

Abstract
Riluzole, the only drug available against amyotrophic lateral sclerosis (ALS), has recently been shown to block muscle ACh receptors (AChRs), raising concerns about possible negative side-effects on neuromuscular transmission in treated patients. In this work we studied riluzole's impact on the function of muscle AChRs in vitro and on neuromuscular transmission in ALS patients, using electrophysiological techniques. Human recombinant AChRs composed of α1β1δ subunits plus the γ or ɛ subunit (γ- or ɛ-AChR) were expressed in HEK cells or Xenopus oocytes. In both preparations, riluzole at 0.5 μm, a clinically relevant concentration, reversibly reduced the amplitude and accelerated the decay of ACh-evoked current if applied before coapplication with ACh. The action on γ-AChRs was more potent and faster than on ɛ-AChRs. In HEK outside-out patches, riluzole-induced block of macroscopic ACh-evoked current gradually developed during the initial milliseconds of ACh presence. Single channel recordings in HEK cells and in human myotubes from ALS patients showed that riluzole prolongs channel closed time, but has no effect on channel conductance and open duration. Finally, compound muscle action potentials (CMAPs) evoked by nerve stimulation in ALS patients remained unaltered after a 1 week suspension of riluzole treatment. These data indicate that riluzole, while apparently safe with regard to synaptic transmission, may affect the function of AChRs expressed in denervated muscle fibres of ALS patients, with biological consequences that remain to be investigated.
Key points
Riluzole is the only drug available against amyotrophic lateral sclerosis (ALS), a fatal disease characterized by death of motor neurones.
Recently it has been shown to block muscle ACh receptors (AChRs), raising concerns about possible side-effects on neuromuscular transmission in patients.
In this work we studied the effect of riluzole on the function of muscle AChRs in vitro and on neuromuscular transmission in ALS patients.
Data indicate that riluzole is apparently safe regarding neuromuscular transmission in patients.
However, riluzole may affect the function of AChRs expressed in denervated muscle fibres of ALS patients, with biological consequences that remain to be investigated.
Introduction
Riluzole is the only drug currently registered to treat patients with amyotrophic lateral sclerosis (ALS), a fatal disease due to selective degeneration of upper and/or lower motoneurones. The compound has several adverse side-effects, among which is asthenia, that further decrease patients’ quality of life, so that a full assessment of its effects might help in devising countermeasures. Riluzole affects the function of a variety of ion channels in neurones and possibly in muscle (Bellingham 2011). In particular, it was reported to impair the function of recombinant mouse acetylcholine receptors (AChRs) at very high concentrations (Mohammadi et al. 2002), and even at clinically used concentrations (0.5 to 5 μm; Bellingham 2011) in human muscle preparations obtained from ALS or denervated patients (Palma et al. 2011). Since AChRs mediate nerve–muscle communication at the neuromuscular junction, the latter report supported the hypothesis that riluzole worsens the compromised neuromuscular transmission in ALS patients, which was further tested in this work.
To elucidate riluzole's action at the molecular level, its effect on both muscle AChR isoforms must be examined. In innervated fibres, receptors are formed by the α1β1ɛδ subunits (ɛ-AChR) and are densely packed at the endplate (Katz & Miledi, 1964). In denervated muscle cells, AChRs are formed by α1β1γδ subunits (γ-AChR), with an almost uniform distribution over the entire sarcolemma (Witzemann et al. 1991). ALS patients experience muscle denervation and reinnervation through collateral sprouting of healthy motor neurones (Eisen & Swash, 2001), so that both ɛ- and γ-AChR are present, reflecting the presence of denervated muscle fibres.
In this work we prove that in vitro riluzole has differential effects on recombinant human γ- or ɛ-AChRs, and provide a characterization of its action at the single channel level in transfected cells and in myotubes derived from ALS patients. Moreover, we analyse the effect of riluzole on nerve–muscle transmission in ALS patients before and during a brief suspension of riluzole treatment, reaching the important conclusion that treatment does not impair compound muscle action potential (CMAP) generation upon repetitive nerve stimulation.
