

Abstract
We pursued studies to determine the effects of the metabolic syndrome (MetS) on brain, and the possibility of modulating these effects by dietary interventions. In addition, we have assessed potential mechanisms by which brain metabolic disorders can impact synaptic plasticity and cognition. We report that high-dietary fructose consumption leads to an increase in insulin resistance index, and insulin and triglyceride levels, which characterize MetS. Rats fed on an n-3 deficient diet showed memory deficits in a Barnes maze, which were further exacerbated by fructose intake. In turn, an n-3 deficient diet and fructose interventions disrupted insulin receptor signalling in hippocampus as evidenced by a decrease in phosphorylation of the insulin receptor and its downstream effector Akt. We found that high fructose consumption with an n-3 deficient diet disrupts membrane homeostasis as evidenced by an increase in the ratio of n-6/n-3 fatty acids and levels of 4-hydroxynonenal, a marker of lipid peroxidation. Disturbances in brain energy metabolism due to n-3 deficiency and fructose treatments were evidenced by a significant decrease in AMPK phosphorylation and its upstream modulator LKB1 as well as a decrease in Sir2 levels. The decrease in phosphorylation of CREB, synapsin I and synaptophysin levels by n-3 deficiency and fructose shows the impact of metabolic dysfunction on synaptic plasticity. All parameters of metabolic dysfunction related to the fructose treatment were ameliorated by the presence of dietary n-3 fatty acid. Results showed that dietary n-3 fatty acid deficiency elevates the vulnerability to metabolic dysfunction and impaired cognitive functions by modulating insulin receptor signalling and synaptic plasticity.
Key points
We provide novel evidence for the effects of metabolic dysfunctions on brain function using the rat model of metabolic syndrome induced by high fructose intake.
We describe that the deleterious consequences of unhealthy dietary habits can be partially counteracted by dietary supplementation of n-3 fatty acid.
High sugar consumption impaired cognitive abilities and disrupted insulin signalling by engaging molecules associated with energy metabolism and synaptic plasticity; in turn, the presence of docosahexaenoic acid, an n-3 fatty acid, restored metabolic homeostasis.
These findings expand the concept of metabolic syndrome affecting the brain and provide the mechanistic evidence of how dietary habits can interact to regulate brain functions, which can further alter lifelong susceptibility to the metabolic disorders.
Introduction
The rise in consumption of high caloric foods has triggered an explosive surge in metabolic syndrome (MetS), known for its effects of increasing morbidity and negatively impacting life expectancy. While the effects of MetS have been characterized in peripheral organ systems, a body of clinical information has started to surface revealing the pervasive effects of MetS on mental health, compromising cognitive functions and emotions. For example, now we know that metabolic disorders such as diabetes and obesity increase the vulnerability to mental illness (Newcomer, 2007); however, the mechanisms that link cellular metabolism and mental health are poorly understood. Therefore, it is imperative to obtain a better understanding of how the action of foods can translate in dysfunctional metabolism and damage the neural substrates of cognition. It is alarming that commonly consumed low-cost foods with high sugar and fat contents, and low essential nutrient levels, have the potential to determine mental health.
Insulin resistance is the hallmark of MetS and is largely a food induced metabolic disorder, defined as decreased sensitivity/or responsiveness to the metabolic action of insulin promoting glucose disposal. Insulin resistance has been characterized in muscle, adipose tissue and liver. Although changes in insulin level can affect brain function, the question remains as whether the insulin resistance seen in peripheral tissue also occurs in the brain tissue of patients with metabolic syndrome. Given that insulin can penetrate the brain–blood barrier, it can have a wide range of brain actions, which may largely depend on the signalling through its receptors. Insulin receptors are found in brain tissue, particularly in areas related to cognitive processing such as the hippocampus, and are involved in synaptic plasticity and behaviour (Agrawal et al. 2009). Brain insulin receptors have been implicated in the pathogenesis of type II diabetes (Gerozissis, 2008) and their desensitization disrupts energy homeostasis (Biessels et al. 2002).
Abundant consumption of fructose is an important contributor to the metabolic syndrome, typically characterized by hyperinsulinaemia, hypertension and hypertriglyceridaemia (Gerrits & Tsalikian, 1993). Studies have shown that rats fed on a high fructose diet display hepatic oxidative damage and altered lipid metabolism as a result of the burden of fructose metabolism (Kelley et al. 2004). We have conducted studies using fructose drinking as an animal model to assess the effects of MetS on insulin signalling, synaptic plasticity, and behaviour.
Unhealthy dietary habits are difficult or almost impossible to completely eliminate. Therefore, the concurrent implementation of healthier dietary components to popular diets can be a productive strategy to counteract metabolic dysfunction and protect mental health. Therefore the present study was planned to study the potential of an n-3 fatty acid (docosahexaenoic acid; DHA) enriched diet to counteract insulin resistance in brain. DHA (C22:6n-3), one of the major n-3 polyunsaturated fatty acids in the brain, is important for brain development and plasticity, and provides support to learning and memory events in animal models of Alzheimer's disease (Hashimoto et al. 2002; Lim et al. 2005) and brain injury (Wu et al. 2004). The action of DHA has been associated with counteracting several aspects of peripheral metabolic disturbances such as reducing the effects of diabetes (Coste et al. 2003). Accordingly, here, we have embarked on studies to test the ability of dietary DHA to counteract the effects of MetS in the central nervous system.
Methods
Animals and experimental design
The experiments were carried out in adult male Sprague–Dawley rats (Charles River Laboratories, Inc., MA, USA) weighing 200–220 g. They were kept in polyacrylic cages and maintained under standard housing conditions (room temperature 22–24°C) with 12 h light/dark cycle. All experiments were performed in accordance with the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of California at Los Angeles (UCLA) Chancellor's Animal Research Committee (ARC).
After acclimatization for 1 week on standard rat chow, rats were trained on the Barnes maze test for 5 days to learn the task, and then randomly assigned to an omega-3 fatty acid diet (n-3 diet) or omega-3 fatty acid deficient diet (n-3 def) diet with or without fructose solution (15%) as drinking water for 6 weeks. Six animals (n = 6) were used in each group. They were kept in individual cages and had free access to their respective diet and drinking solution. The diets were provided in powder in a bowl.
Diet composition
The two custom diets used were based on the composition of the American Institute of Nutrition diet and prepared commercially (Dyets, Bethlehem, PA, USA) as previously described (Greiner et al. 2003). Both diets had the same basal macronutrients, vitamins, minerals and basal fats (hydrogenated coconut and safflower oils). For the casein source, vitamin-free casein Alacid 710 (NZMP North America Inc., CA, USA) was used. Dextrose, maltose-dextrin, cornstarch and sucrose were used as carbohydrate sources. The only difference between the n-3 def and n-3 diets was the amount of n-3 fatty acids, which was achieved by adding 0.5% of flaxseed oil and 1.2% of docosahexaenoic acid capsule oil (Nordic Naturals, Inc., Watsonville, CA, USA) to the n-3 diet. These fats supply LNA (alpha-linolenic acid) and DHA, respectively, as their principal component. The total fat content in both diets was 10 g (100 g)−1 of diet.
Barnes maze test
All rats were tested on the Barnes maze to assess learning and memory functions before and after experimental diets (Barnes, 1979). In brief, animals were trained to locate a dark escape chamber hidden underneath a hole positioned around the perimeter of a disk. The disk was brightly illuminated by four overhead halogen lamps to provide a aversive stimulus. Our maze was manufactured from acrylic plastic to form a disk 1.5 cm thick and 115 cm in diameter, with 18 evenly spaced 7 cm holes at its edges. All trials were recorded simultaneously by a video camera installed directly overhead at the centre of the maze. Animals were trained with two trials per day for five consecutive days before the experimental diet. A trial was started by placing the animal in the centre of the maze covered under a cylindrical start chamber; after a 10 s delay, the start chamber was raised. A training session ended after the animal had entered the escape chamber or when a pre-determined time (5 min) had elapsed, whichever came first. In order to assess memory retention, two trials were given after 6 weeks of experimental diets. All surfaces were routinely cleaned before and after each trial to eliminate possible olfactory cues from preceding animals.
