humans. Med Sci Sports Exerc. 2010b;42:739–744. doi: 10.1249/MSS.0b013e3181bcab6c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober AK, Neurath MF, Harsch IA. Prevalence of sleep apnoea in diabetic patients. Clin Respir J. 2011;5:165–172. doi: 10.1111/j.1752-699X.2010.00216.x. [DOI] [PubMed] [Google Scholar]
- Schwartz RS, Jaeger LF, Veith RC. The thermic effect of feeding in older men: the importance of the sympathetic nervous system. Metabolism. 1990a;39:733–737. doi: 10.1016/0026-0495(90)90109-p. [DOI] [PubMed] [Google Scholar]
- Schwartz RS, Jaeger LF, Veith RC, Lakshminarayan S. The effect of diet or exercise on plasma norepinephrine kinetics in moderately obese young men. Int J Obes. 1990b;14:1–11. [PubMed] [Google Scholar]
- Stanley TL, Zanni MV, Johnsen S, Rasheed S, Makimura H, Lee H, Khor VK, Ahima RS, Grinspoon SK. TNF-α antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J Clin Endocrinol Metab. 2011;96:E146–E150. doi: 10.1210/jc.2010-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicini P, Avogaro A, Spilker ME, Gallo A, Cobelli C. Epinephrine effects on insulin-glucose dynamics: the labeled IVGTT two-compartment minimal model approach. Am J Physiol Endocrinol Metab. 2002;283:E78–E84. doi: 10.1152/ajpendo.00530.2001. [DOI] [PubMed] [Google Scholar]
- Wehrwein EA, Basu R, Basu A, Curry TB, Rizza RA, Joyner MJ. Hyperoxia blunts counterregulation during hypoglycaemia in humans: possible role for the carotid bodies? J Physiol. 2010;588:4593–4601. doi: 10.1113/jphysiol.2010.197491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk R, Somers VK. Sleep and the metabolic syndrome. Exp Physiol. 2007;92:67–78. doi: 10.1113/expphysiol.2006.033787. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- Zoccal DB, Machado BH. Coupling between respiratory and sympathetic activities as a novel mechanism underpinning neurogenic hypertension. Curr Hypertens Rep. 2011;13:229–236. doi: 10.1007/s11906-011-0198-7. [DOI] [PubMed] [Google Scholar]