Methods
Patients recruitment and analysis
Thirty-six patients (14 females) with probable or definite ALS according to El Escorial criteria were recruited and analysed at the ALS centre of Policlinico Umberto I, Università Sapienza of Rome. Their mean age was 64.5 ± 7.8 years (mean ± SD; range: 47–78 years). CMAPs were recorded from the abductor digiti minimi and deltoid muscles, by ulnar nerve stimulation at the wrist and axillary nerve at Erb's point, respectively. The amplitudes of the initial negative peaks of the CMAPs were measured and the changes in CMAPs amplitude were analysed. Repetitive nerve stimulation test was performed on the axillary nerve and the ulnar nerve, with the recording electrode over the deltoid muscle and the abductor digiti minimi muscle, respectively. For each train of repetitive stimuli, the amplitudes of the first and fifth CMAPs were compared, and the resulting decrement of the latter expressed as a percentage of the first. All patients were examined before and after a 1 week suspension of riluzole treatment; data values were compared using Student's paired t test. Results were considered to be significantly different when P < 0.05.
Needle biopsy was performed as previously described (Palma et al. 2011). Written informed consent to participate in the study was given by all patients. The study was authorized by the Ethical Committee of Policlinico Umberto I – Sapienza University (Rome) and conducted in accordance with the guidelines of the Declaration of Helsinki. Written informed consent to participate in the study was given by all patients.
Cell culture and AChR expression in cells and oocytes
AChR subunit cDNAs in pRBG4 were kindly provided by Dr A. G. Engel (Mayo Clinic, Rochester, MN, USA).
Human embryonic kidney 293 (HEK) cells were grown in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal bovine serum (both from Invitrogen, Carlsbad, CA, USA) and 1% penicillin/streptomycin. Cells were plated on poly-l-lysine-coated 35 mm Petri dishes and transfected 24 h later using Lipofectamine 2000 (Invitrogen), adding to each dish 1 μg of each subunit cDNA plus 0.5 μg cDNA encoding enhanced green fluorescent protein (EGFP). HEK cells were mechanically dissociated and replated onto glass coverslips 24 h before measurements. Routinely, cells were used for electrophysiological experiments 48–72 h after transfection, as previously reported (Di Angelantonio et al. 2011). Cultures were maintained in a humidified incubator with 5% CO2 at 37°C.
Human satellite cells were derived from muscle needle biopsies performed on ALS patients. Cells were cultured as previously described (Fucile et al. 2006; Palma et al. 2011); differentiation was induced at approximately 50% confluence by switching to a low-serum medium (DMEM plus 2% horse serum and penicillin/streptomycin).
Preparation of Xenopus laevis oocytes and nuclear injection procedures were as detailed elsewhere (Miledi et al. 2006). Equal amounts of cDNA (1 μg μl−1) encoding each AChR subunit were injected. Oocytes were collected under anaesthesia from frogs that were humanely killed after the final collection.
Patch clamp recordings in cultured cells
Currents were recorded at room temperature (23–27°C) via an Axopatch 200B amplifier (Molecular Devices, Union City, CA, USA), using pCLAMP9 (Molecular Devices). In whole-cell and outside-out recordings cells were continuously superfused using a gravity-driven fast exchanger perfusion system (RSC-200, Bio-Logic, France). Whole-cell recordings were performed in standard external solution containing (mm): 140 NaCl, 2.8 KCl, 2 CaCl2, 2 MgCl2, 10 Hepes/NaOH, 10 glucose, pH 7.3. Patch pipettes (2–5 MΩ tip resistance) contained (mm): 140 CsCl, 5 BAPTA, 10 Hepes-KOH, 2 Mg-ATP, 2 MgCl2, pH 7.3. The patch series resistance was compensated by 80–95% and measurements were performed at a holding potential of −60 mV, unless otherwise indicated. Outside-out recordings were performed at −80 mV using standard external medium; patch pipettes (Sylgard-coated for single-channel recordings) contained (mm): 90 CsCl 90, 50 CsF, 5 BAPTA, 10 Hepes-KOH, 2 Mg-ATP, 2 MgCl2, pH 7.3. The rise time of the ACh-evoked currents, measured as the interval between 10% and 90% of peak amplitude, ranged between 0.8 and 3 ms in outside-out recordings.