Biochemical analysis
For biochemical analysis, blood was collected from rat tail vein after overnight fasting and then centrifuged at 3000 g for 15 min at 4°C to obtain serum samples. Glucose level was measured using a glucometer (Bayer's Contour meter) and insulin levels were determined by an ELISA kit (Millipore, MA, USA) as per manufacturer's instructions. The homeostasis model assessment ratio (HOMA-R), which is an index of insulin resistance (Matthews et al. 1985), was calculated using the formula: HOMA-R = fasting glucose (mmol l−1) × fasting insulin (μIU ml−1)/22.5. Serum triglyceride was assayed enzymatically by ACE triglycerides reagent (Alfa Wassermann, NJ, USA) using VetACE chemistry analyser (Alfa Wassermann).
Tissue collection
After the memory test the animals were killed by decapitation and the fresh brains were dissected out, frozen in dry ice and stored at −70°C until use.
Fatty acid analysis
Total lipids were extracted from brain tissue according to the method of Bligh & Dyer (1959). Briefly, frozen brains were homogenized in chloroform/methanol (2:1 v/v), containing 50 μg ml−1 of butylated hydroxytoluene to prevent lipid oxidation during the procedure. Tricosanoic acid methylester (C23:0) was used as an internal standard. Tissues were ground to powder under liquid nitrogen and subjected to extraction of total lipids. Fatty acid methylation was done by heating at 90°C for 1 h under 14% (w/v) boron trifluoride–methanol reagent.
Extracted lipids were analysed on a Clarus 500 gas chromatograph (GC; PerkinElmer) with a built-in autosampler. An Elite-WAX column (60 m, 0.32 mm internal diameter, PerkinElmer) was used, with hydrogen as the carrier gas. GC oven temperature was initially held at 140°C for 2 min and raised with a gradient of 5°C min−1 until 250°C and held for 10 min. The total run time was 34 min. The injector and detector were maintained at 250°C and 300°C, respectively. A 1 μl sample of fatty acid methyl esters (FAME) was injected in split injection mode with a 100:1 split ratio. Peaks of resolved fatty acid methyl esters were identified and quantified by comparison with standards (Supelco 37-component FAME Mix).
Immunoblotting
Hippocampal tissues were homogenized in a lysis buffer containing 137 mm NaCl, 20 mm Tris–HCl pH 8.0, 1% NP40, 10% glycerol, 1 mm phenylmethylsulfonylfluoride (PMSF), 10 μg ml−1 aprotinin, 0.1 mm benzethonium chloride, and 0.5 mm sodium vanadate. The homogenates were then centrifuged, the supernatants were collected and total protein concentration was determined according to the MicroBCA procedure (Pierce, IL, USA), using bovine serum albumin (BSA) as standard. Briefly, protein samples were separated by electrophoresis on a 10% polyacrylamide gel and electrotransferred to a PVDF membrane (Millipore, MA, USA). Non-specific binding sites were blocked in Tris-buffered saline (TBS), pH 7.6, containing 5% non-fat dry milk. Membranes were rinsed in buffer (0.05% Tween-20 in TBS) and then incubated with primary antibodies: anti-actin, anti-pLKB1, anti-LKB1, anti-pAMPK, anti-p-synapsin, anti-synapsin, anti-4-hydroxynonenal, anti-insulin receptor (IR)-β (1:500; Santa Cruz Biotechnology, CA, USA), anti-pCREB, anti-CREB, anti-Sir2, anti-synaptophysin (1:1000, Millipore), anti-AMPK, anti-pAkt, anti-Akt (1:1000; Cell Signaling Technology, MA, USA) followed by anti-rabbit or anti-goat IgG horseradish peroxidase conjugate (1:10,000; Santa Cruz Biotechnology). After rinsing with buffer, the immunocomplexes were visualized by chemiluminescence using the ECL kit (Amersham Pharmacia Biotech Inc., NJ, USA) according to the manufacturer's instructions. The film signals were digitally scanned and then quantified using ImageJ software. Actin was used as an internal control for western blot such that data were standardized according to actin values.
Immunoprecipitation
For immunoprecipitation, hippocampal lysate (500 μg protein) was precleared with 20 μl of protein A/G plus-agarose beads (Santa Cruz Biotechnology). After centrifugation, the supernatants were incubated with 2 μg monoclonal anti-phosphotyrosine (PY20) (Millipore) or normal rabbit IgG for 4 h at 4°C followed by overnight incubation with protein A/G-agarose beads. Immunoprecipitates were collected by centrifugation at 2000 g for 5 min at 4°C. The immune complexes were washed 3 times with PBS containing 0.02% Tween-20 and pelleted by centrifugation at 2000 g for 2 min, which were resuspended in 1x electrophoresis buffer for immunoblotting. Expression of insulin receptor was detected in the resulting immunoprecipitates by western blot analysis as mentioned above.
Statistical analysis
The results are expressed as mean ± standard error of the mean (SEM). The data were analysed by one-way analysis of variance (ANOVA) followed by Newman-Keuls test to determine the significance of difference among various groups. A P value < 0.05 was considered as statistically significant. Analysis of correlation (linear regression) was performed on individual samples to evaluate the association between variables.
Results
Effects of fructose and n-3 fatty acid treatments on body weight, caloric intake, food and water consumption
As shown in Table 1, no significant differences were observed in body weight (F3,20 = 0.6932, P > 0.05), food intake (F3,20 = 3.419, P > 0.05) and water intake (F3,20 = 2.043, P > 0.05) among any of the groups. Animals showed a preference towards fructose drinking in comparison to the food intake; however, total caloric intake (F3,20 = 1.832, P > 0.05) was similar in all the groups.
Table 1.
Body weight, caloric intake, food and water consumption in groups subjected to n-3 and n-3 deficient diets with or without fructose water
| Body weight (g) | Food intake (g day−1) | Water intake (ml day−1) | Caloric intake (kcal day−1) | |
|---|---|---|---|---|
| n-3 diet | 508.2 ± 13.51 | 26.18 ± 1.28 | 30.77 ± 1.49 | 109.6 ± 4.14 |
| n-3 def | 492.5 ± 5.43 | 25.72 ± 0.605 | 33.18 ± 2.07 | 102.8 ± 2.17 |
| n-3 def/Fru | 512.8 ± 9.72 | 22.0 ± 1.52 | 45.72 ± 8.21 | 110.2 ± 7.14 |
| n-3 diet/Fru | 522.5 ± 24.44 | 22.58 ± 0.993 | 41.91 ± 4.90 | 117.4 ± 2.17 |
Values are expressed as mean ± SEM.
Influence of dietary n-3 fatty acid manipulation and fructose on cognitive functions
All animals were assessed for spatial learning in the Barnes maze for 5 days before being exposed to experimental diets. All groups showed a decrease in latency time to find the escape hole from first to last day of training, and showed similar latency time (F3,20 = 0.0547, P > 0.05) on last day (day 5), indicating the rats were in the same cognitive condition prior to experimental diets (Fig. 1A).
Figure 1.
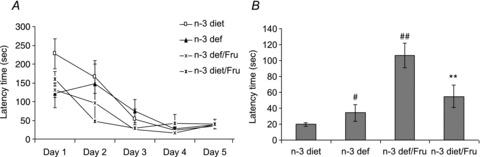
Comparison of latency times in Barnes maze test in groups subjected to n-3 (diet) and n-3 deficient (def) diets with (Fru) or without fructose water before (A) and after (B) experimental diet to assess spatial learning and memory retention, respectively. Values are expressed as mean ± SEM. #P < 0.05, ##P < 0.01: significant difference from n-3 diet; **P < 0.01: significant difference from n-3 def/Fru; ANOVA (one-way) followed by Newman–Keuls test.