Cell-attached recordings were performed on cells bathed in standard external solution or a KCl-based solution (mm): 140 KCl, 2.8 NaCl, 2 CaCl2, 2 MgCl2, 10 glucose, 10 Hepes-KOH, pH 7.3. Sylgard-coated patch pipettes were filled with the extracellular solution used plus ACh (100 nm) alone or with riluzole (0.5 μm) added, as indicated.
The decay phase of macroscopic ACh-evoked current (IACh) was fitted using pCLAMP to a single exponential function:
| (1) |
Single channel recordings obtained under outside-out or cell-attached conditions were filtered at 5 kHz and sampled at 25 kHz. Data were analysed with pCLAMP 9, using 50% threshold criterion, omitting events shorter than 0.12 ms, as previously described (Di Castro et al. 2007). In cell-attached recordings, slope conductance was calculated by linear fitting of the unitary amplitudes recorded at least at three different pipette potentials for each cell. For outside-out recordings, channel conductance was calculated dividing unitary channel amplitude by pipette potential, taking into account a mean reversal potential of 13 ± 3 mV (mean ± SEM, 14 patches).
All salts were purchased from Sigma Italia (Milano, Italy). Two data sets were considered statistically different when P < 0.05 by ANOVA or Student's paired t test.
Voltage clamp recordings in oocytes
Membrane currents were recorded from voltage clamped oocytes 2–4 days after injection using two microelectrodes filled with 3 m KCl. The oocytes were placed in a recording chamber (volume, 0.1 ml) and perfused continuously, 8–10 ml min−1, with oocyte Ringer solution (Miledi et al. 2006) at room temperature (20–22°C). Unless otherwise specified, oocytes were voltage clamped at −60 mV.
The half-inhibitory concentration (IC50) of riluzole and the ACh concentration producing half-maximal effect (EC50) were estimated by fitting the data to Hill equations, using least-square routines as previously described (Palma et al. 2002). IACh desensitization was measured by fitting ACh currents with eqn (1).
Data were analysed using SigmaPlot software (OriginLab Corporation, Northampton, MA, USA) and are given as means ± SEM; data sets are considered statistically different when P < 0.05 (ANOVA).
Results
Riluzole effect on whole-cell ACh-evoked currents in HEK cells
We first examined the influence of riluzole on the function of recombinant human γ- or ɛ-AChRs expressed in HEK cells. ACh (100 μm, 0.5 s) elicited whole cell current (IACh) responses in virtually all EGFP-positive cells. No consistent difference in current amplitude was observed between cells expressing γ- or ɛ-AChRs, in spite of large cell-to-cell variations (range: from −45 to – 0.5 nA at –60 mV for both AChR types). These currents had an exponential decay during sustained ACh applications (Fig. 1A) with time constants of 117 ± 10 ms (γ-AChR, n = 32) and 113 ± 6 ms (ɛ-AChR, n = 23). At a concentration routinely attained in patients’ serum (0.5 μm), riluzole coapplied with ACh had almost no effect on IACh. However, when cells expressing γ-AChRs were pretreated with riluzole before it was co-applied with ACh, current amplitude was decreased and its decay accelerated (Fig. 1A; see Supplementary Table 1 for details), so that the current integral (QACh), which depends on both parameters, was reduced. Even the briefest pretreatment tested (0.5 s) enhanced riluzole's effect, which reached a plateau with 5 s preapplications, when QACh was reduced to about 50% of control value (Fig. 1B and E). In cells expressing ɛ-AChRs, riluzole was less effective, as QACh was reduced at most to about 70% of control value (Fig. 1B and E and Supplementary Table 1). For both γ- and ɛ-AChRs, IACh block was enhanced when the riluzole concentration was increased to 50 μm (Fig. 1C and E and Supplementary Table 1), without major qualitative differences as compared to its action at 0.5 μm. Also at this concentration, the effect was stronger on γ-AChRs than on ɛ-AChRs.
Figure 1. Effect of riluzole on ACh-evoked whole-cell currents in HEK cells.
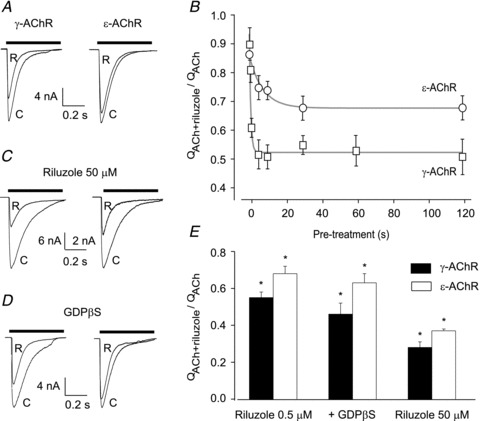
A, typical whole-cell currents evoked by ACh (100 μm, horizontal bar) alone (C) or in the presence of riluzole (R; 0.5 μm, 30 s pre-treatment) in cells expressing the indicated AChR type. B, duration of pre-treatment with riluzole (0.5 μm) enhances its effect in cells expressing γ- or ɛ-AChRs. The integral of current response (QACh or QACh+riluzole) was calculated over the entire duration of ACh (100 μm) application; riluzole's effect was quantified by the ratio QACh+riluzole/QACh in each cell (4–10 for each point). Grey lines represent the exponential best fit of the experimental points. C, typical whole-cell currents evoked by ACh alone or plus riluzole 50 μm (as in A). D, currents as in A, with GDPβS (100 μm) included in the patch pipette. Notice that riluzole's effect is preserved. E, summary of riluzole's effect on ACh-evoked responses, quantified as in B. *QACh+riluzole significantly different from QACh (P < 0.001; Student's paired t test). In all panels, holding potential, −60 mV.
The increase of riluzole-induced effect on IACh after pretreatment might be due to its interference with intracellular signalling pathways that in turn modulate IACh. We therefore assessed the effect of riluzole in cells internally dialysed with GDPβS, a non-phosphorylable analogue of GDP, which prevents G-protein activation. However, inclusion of GDPβS (100 μm) into the patch pipette did not interfere with the effect of riluzole (0.5 μm, 30 s pretreatment, Fig. 1D and E) in transfected cells expressing γ- or ɛ-AChRs, arguing against a G protein-mediated action of riluzole on AChRs.
Block of ACh-evoked currents in oocytes
Riluzole-induced reduction of IACh in cells expressing γ-AChRs was similar to that observed in human myotubes (Palma et al. 2011), suggesting that the expression system used does not influence riluzole's action. This point was further tested in Xenopus oocytes expressing γ- or ɛ-AChRs (Fig. 2). Application of ACh to these cells (1 μm to 1 mm for 4 s) elicited inward currents (IACh) with peak amplitude depending on transmitter concentration, whereas non-injected oocytes showed no detectable responses to ACh. In oocytes expressing ɛ-AChRs, ACh at 50 μm elicited a current with mean peak amplitude of −2.8 ± 0.9 μA (−60 mV; 24 oocytes/4 frogs; 24/4). Currents of similar amplitude (−1.7 ± 0.4 μA; 22/4) were recorded from oocytes expressing γ-AChRs. As in HEK cells, riluzole affected IACh and the block was enhanced if the drug was applied before ACh. In oocytes, the plateau was attained at 120 s of pretreatment, which was used in all experiments described below, including the concentration–response curve, while a shorter pretreatment was used in our previous study (Palma et al. 2011). Riluzole-induced inhibition of IACh was more potent in oocytes expressing γ-AChRs than ɛ-AChRs, as shown by concentration–response curves (Fig. 2) that yielded IC50 values of 8.0 ± 1.1 μm (n = 8/3) and 55.6 ± 0.7 μm (n = 10/3), respectively (P = 0.00001); nH was about 1.5 for both AChR isoforms. For both AChR types, the effect of riluzole was voltage independent in the range −100 to +20 mV (8 oocytes each; data not shown).