Six weeks after experimental diets, a Barnes maze test was performed for 1 day to evaluate memory retention. The deficiency of n-3 fatty acid resulted in a significant increase in latency time indicating memory impairment, which was further enhanced by fructose intake. The effect of fructose on memory in n-3 deficiency was found to be ameliorated by the n-3 diet, indicating that dietary n-3 deficiency influences the vulnerability for fructose induced changes (F3,20 = 10.49, P < 0.01) (Fig. 1B).
Dietary n-3 fatty acid and fructose induced changes in metabolic markers
To assess the metabolic dysfunction, we measured the fasting blood glucose, insulin and triglyceride levels in groups subjected to n-3 fatty acid and fructose treatments (Table 2). Dietary n-3 fatty acid deficiency significantly increased triglyceride level (F3,20 = 10.53, P < 0.01), which was further increased with fructose treatment. With an n-3 deficient diet, fructose elevated the levels of glucose (F3,20 = 9.83, P < 0.01) and insulin (F3,20 = 13.15, P < 0.01); however, the presence of n-3 in the diet reduced the fructose induced increase in insulin and triglyceride levels.
Table 2.
Blood glucose, insulin and triglyceride levels in groups subjected to n-3 and n-3 deficient diets with or without fructose water
| Glucose level (mg dl−1) | Insulin level (ng ml−1) | Triglyceride level (mg dl−1) | |
|---|---|---|---|
| n-3 diet | 81.17 ± 3.02 | 1.46 ± 0.24 | 91.17 ± 10.69 |
| n-3 def | 77.17 ± 4.26 | 1.56 ± 0.30 | 142.0 ± 10.60# |
| n-3 def/Fru | 106.0 ± 6.55## | 3.28 ± 0.21## | 218.8 ± 23.04## |
| n-3 diet/Fru | 99.83 ± 3.13 | 2.54 ± 0.16* | 166.2 ± 17.65* |
Values are expressed as mean ± SEM. #P < 0.05, ##P < 0.01: significant difference from n-3 diet; *P < 0.05: significant difference from n-3 def/Fru; ANOVA (one-way) followed by Newman–Keuls test.
Effect of dietary n-3 fatty acid on fructose induced insulin resistance
We have calculated the HOMA-R to assess the insulin resistance index (Fig. 2A) and found no significant change in insulin resistance index with n-3 fatty acid deficiency. Fructose rats showed a significant increase in insulin resistance index with an n-3 deficient diet, indicating that insulin resistance had developed in high fructose intake rats. Insulin resistance was found to be ameliorated by the presence of n-3, which indicates improved insulin sensitivity (F3,20 = 21.48, P < 0.01).
Figure 2.
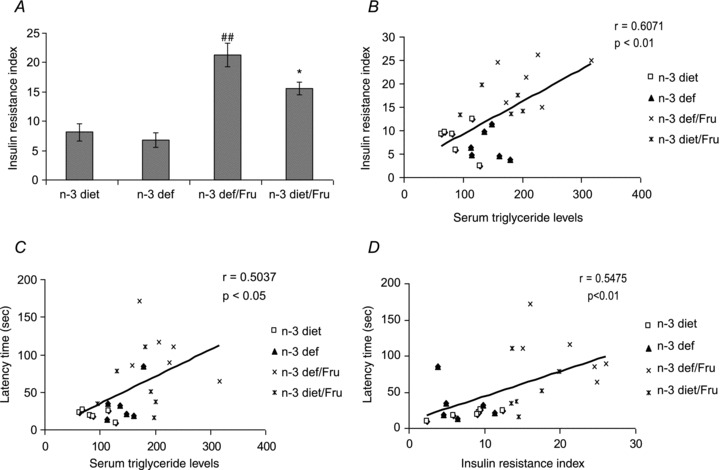
A, Insulin resistance index in groups subjected to n-3 and n-3 deficient diet with or without fructose water. B-D, Correlation analysis revealed a positive correlation between serum triglyceride levels and insulin resistance index (B), serum triglyceride levels and latency time (C), and insulin resistance index and latency time (D) in Barnes maze test. Values are expressed as mean ± SEM. ##P < 0.01 significant difference from n-3 diet, *P < 0.05 significant difference from n-3 def/Fru; ANOVA (one-way) followed by Newman-Keuls test.
Association between metabolic changes and cognitive behaviour
To evaluate a possible association between fructose mediated metabolic changes and cognitive behaviour, we assessed the correlation of serum triglyceride and insulin resistance levels with memory. We found a positive correlation between serum triglyceride levels and insulin resistance index (r = 0.6071, P < 0.01), which indicates that increased serum triglyceride levels may contribute to increase insulin resistance (Fig. 2B). There was a positive correlation (r = 0.5037, P < 0.05) between fructose induced memory deficits and triglyceride levels, indicating a triglyceride association with memory functions (Fig. 2C). Furthermore, we found that the latency time varied in proportion to the insulin resistance (r = 0.5475, P < 0.01), which suggests that memory performance may rely on levels of insulin resistance index (Fig. 2D).
Dietary n-3 fatty acid influences the fructose induced changes in insulin receptor signalling
To measure the changes in insulin receptor signalling, we assessed the levels of insulin receptor tyrosine phosphorylation (pTyrIR) and Akt phosphorylation in groups subjected to n-3 and n-3 deficient diets with or without fructose. Deficiency of dietary n-3 fatty acid in combination with fructose influenced the insulin receptor signalling as evidenced by a decrease in pTyrIR levels in hippocampus, which was found to be reversed in the presence of the n-3 diet (F3,20 = 6.39, P < 0.01) (Fig. 3A). We used immunoprecipitation followed by immunoblotting to assess the levels of pTyrIR in normal and insulin resistant conditions. In addition, the negative correlation found between insulin resistance index and pTyrIR levels (r = −0.5874, P < 0.01), suggests that the increased insulin resistance in the body may disrupt insulin receptor signalling in brain (Fig. 3B). The Akt phosphorylation was found to be decreased with n-3 fatty acid deficiency, which was exacerbated by fructose intake. The presence of n-3 in the diet alleviates the fructose induced changes in Akt phosphorylation (F3,20 = 18.17, P < 0.01) (Fig. 3C).
Figure 3.
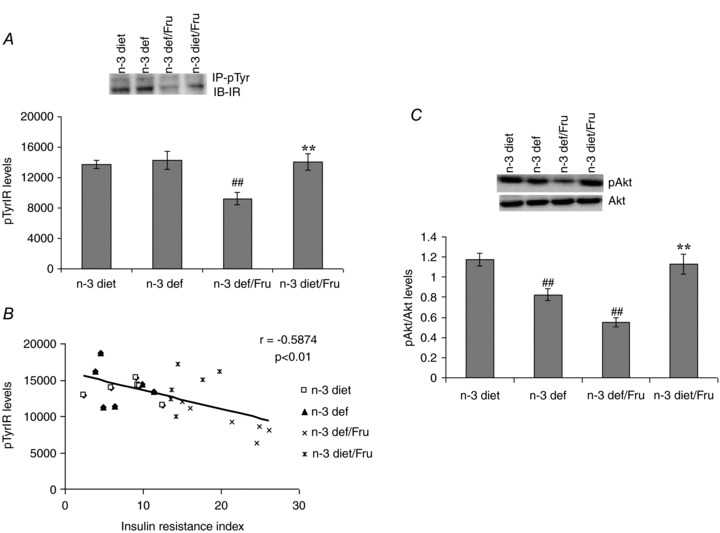
Tyrosine phosphorylation of insulin receptor (pTyrIR; A) and Akt phosphorylation (C) in groups subjected to n-3 and n-3 deficient diets with or without fructose water. B, negative correlation between insulin resistance index and pTyrIR levels. Values are expressed as mean ± SEM. #P < 0.05, ##P < 0.01: significant difference from n-3 diet; **P < 0.01: significant difference from n-3 def/Fru; ANOVA (one-way) followed by Newman–Keuls test.