Figure 2. Inhibition of ACh-evoked currents by riluzole in oocytes.
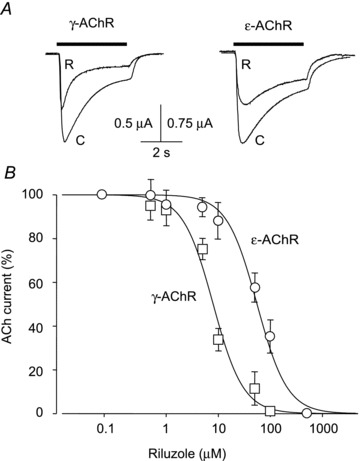
A, typical currents evoked by ACh (horizontal bars) alone (C) or together with riluzole (R; 50 μm for ɛ-AChR; 10 μm for γ-AChR; 120 s pretreatment for both) in two oocytes expressing AChRs as indicated. B, plot of relative amplitude of currents evoked by ACh (50 μm, −60 mV) plus riluzole at various concentrations (120 s pretreatment), expressed as a percentage of control current (ACh alone) in the same cell, for oocytes expressing γ-AChRs (□, 8/3) or ɛ-AChRs (◯, 10/3). IC50 and nH were respectively 8.0 ± 1.1 μm and 1.5 ± 0.2 (γ-AChR), and 55.6 ± 0.7 μm and 1.5 ± 0.3 (ɛ-AChR).
In oocytes expressing ɛ-AChRs the decay of IACh was not modified by riluzole at its IC50 (50 μm), as τdecay was 1.7 ± 0.3 s and 1.5 ± 0.4 s (n = 8/3; P = 0.3) before and in the presence of riluzole, respectively. By contrast, in oocytes expressing γ-AChRs the riluzole-induced decrease of IACh was accompanied by a significant change in current decay, as τdecay was 1.8 ± 0.2 s (n = 8/3) under control conditions and, in the same oocytes, it became 0.7 ± 0.1 s (P = 0.002) during treatment with riluzole (10 μm), resembling the effect described in transfected HEK cells.
The block of IACh was independent of ACh concentration (1 μm to 1 mm) and riluzole (50 μm) did not affect the potency of ACh on γ- or ɛ-AChRs, since the ACh concentration–current response relationships yielded similar values of EC50 and nH (16.7 ± 1.1 μm and 1.1 ± 0.1, n = 6/2 for ɛ-AChR; 21.5 ± 0.2 μm and 1.6 ± 0.2, n = 6/2 for γ-AChR) before and during riluzole treatment (data not shown). These observations suggest that the riluzole-induced acceleration of current decay is not related to a change in neurotransmitter potency.
Effect on unitary ACh-evoked events in HEK cells and human myotubes
To understand at a molecular level the mechanism of riluzole action on IACh, we analysed unitary ACh-evoked events in the cell attached or outside-out configuration, both in HEK cells and in myotubes from ALS patients. Outside-out recordings were performed on HEK cells expressing γ- or ɛ-AChRs, subsequently exposed to ACh (100 nm at −80 mV) or ACh plus riluzole (0.5 μm), with at least 10 s wash between applications. In each patch, unitary events evoked by ACh alone or plus riluzole had identical conductance (39.6 ± 0.7 pS, n = 11 for γ-AChR; 50.0 ± 1.0 pS, n = 7 for ɛ-AChR) and open time distribution (Table 1). However, in the presence of riluzole, closed time histograms were shifted to the right in all patches, as compared to control conditions (Fig. 3), although only the third and fourth (for γ-AChR) best fitting exponential components were significantly prolonged (Table 1). The riluzole-induced increase of channel closed duration was not an artefact due to channel run-down, as in most patches closed times reversed towards control values when ACh was applied alone for a second time (data not shown). It must be noted that in these experiments, riluzole preapplication was not used, but the initial 5–10 s of the recordings, when multiple ACh-induced openings occurred, were not considered for analysis.
Table 1.
Riluzole action on AChR-channel kinetics in outside-out patches
| ɛ-AChR (7 patches) | γ-AChR (11 patches) | |||
|---|---|---|---|---|
| Control | Riluzole | Control | Riluzole | |
| τopen1 (ms) | 0.50 ± 0.05 | 0.50 ± 0.02 | 0.7 ± 0.1 | 0.7 ± 0.1 |
| (aopen1) (%) | (55 ± 5) | (45 ± 5) | (40 ± 2) | (40 ± 2) |
| τopen2 (ms) | 1.8 ± 0.2 | 1.7 ± 0.1 | 3.5 ± 0.4 | 3.9 ± 0.5 |
| (aopen2) (%) | (45 ± 5) | (55±5) | (60 ± 2) | (60 ± 2) |
| τclosed1 (ms) | 0.20 ± 0.02 | 0.30 ± 0.06 | 0.20 ± 0.08 | 0.5 ± 0.2 |
| (aclosed1) (%) | (16 ± 1) | (20 ± 3) | (21 ± 3) | (8 ± 1) |
| τclosed2 (ms) | 1.8 ± 0.3 | 2.8 ± 0.5 | 3.9 ± 0.9 | 6.2 ± 1.1 |
| (aclosed2) (%) | (34 ± 3) | (31 ± 5) | (22 ± 4) | (21 ± 4) |
| τclosed3 (ms) | 8.7 ± 1.9 | 18 ± 5* | 33 ± 6 | 61 ± 12* |
| (aclosed3) (%) | (48 ± 2) | (47 ± 4) | (39 ± 3) | (43 ± 3) |
| τclosed4 (ms) | 219 ± 50 | 420 ± 100* | ||
| (aclosed4) (%) | (23 ± 4) | (31 ± 7) | ||
τopen (aopen) and τclosed (aclosed): time constants (weight) of the exponential components best fitting the distribution of open and closed durations, respectively. Data are given as means ± SEM for the indicated number of patches, each exposed to ACh (100 nm, −80 mV) alone and to ACh plus riluzole (0.5 μM).
Significantly larger than control (P < 0.04, Student's paired t test).
Figure 3. Effect of riluzole on unitary ACh-evoked currents in outside-out patches.
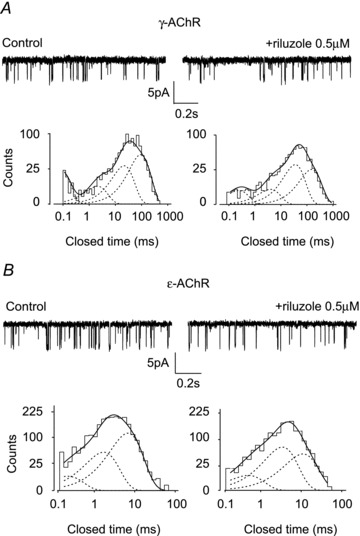
A and B, unitary events evoked by ACh (100 nm, −80 mV) in outside-out patches expressing γ-AChRs (A) or ɛ-AChRs (B), sequentially exposed to ACh alone (control) or plus riluzole (0.5 μm), as indicated. Bottom panels represent the histograms of channel closed times, obtained in the recordings shown above, best fitted with four (γ-AChR) or three (ɛ-AChR) exponential components. Notice the lengthening of the closed times in the presence of riluzole, as compared to control conditions. Channel openings represented by downward deflections.