Dietary n-3 fatty acid and fructose induced changes in molecules involved in energy metabolism
AMPK is activated when AMP and ADP levels in the cells rise owing to a variety of physiological stresses, or the presence of pharmacological inducers. LKB1 is the upstream kinase activating AMPK in response to AMP or ADP increase. An n-3 deficient diet showed a significant decrease in phosphorylation of LKB1, whereas an n-3 diet increased the level of LKB1 phosphorylation (F3,20 = 5.12, P < 0.01) (Fig. 4A). We observed a positive correlation between phosphorylated LKB1 and DHA (r = 0.4398, P < 0.05) (Fig. 4B), and a negative correlation with arachidonic acid (AA; r = −0.7015, P < 0.01) (Fig. 4C), pointing towards a concomitant alteration of LKB1 after diet treatment with the altered lipid composition in brain.
Figure 4.
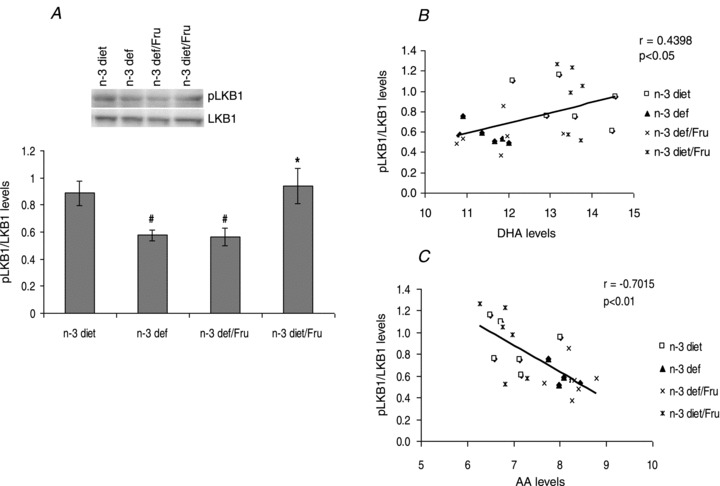
A, phosphorylation of LKB1 in groups subjected to n-3 and n-3 deficient diet with or without fructose water. B, positive correlation between levels of phosphorylated LKB1 and DHA. C, negative correlation between levels of phosphorylated LKB1 and AA. Values are expressed as mean ± SEM. #P < 0.05: significant difference from n-3 diet, *P < 0.05 significant difference from n-3 def/Fru; ANOVA (one-way) followed by Newman–Keuls test.
Omega-3 fatty acid deficiency resulted in a reduction in energy metabolism, as evidenced by the decrease in AMPK phosphorylation, whereas the presence of n-3 in the diet, with or without fructose, increased the level of AMPK phosphorylation (F3,20 = 16.52, P < 0.01) (Fig. 5A). Fructose intake decreased the level of Sir2 in animals deficient in n-3, but not in the animals exposed to the n-3 diet (F3,20 = 12.01, P < 0.01) (Fig. 5B).
Figure 5.
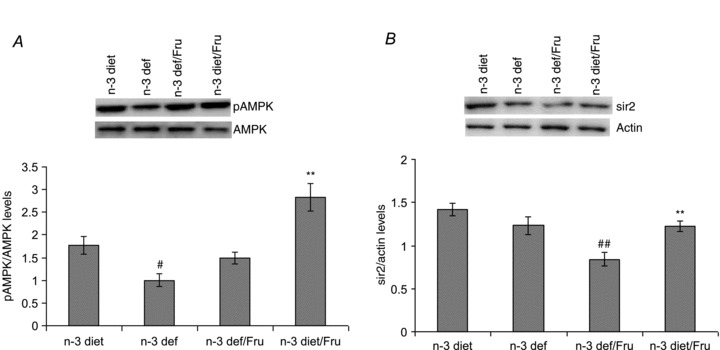
Phosphorylation of AMPK (A) and levels of Sir2 (B) in groups subjected to n-3 and n-3 deficient diets with or without fructose water. Values are expressed as mean ± SEM. #P < 0.05, ##P < 0.01: significant difference from n-3 diet; **P < 0.01: significant difference from n-3 def/Fru; ANOVA (one-way) followed by Newman–Keuls test.
Influence of dietary n-3 fatty acid manipulation and fructose on molecules associated with synaptic plasticity
We assessed cAMP-response element binding (CREB) protein, a family of transcription factors that play a major role in synaptic plasticity and cognitive functions (Benito & Barco, 2010), to study the involvement of metabolic pathways in regulating cognitive function. The deficiency of n-3 fatty acid showed a significant decrease in phosphorylation of CREB (F3,20 = 10.38, P < 0.01), which was further exacerbated by fructose treatment (Fig. 6A). In the fructose drinking group, the presence of dietary n-3 fatty acid increased the level of CREB phosphorylation, suggesting that the presence of n-3 can counter-regulate the fructose induced alterations in synaptic plasticity via CREB. The positive correlation found between Sir2 and CREB indicates the involvement of Sir2 in plasticity and cognitive function in hippocampus (r = 0.6588, P < 0.01) (Fig. 6B). In addition, we also measured synapsin I, a synaptic marker that regulates neurotransmitter release at the synapse, and synaptophysin (SYP), a marker for synaptic growth. There was a significant decrease in phosphorylation of synapsin I and synaptophysin levels with n-3 deficiency. The consumption of fructose also decreased the activation of synapsin I (F3,20 = 11.60, P < 0.01) (Fig. 6C) and synaptophysin level (F3,20 = 8.837, P < 0.01) (Fig. 6D) in the presence of n-3 deficiency; however, with the n-3 diet it shows the opposite effect. We have expressed the level of phosphorylated proteins relative to total protein levels, as there were no significant differences in total protein levels in any of these molecules.
Figure 6.
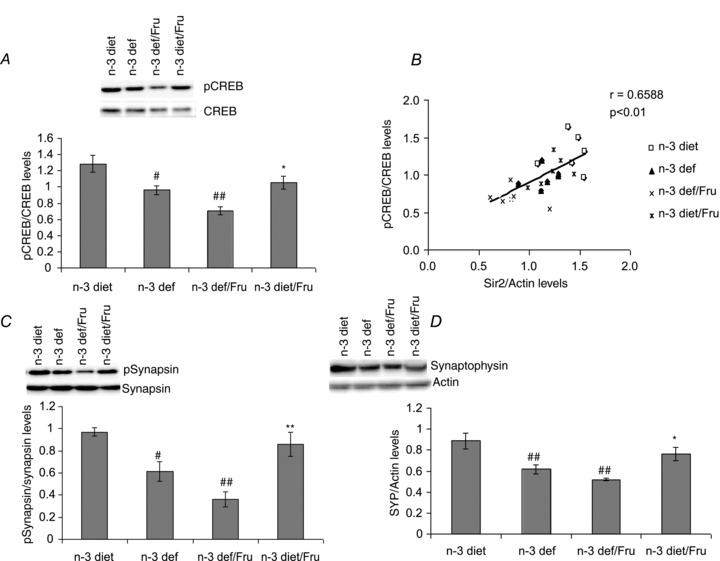
A, CREB phosphorylation. B, positive correlation between Sir2 levels and phosphorylated CREB. C, phosphorylation of synapsin. D, levels of synaptophysin (SYP) in groups subjected to n-3 and n-3 deficient diets with or without fructose water. Values are expressed as mean ± SEM. #P < 0.05, ##P < 0.01: significant difference from n-3 diet, *P < 0.05, **P < 0.01: significant difference from n-3 def/Fru; ANOVA (one-way) followed by Newman–Keuls test.
Effect of n-3 fatty acid dietary manipulation on lipid peroxidation induced by high fructose intake
Free radical attack to unsaturated fatty acids has been shown to increase 4-hydroxynonenal (4-HNE), a marker for lipid peroxidation (Subramaniam et al. 1997). The level of 4-HNE was increased significantly with fructose intake in n-3 fatty acid deficiency as compared to the n-3 diet, whereas rats fed on the n-3 diet showed an increase in 4-HNE level (F3,20 = 6.332, P < 0.01) (Fig. 7). These results indicate that the n-3 fatty acid deficient diet makes the brain more vulnerable to fructose induced free radical attack.
Figure 7.
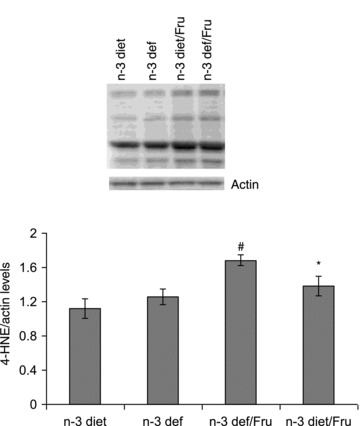
Level of 4-HNE in groups subjected to n-3 and n-3 deficient diets with or without fructose water. Values are expressed as mean ± SEM. #P < 0.05: significant difference from n-3 diet; *P < 0.05: significant difference from n-3 def/Fru; ANOVA (one-way) followed by Newman–Keuls test.
Fatty acid composition in brain
We performed a profiling of various fatty acids in brain in response to the diets by using gas chromatography. Results of a detailed analysis of the brain fatty acid composition are shown in Table 3. The n-3 deficient diet with or without fructose did not alter saturated or mono-unsaturated fatty acids levels, but specifically decreased the levels of DHA (22:6n-3) (F3,20 = 14.25, P < 0.001), and increased the n-6 polyunsaturated fatty acids (PUFAs) docosapentanoic acid (DPA; 22:5n-6) (F3,20 = 253.9, P < 0.001) and AA (20:4n-6) (F3,20 = 21.45, P < 0.001). The exposure to the n-3 diet reversed the changes induced by n-3 deficiency and fructose. Clinical studies (Griffin, 2008) indicate that the ratio of n-6 to n-3 fatty acids is important to maintain health. We found an increased ratio of n-6 to n-3 during n-3 deficiency and/or fructose and this ratio can be counter-regulated by dietary n-3 fatty acid.
Table 3.
Fatty acid composition in groups subjected to n-3 and n-3 deficient diets with or without fructose water
| Fatty acids | n-3 diet | n-3 def | n-3 def/Fru | n-3 diet/Fru |
|---|---|---|---|---|
| 14:0 | 0.338 ± 0.019 | 0.299 ± 0.039 | 0.311 ± 0.004 | 0.391 ± 0.018 |
| 16:0 | 20.27 ± 0.274 | 20.57 ± 0.552 | 20.55 ± 0.377 | 21.00 ± 0.298 |
| 18:0 | 18.72 ± 0.150 | 19.32 ± 0.239 | 18.84 ± 0.199 | 18.81 ± 0.295 |
| 18:1 | 15.10 ± 0.214 | 14.34 ± 0.351 | 14.55 ± 0.208 | 14.84 ± 0.225 |
| 18:2n-6 (LA) | 0.353 ± 0.020 | 0.310 ± 0.063 | 0.250 ± 0.012 | 0.340 ± 0.016 |
| 20:0 | 0.254 ± 0.012 | 0.246 ± 0.018 | 0.236 ± 0.017 | 0.238 ± 0.013 |
| 20:1 | 1.028 ± 0.020 | 0.963 ± 0.076 | 0.997 ± 0.045 | 0.953 ± 0.020 |
| 20:4n-6 (AA) | 7.017 ± 0.228 | 8.149 ± 0.107## | 8.265 ± 0.149## | 6.821 ± 0.136** |
| 22:0 | 0.290 ± 0.023 | 0.265 ± 0.017 | 0.285 ± 0.022 | 0.264 ± 0.010 |
| 22:5n-6 (DPA) | 0.212 ± 0.010 | 0.968 ± 0.032## | 0.912 ± 0.036## | 0.222 ± 0.014** |
| 22:6n-3 (DHA) | 13.48 ± 0.388 | 11.44 ± 0.199## | 11.77 ± 0.375## | 13.52 ± 0.089** |
| 24:0 | 0.652 ± 0.048 | 0.578 ± 0.035 | 0.679 ± 0.040 | 0.629 ± 0.023 |
| 24:1n-9 | 1.254 ± 0.078 | 1.167 ± 0.088 | 1.260 ± 0.064 | 1.212 ± 0.044 |
| n-6/n-3 | 0.562 ± 0.010 | 0.824 ± 0.017## | 0.803 ± 0.020## | 0.546 ± 0.011** |
Values are expressed as mean ± SEM. ##P < 0.01: significant difference from n-3 diet; **P < 0.01: significant difference from n-3 def/Fru; ANOVA (one-way) followed by Newman–Keuls test. LA, linoleic acid; AA, arachidonic acid; DPA, docosapentaenoic acid; DHA, docosahexaenoic acid.
Discussion
It is becoming an alarming public health issue that unhealthy dietary habits can lead to metabolic disorders with a heavy toll on mental health. We have embarked on studies to determine crucial mechanisms by which aberrant body metabolism can disrupt brain plasticity and cognition. In addition, we have investigated the ability of dietary n-3 fatty acids to counteract metabolic disorders. We found that the lack of n-3 fatty acids in the diet elevated parameters of peripheral insulin resistance, and resulted in disrupted insulin signalling in brain, and these effects were aggravated by fructose treatment. Moreover, dysfunctional insulin receptor signalling was associated with lowered learning performance in the Barnes maze. These results illustrate a potential mechanism by which metabolic disorders can influence cognitive abilities. Furthermore, according to our results, n-3 fatty acids appear as a crucial dietary component to maintain metabolic homeostasis and mental health, particularly under challenging situations.
Metabolic dysfunction and cognitive performance
We found that dietary n-3 fatty acid deficiency compromised molecular mechanisms important for the maintenance of metabolic homeostasis, with subsequent effects on cognitive abilities. In particular, the deficiency of n-3 reflected a decline in spatial memory in proportion to the intensity of the index of insulin resistance, and all parameters were further aggravated by an increase in the fructose intake. Although there was a preference towards fructose drinking in comparison to the food intake, no differences were observed in body weight and total caloric intake, thus suggesting that obesity is not a major contributor to altered memory functions in this model. Based on current results, it appears that an increase in peripheral insulin resistance is pivotal for the protracted cognitive function as observed in the current study. As an attempt to understand how peripheral metabolic events can alter the brain, we assessed several markers of metabolic function in serum. We found that increased consumption of fructose, particularly when combined with the DHA deficiency, resulted in hyperinsulinaemia, hyperglycaemia and an increase in triglyceride (TG) levels.
The fact that the insulin resistance index increased in proportion to TG levels, and that cognitive impairment is associated with TG levels, raises the possibility that fructose may predispose the brain towards insulin resistance via its effects on TGs. Indeed, it has been shown that application of TGs to liver cells decreases the ability of insulin to activate its signalling cascade (Kim et al. 2007), and that TGs can penetrate the blood–brain barrier (Drew et al. 1998). Insulin resistance in humans is commonly accompanied by elevated levels of circulating TGs (Lêet al. 2009). The association between TG levels and cognitive function is supported by previous reports that an injection of TGs directly into the brain ventricles impairs memory (Farr et al. 2008), and recent evidence indicates that hippocampal insulin signalling facilitates memory (Agrawal et al. 2011). Based on these studies, we hypothesize that metabolic dysfunction leading to insulin resistance can affect memory performance through regulation of the insulin signalling system. It is also possible that fructose can directly affect brain function. Evidence is accumulating that neuronal cells can metabolize fructose (Funari et al. 2007), and that fructose feeding increases the expression of fructose sensitive glucose transporters (glut5) in the hippocampus (Shu et al. 2006). Thus, it is possible that fructose, or one of its brain metabolites, directly induced the memory deficits that were observed here.