To check that riluzole's effect was not influenced by patch excision, we performed cell-attached recordings on HEK cells expressing γ-AChRs, including riluzole (0.5 μm) in the patch pipette. Again, unitary channel conductance was identical in control and riluzole-exposed patches (34.0 ± 3.1 pS and 32.2 ± 0.9 pS, respectively; n = 7 for both). The distribution of open durations was adequately fitted by two exponential components with similar time constants in the two experimental groups (Fig. 4A; details in Supplementary Table 2). Closed time distribution was widely variable for both control and treated cells, and could not be reliably compared. Cell-attached recordings were performed also on myotubes from ALS patients (Fig. 4B) and yielded analogous results. In these cells, most experiments were performed bathing cells in KCl-based extracellular solution. Channel conductance was 35.7 ± 1.1 pS (n = 13) under control conditions and 33.0 ± 3.1 pS (n = 7) with riluzole. In both control and riluzole-exposed patches, open time distributions were fitted by two exponential components with time constants similar to those observed in HEK cells, and not influenced by the drug (Fig. 4B; details in Supplementary Table 2).
Figure 4. Effect of riluzole in cell-attached recordings.
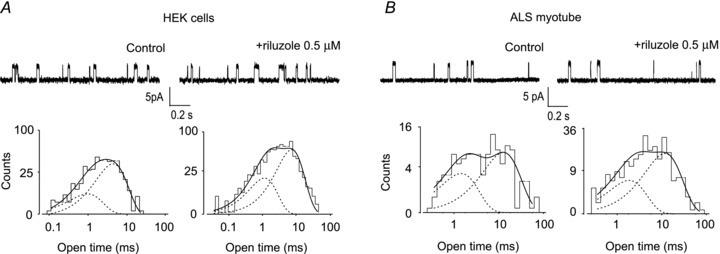
A, typical cell-attached recordings in two different HEK cells expressing γ-AChR, including in the patch pipette ACh (100 nm) alone (slope conductance, 31 pS) or plus riluzole (0.5 μm; slope conductance, 34 pS), as indicated. Bottom, the histograms of channel open durations, obtained in the same recordings, were best fitted by two exponential components, with time constants τop1 = 1.5 ms (33%) and τop2 = 9.5 ms (67%) for control; 1.1 ms (35%) and 7.4 ms (65%) in presence of riluzole. B, cell-attached recording of unitary events in two different patches on the same myotube from an ALS patient, using ACh (100 nm) alone or plus riluzole, as indicated. Channel conductance was 30 pS (control) or 34 pS (+ riluzole). Bottom, the corresponding histograms of channel opening durations, best fitted by two exponential components with τop1 = 1.4 ms (39%) and τop2 = 12.7 ms (61%) in control conditions, 1.8 ms (36%) and 11.7 ms (64%) in the presence of riluzole. In all panels, channel openings are represented by upward deflections; cells were bathed in standard external solution.
How fast is riluzole action?
During neuromuscular transmission, nerve-released ACh is present in the synaptic cleft only for a few milliseconds before being hydrolysed by acetylcholinesterase. The action of riluzole on such a fast time scale cannot be inferred from whole-cell recordings, as the rise time of ACh-evoked currents is in the order of 10–20 ms. Macroscopic ACh-evoked currents from outside-out patches have a much shorter rise time, and we could assess the effect of riluzole (0.5 μm, 30 s pretreatment) within few milliseconds of ACh (100 μm) application (Fig. 5). In membrane patches expressing ɛ-AChRs, in the presence of riluzole QACh was 96 ± 3% (n = 8) of control value after 10 ms of ACh application, but it was significantly reduced to 83 ± 6% (n = 8, P = 0.02) at 100 ms, indicating that the action of the drug develops slowly. For γ-AChR, QACh was significantly reduced to 79 ± 4% of control value (n = 6, P = 0.002) at 10 ms and to 66 ± 3% (n = 6, P = 0.0001) at 100 ms.
Figure 5. Effect of riluzole during fast ACh applications.