Insulin signalling in brain and metabolic dysfunction
Our results showing a correlation between insulin resistance index and pTyrIR indicate a possible association between peripheral insulin and disruption of insulin signalling in brain. In addition, the fact that memory deficits were positively correlated with increases in insulin resistance index suggests the possibility that insulin signals neurons directly, as insulin can go across the blood–brain barrier (Banks et al. 1997). We found that fructose and DHA deficiency increased hippocampal insulin resistance, as evidenced by a decrease in the insulin receptor signalling. Insulin resistance is the consequence of impaired signalling at the level of the insulin receptor and its downstream effectors as a result of post-translational modifications such as altered phosphorylation. In the present study, we evaluated the insulin receptor phosphorylation by immunoprecipitation of phosphotyrosine followed by immunoblotting with the insulin receptor, which provides an indication of the activation of the insulin receptor. We found that phosphorylation of the insulin receptor and its signalling molecules Akt were diminished after the n-3 deficient diet, and these effects were aggravated after fructose treatment. These results indicate the importance of dietary DHA for maintaining proper insulin signalling in brain. In addition, these results show the necessity of adequate levels of n-3 in the diet to cope with challenges imposed by fructose.
Influences of metabolic disturbances on neuronal signalling
Energy metabolism and membrane function are tightly related events. Our results show that metabolic dysfunction potentiates pathways that can lead to the disruption of membrane homeostasis, and this may have detrimental consequences for neuronal function. We have found that fructose intake disrupts plasma membranes as evidenced by an increase in the levels of 4-HNE, a marker of lipid peroxidation. In turn, the deficiency in DHA exacerbated the deleterious effects of fructose on the stability of plasma membranes, as reflected by decreases in DHA levels and increases in AA levels. The relationship between n-3 and n-6 is important for the function of the plasma membrane, which is also required for synaptic plasticity, growth, and repair. Peroxidation of membrane bound n-6 AA results in the generation of 4-HNE, which produces alterations in the function of key membrane proteins including glucose transporter, glutamate transporter, and sodium potassium ATPases (Mark et al. 1997; Lauderback et al. 2001). Membrane peroxidation also produces alterations in insulin receptor signalling via Akt, as it has recently been reported that 4-HNE inhibits insulin-dependent Akt signalling in HepG2 cells (Shearn et al. 2011). The overall evidence provides an indication of the influence of insulin resistance on lipid peroxidation and membrane homeostasis disruption.
PUFA precursors of the n-3 or n-6 families are essential nutriments that cannot be synthesized de novo in mammals. They exist in plants as precursors 18:2n-6 (linoleic acid) and 18:3n-3 (α-linolenic acid) and are metabolized by elongations and desaturations into arachidonic acid, EPA (eicosapentaenoic acid) and DHA in mammals (Igarashi et al. 2007). Because the two series of PUFAs compete for their biosynthetic enzymes, and because they have distinct physiological properties, the dietary n-6/n-3 ratio is of fundamental importance. Here, the n-3 deficient diet elicited a significant increase in n-6/n-3 ratio alone or in the presence of fructose. The n-3 diet in the presence of fructose was able to maintain this ratio within the normal range, hence suggesting its efficacy in balancing the deleterious effects of a high sugar diet. The increase in the ratio n-6/n-3 observed in the group of animals fed on n-3 def and fructose might be indicative of a substitution of n-3 by the n-6 in the membrane, which may alter membrane fluidity. A reduction in membrane fluidity may be responsible for disrupting membrane insulin receptor signalling in conjunction with its downstream cascades such as IRS-1 and Akt, thereby altering synaptic plasticity and cognition.
Dietary influences on energy homeostasis
Brain energy metabolism is central to all cellular processes that maintain neuronal functionality and has the capacity to modulate the function of the plasma membrane and neuronal signalling. Accordingly, we assessed the effects of the dietary interventions on molecular markers of cellular energy metabolism. ATP and NAD are small molecules involved in all energy transactions in cells, and they can be sensed by regulatory proteins, such as AMP-activated protein kinase (AMPK, which senses the AMP/ATP ratio) and sirtuins (which require NAD to deacetylate protein substrates). We assessed AMPK, a serine-threonine kinase, which has the ability to sense low energy levels and activate or inhibit the appropriate molecules to re-establish the proper energy balance of the cell. The rise in AMPK phosphorylation in n-3 fed rats advocates that n-3 may activate mechanisms to conserve ATP levels in the hippocampus. In turn, a decrease in AMPK activation in the n-3 deficient diet may indicate a disturbance in energy homeostasis. Studies have supported a mechanistic association between Sir2 and AMPK, as they showed that NAD, a critical substrate for Sir2 function, is activated by AMPK in a dose-dependent manner (Rafaeloff-Phail et al. 2004). Accordingly, we assessed Sir2 based on its implications in cellular homeostasis and energy metabolism (Starai et al. 2002; Hallows et al. 2006). It has previously been shown that consumption of a high caloric diet rich in saturated fat and sugars has harmful consequences for synaptic plasticity and reduces the expression of Sir2 in the hippocampus and cerebral cortex (Wu et al. 2006). Here we found that fructose intake decreases Sir2 levels, indicating the association between energy metabolism and Sir2. The fact that the diet rich in n-3 fatty acids normalized Sir2 levels emphasizes the salutary effects of DHA on maintaining energy homeostasis.
To further explore the effects of dietary interventions on the signalling of metabolic sensors, we studied the phosphorylation status of LKB1, an upstream kinase that activates AMPK in response to AMP or ADP increase. Our data showed that deficiency of n-3 with or without fructose promoted a decrease in phosphorylation of LKB1, indicating that activation of LKB1 may rely on levels of n-3 fatty acid in brain. Also the changes in LKB1 phosphorylation varied in direct proportion to DHA level and inverse proportion to AA level, which suggests that a decline in the ratio n-6/n-3 contributes to maintaining energy homeostasis. Overall, the alterations in metabolic sensors (LKB1, AMPK and Sir2) seem to be a consequence of altered lipid composition.
Implications for synaptic plasticity
We show the effects of dietary manipulations on several markers of synaptic plasticity. It has been reported that AMPK regulates cAMP-response element binding (CREB) proteins (Thomson et al. 2008), which is a family of transcription factors playing a major role in synaptic plasticity and cognitive functions (Benito & Barco, 2010). Recently SIRT1 (mammalian Sir2 homologue) has also been shown to modulate synaptic plasticity and memory formation via post-transcriptional regulation of CREB (Gao et al. 2010). The positive correlation between Sir2 and CREB in our study also reflects some interaction between Sir2 and the regulation of plasticity and cognitive function in the hippocampus. We have also assessed synapsin I and synaptophysin, the markers for synaptic plasticity, to examine the modulatory role of dietary factors on synaptic functions. Synapsin I is a nerve terminal protein implicated in the regulation of neurotransmitter release during synaptic plasticity (Cesca et al. 2010) as well as synaptogenesis and neurite outgrowth (Han et al. 1991; Lu et al. 1992). Interestingly, the synapsin I gene is also thought to be regulated by CREB (Silva et al. 1998). Results indicate that n-3 deficiency decreases phosphorylation of CREB and synapsin I, and fructose consumption potentiated this effect. Synaptophysin, a marker for synaptic growth, was also decreased with n-3 deficiency and fructose treatment. It is suggested to be involved in calcium binding (Rehm et al. 1986), channel formation (Thomas et al. 1988), exocytosis (Alder et al. 1992) and synaptic vesicle recycling via endocytosis (Evans & Cousin, 2005); makes it an important player for regulating synaptic plasticity. The n-3 supplementation was found to be obligatory for normalizing this effect even in the presence of fructose, suggesting that n-3 fatty acids can restore the cognitive function under challenging conditions by normalizing the action of insulin resistance on synaptic plasticity via CREB, synapsin I and synaptophysin.
Health implications: dietary n-3 fatty acid and vulnerability to brain disorders
Our results suggest that the lack of dietary n-3 fatty acids predisposes the organism to MetS, promotes brain insulin resistance, and increases the vulnerability to cognitive dysfunction. DHA is a key component of neuronal membranes at sites of signal transduction at the synapse, suggesting that its action is vital for brain function (Gomez-Pinilla, 2008). Because mammals are inefficient at producing DHA from precursors, supplementation of DHA in the diet is crucial for ensuring the proper function of neurons during homeostatic conditions. In the present study, we found that n-3 deficiency increases the vulnerability to the effects of fructose, as evidenced by disruptions of insulin signalling, membrane homeostasis and cognitive functions. This implies that adequate levels of DHA are particularly necessary under challenging conditions. Based on the abundant consumption of sugars in Western society, proper consumption of DHA emerges as a primary necessity to foster protection against the effects of metabolic syndrome in the brain. Evidence suggests that DHA serves to improve neuronal function by supporting synaptic membrane fluidity (Suzuki et al. 1998), and regulating gene expression and cell signalling (Salem et al. 2001). This implies that insufficient DHA can result in neuronal dysfunction affecting a broad array of functional modalities. For example, it has been recently reported that n-3 deficiency during brain maturation results in elevated anxiety-like behaviour during adulthood (Bhatia et al. 2011).
Conclusions
The concept of metabolic syndrome has been mainly associated with the body, and here we introduce this concept with respect to the brain. We provided evidence supporting the harmful impact of the metabolic syndrome on the brain, impacting synaptic plasticity and cognitive function. Our results show the impact of dietary n-3 deficiency on brain function, using a mechanism centred on the action of insulin signalling, energy metabolism and membrane homeostasis. The deficiency of dietary n-3 increases vulnerability to impaired cognitive functions, and intake of a high fructose diet exacerbates this condition. It is encouraging that the presence of the n-3 diet was sufficient to buffer the effects of metabolic dysfunction. Overall, our results provide mechanistic evidence for how dietary factors can interact to regulate brain plasticity, which can further alter lifelong susceptibility to metabolic disorders (Fig. 8). In terms of public health, these results support the encouraging possibility that healthy diets can attenuate the action of unhealthy diets such that the right combination of foods is crucial for a healthy brain.
Figure 8.
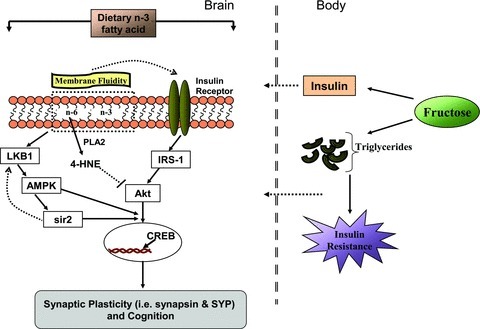
Proposed mechanism by which insulin resistance leads to disruption of brain metabolism with subsequent effects on synaptic plasticity and cognition. It is also depicted how dietary n-3 fatty acid content in the diet may influence the vulnerability to metabolic dysfunction. Abundant consumption of fructose leads to an increase in triglyceride and insulin levels in the body, which can affect brain function after crossing the blood–brain barrier. The changes in membrane n-3 and n-6 fatty acids may alter the membrane fluidity, thereby disrupting membrane insulin receptor function. This, in turn, can influence downstream insulin receptor cascades such as IRS-1, Akt and CREB, leading to alteration in synaptic plasticity and cognition. Release of n-6 arachidonic acid from the phospholipid membrane by phospholipase A2 (PLA2) and subsequent peroxidation result in the generation of 4-HNE, which produces alterations in insulin receptor signalling via inhibiting Akt signalling. These alterations can result in abnormal neuronal signalling, which can reduce learning capacity and other functions that rely on synaptic plasticity and neuronal excitability. Dietary components can also affect mitochondrial energy production by modulating energy molecules LKB1, AMPK and Sir2, which are important for maintaining neuronal excitability and synaptic function via CREB. Regulation of synaptic functions by dietary intervention can also be directly mediated by synapsin I and synaptophysin (SYP). These events are important for our understanding of how dietary factors can interact to regulate brain plasticity, and how dietary management can be used to promote brain health.
Acknowledgments
This work was supported by National Institutes of Health Grants NS50465 and NS56413.
Glossary
Abbreviations
- AA
arachidonic acid
- DHA
docosahexaenoic acid
- 4-HNE
4-hydroxynonenal
- HOMA-R
homeostasis model assessment ratio
- MetS
metabolic syndrome
- pTyrIR
insulin receptor tyrosine phosphorylation
- PUFA
polyunsaturated fatty acid
- TGs
triglycerides
Author contributions
Experiments were performed by R.A. at the Department of Integrative Biology and Physiology, UCLA. Study design, data analysis, data interpretation and manuscript writing: R.A. Study concept, manuscript editing and critical revision for intellectual content: F.G.-P. Both authors approved the final version of the manuscript. Conflicts of interest: none.
References
- Agrawal R, Tyagi E, Shukla R, Nath C. A study of brain insulin receptors, AChE activity and oxidative stress in rat model of ICV STZ induced dementia. Neuropharmacology. 2009;56:779–787. doi: 10.1016/j.neuropharm.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Agrawal R, Tyagi E, Shukla R, Nath C. Insulin receptor signaling in rat hippocampus: a study in STZ (ICV) induced memory deficit model. Eur Neuropsychopharmacol. 2011;21:261–273. doi: 10.1016/j.euroneuro.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Alder J, Xie ZP, Valtorta F, Greengard P, Poo M. Antibodies to synaptophysin interfere with transmitter secretion at neuromuscular synapses. Neuron. 1992;9:759–768. doi: 10.1016/0896-6273(92)90038-f. [DOI] [PubMed] [Google Scholar]
- Banks WA, Jaspan JB, Kastin AJ. Selective, physiological transport of insulin across the blood-brain barrier: novel demonstration by species-specific radioimmunoassays. Peptides. 1997;18:1257–1262. doi: 10.1016/s0196-9781(97)00198-8. [DOI] [PubMed] [Google Scholar]
- Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- Benito E, Barco A. CREB's control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 2010;33:230–240. doi: 10.1016/j.tins.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Bhatia HS, Agrawal R, Sharma S, Huo YX, Ying Z, Gomez-Pinilla F. Omega-3-fatty acid deficiency during brain maturation reduces neuronal and behavioral plasticity in adulthood. PLos ONE. 2011;6(12):e28451. doi: 10.1371/journal.pone.0028451. (1–9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biessels GJ, van der Heide LP, Kamal A, Bleys RL, Gispen WH. Ageing and diabetes: implications for brain function. Eur J Pharmacol. 2002;441:1–14. doi: 10.1016/s0014-2999(02)01486-3. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol. 2010;91:313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Coste TC, Gerbi A, Vague P, Pieroni G, Raccah D. Neuroprotective effect of docosahexaenoic acid-enriched phospholipids in experimental diabetic neuropathy. Diabetes. 2003;52:2578–2585. doi: 10.2337/diabetes.52.10.2578. [DOI] [PubMed] [Google Scholar]
- Drew PA, Smith E, Thomas PD. Fat distribution and changes in the blood brain barrier in a rat model of cerebral arterial fat embolism. J Neurol Sci. 1998;156:138–143. doi: 10.1016/s0022-510x(98)00039-2. [DOI] [PubMed] [Google Scholar]
- Evans GJ, Cousin MA. Tyrosine phosphorylation of synaptophysin in synaptic vesicle recycling. Biochem Soc Trans. 2005;33:1350–1353. doi: 10.1042/BST20051350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr SA, Yamada KA, Butterfield DA, Abdul HM, Xu L, Miller NE, Banks WA, Morley JE. Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology. 2008;149:2628–2636. doi: 10.1210/en.2007-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funari VA, Crandall JE, Tolan DR. Fructose metabolism in the cerebellum. Cerebellum. 2007;6:130–140. doi: 10.1080/14734220601064759. [DOI] [PubMed] [Google Scholar]
- Gao J, Wang WY, Mao YW, Gräff J, Guan JS, Pan L, Mak G, Kim D, Su SC, Tsai LH. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature. 2010;466:1105–1109. doi: 10.1038/nature09271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerozissis K. Brain insulin, energy and glucose homeostasis; genes, environment and metabolic pathologies. Eur J Pharmacol. 2008;585:38–49. doi: 10.1016/j.ejphar.2008.01.050. [DOI] [PubMed] [Google Scholar]
- Gerrits PM, Tsalikian E. Diabetes and fructose metabolism. Am J Clin Nutr. 1993;58:796S–799S. doi: 10.1093/ajcn/58.5.796S. [DOI] [PubMed] [Google Scholar]
- Gomez-Pinilla F. Brain foods: the effects of nutrients on brain function. Nat Rev Neurosci. 2008;9:568–578. doi: 10.1038/nrn2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner RS, Catalan JN, Moriguchi T, Salem N., Jr Docosapentaenoic acid does not completely replace DHA in n-3 FA-deficient rats during early development. Lipids. 2003;38:431–435. doi: 10.1007/s11745-003-1080-2. [DOI] [PubMed] [Google Scholar]
- Griffin BA. How relevant is the ratio of dietary n-6 to n-3 polyunsaturated fatty acids to cardiovascular disease risk? Evidence from the OPTILIP study. Curr Opin Lipidol. 2008;19:57–62. doi: 10.1097/MOL.0b013e3282f2e2a8. [DOI] [PubMed] [Google Scholar]
- Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A. 2006;103:10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han HQ, Nichols RA, Rubin MR, Bähler M, Greengard P. Induction of formation of presynaptic terminals in neuroblastoma cells by synapsin IIb. Nature. 1991;349:697–700. doi: 10.1038/349697a0. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Hossain S, Shimada T, Sugioka K, Yamasaki H, Fujii Y, Ishibashi Y, Oka J, Shido O. Docosahexaenoic acid provides protection from impairment of learning ability in Alzheimer's disease model rats. J Neurochem. 2002;81:1084–1091. doi: 10.1046/j.1471-4159.2002.00905.x. [DOI] [PubMed] [Google Scholar]
- Igarashi M, Ma K, Chang L, Bell JM, Rapoport SI. Dietary n-3 PUFA deprivation for 15 weeks upregulates elongase and desaturase expression in rat liver but not brain. J Lipid Res. 2007;48:2463–2470. doi: 10.1194/jlr.M700315-JLR200. [DOI] [PubMed] [Google Scholar]
- Kelley GL, Allan G, Azhar S. High dietary fructose induces a hepatic stress response resulting in cholesterol and lipid dysregulation. Endocrinology. 2004;145:548–555. doi: 10.1210/en.2003-1167. [DOI] [PubMed] [Google Scholar]
- Kim DS, Jeong SK, Kim HR, Chae SW, Chae HJ. Effects of triglyceride on ER stress and insulin resistance. Biochem Biophys Res Commun. 2007;363:140–145. doi: 10.1016/j.bbrc.2007.08.151. [DOI] [PubMed] [Google Scholar]
- Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of Aβ1–42. J Neurochem. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- Lim GP, Calon F, Morihara T, Yang F, Teter B, Ubeda O, Salem N, Jr, Frautschy SA, Cole GM. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J Neurosci. 2005;25:3032–3040. doi: 10.1523/JNEUROSCI.4225-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê KA, Ith M, Kreis R, Faeh D, Bortolotti M, Tran C, Boesch C, Tappy L. Fructose overconsumption causes dyslipidemia and ectopic lipid deposition in healthy subjects with and without a family history of type 2 diabetes. Am J Clin Nutr. 2009;89:1760–1765. doi: 10.3945/ajcn.2008.27336. [DOI] [PubMed] [Google Scholar]
- Lu B, Greengard P, Poo MM. Exogenous synapsin I promotes functional maturation of developing neuromuscular synapses. Neuron. 1992;8:521–529. doi: 10.1016/0896-6273(92)90280-q. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid β-peptide. J Neurochem. 1997;68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- Newcomer JW. Metabolic syndrome and mental illness. Am J Manag Care. 2007;13:S170–177. [PubMed] [Google Scholar]
- Rafaeloff-Phail R, Ding L, Conner L, Yeh WK, McClure D, Guo H, Emerson K, Brooks H. Biochemical regulation of mammalian AMP-activated protein kinase activity by NAD and NADH. J Biol Chem. 2004;279:52934–52939. doi: 10.1074/jbc.M409574200. [DOI] [PubMed] [Google Scholar]
- Rehm H, Wiedenmann B, Betz H. Molecular characterization of synaptophysin, a major calcium-binding protein of the synaptic vesicle membrane. EMBO J. 1986;5:535–541. doi: 10.1002/j.1460-2075.1986.tb04243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem N, Jr, Litman B, Kim HY, Gawrisch K. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids. 2001;36:945–959. doi: 10.1007/s11745-001-0805-6. [DOI] [PubMed] [Google Scholar]
- Shearn CT, Fritz KS, Reigan P, Petersen DR. Modification of Akt2 by 4-hydroxynonenal inhibits insulin-dependent Akt signaling in HepG2 cells. Biochemistry. 2011;50:3984–3996. doi: 10.1021/bi200029w. [DOI] [PubMed] [Google Scholar]
- Shu HJ, Isenberg K, Cormier RJ, Benz A, Zorumski CF. Expression of fructose sensitive glucose transporter in the brains of fructose-fed rats. Neuroscience. 2006;140:889–895. doi: 10.1016/j.neuroscience.2006.02.071. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science. 2002;298:2390–2392. doi: 10.1126/science.1077650. [DOI] [PubMed] [Google Scholar]
- Subramaniam R, Roediger F, Jordan B, Mattson MP, Keller JN, Waeg G, Butterfield DA. The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortical synaptosomal membrane proteins. J Neurochem. 1997;69:1161–1169. doi: 10.1046/j.1471-4159.1997.69031161.x. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Park SJ, Tamura M, Ando S. Effect of the long-term feeding of dietary lipids on the learning ability, fatty acid composition of brain stem phospholipids and synaptic membrane fluidity in adult mice: a comparison of sardine oil diet with palm oil diet. Mech Ageing Dev. 1998;101:119–128. doi: 10.1016/s0047-6374(97)00169-3. [DOI] [PubMed] [Google Scholar]
- Thomas L, Hartung K, Langosch D, Rehm H, Bamberg E, Franke WW, Betz H. Identification of synaptophysin as a hexameric channel protein of the synaptic vesicle membrane. Science. 1988;242:1050–1053. doi: 10.1126/science.2461586. [DOI] [PubMed] [Google Scholar]
- Thomson DM, Herway ST, Fillmore N, Kim H, Brown JD, Barrow JR, Winder WW. AMP-activated protein kinase phosphorylates transcription factors of the CREB family. J Appl Physiol. 2008;104:429–438. doi: 10.1152/japplphysiol.00900.2007. [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gómez-Pinilla F. Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. J Neurotrauma. 2004;21:1457–1467. doi: 10.1089/neu.2004.21.1457. [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F. Oxidative stress modulates Sir2α in rat hippocampus and cerebral cortex. Eur J Neurosci. 2006;23:2573–2580. doi: 10.1111/j.1460-9568.2006.04807.x. [DOI] [PubMed] [Google Scholar]