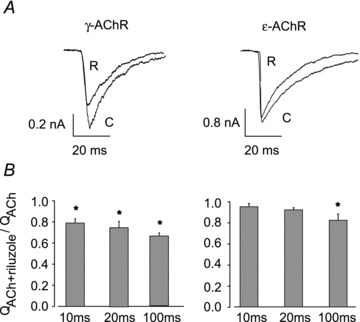
A, superimposed currents evoked by sequential applications of ACh (100 μm, 100 ms) alone (C) or together with riluzole (R; 0.5 μm, 30 s pre-treatment) to outside-out patches from HEK cells expressing γ- or ɛ-AChRs, as indicated. B, bar graphs representing relative current integral during 10, 20 and 100 ms from ACh application. For γ-AChRs, riluzole-induced reduction of QACh was significant already at 10 ms (*P < 0.002); for ɛ-AChR, current integral was significantly reduced only at 100 ms (*P = 0.02). In all panels, holding potential, −60 mV.
Riluzole does not affect CMAPs in ALS patients
To examine if riluzole as used in clinical practice affects neuromuscular transmission, we examined 36 ALS patients, measuring the amplitude of compound muscle action potentials (CMAPs) elicited by repetitive nerve stimulation (RNS), which provides a sensitive and non-invasive assay of neuromuscular transmission. Patients were tested immediately before and after 1 week suspension of riluzole treatment, a time that allows for adequate riluzole washout, which has been estimated at about 40 h (Le Liboux et al. 1999; Chandu et al. 2010). The test was performed on two different nerve-muscle groups. After the week of riluzole washout, CMAP amplitude did not change significantly in any of the patients examined, showing that no further axonal depletion occurred between the two neurophysiological evaluations. The decrease of CMAP amplitude during RNS was also not statistically different at the end of the riluzole washout period as compared to the control value, for both the ulnar and the axillary nerves (Fig. 6). Thus, riluzole treatment appears to be devoid of detrimental effects at least on this estimator of neuromuscular transmission.
Figure 6. Riluzole has no effect on neuromuscular transmission in ALS patients.
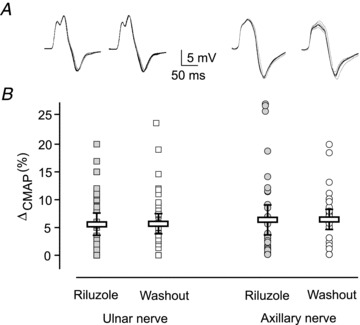
A and B, superimposed traces representing 10 CMAPs evoked by repetitive stimulation of the ulnar (A) and axillary nerve (B) in a patient, during riluzole treatment (left) and at the end of a 1 week medicament washout. Notice that CMAP amplitude remains constant in this individual. C, dispersion plot of the difference between CMAP amplitude evoked by the 1st and 5th stimulus, expressed as a percentage of the amplitude of the 1st response, for all 36 patients considered in the study. Boxes represent mean ± 95% confidence interval of all values. For both ulnar and axillary nerves, values were not modified by riluzole washout (P > 0.9).
Discussion
The results of this paper disclose a differential effect of riluzole on recombinant human γ-AChRs and ɛ-AChRs, expressed in HEK cells or oocytes. In both cell systems, riluzole-induced block was faster and stronger for γ-AChRs than for ɛ-AChRs, although with kinetic differences between the two cell types, likely to be due to the large size and complex membrane of oocytes as compared to HEK cells. Outside-out recordings showed that the synaptic AChR isoform was blocked by riluzole only with ACh applications far outlasting the typical duration of synaptic events, allowing the hypothesis that riluzole at clinical concentrations should not impair endplate function. This supposition was verified in vivo, measuring CMAPs elicited by nerve stimulation in ALS patients, to determine the efficiency of neuromuscular transmission. In agreement with our hypothesis, muscle response to nerve stimulation was not influenced by 1 week suspension of riluzole treatment, which produces complete washout of plasma riluzole (Le Liboux et al. 1999; Chandu et al. 2010). Furthermore, the absence of effects on CMAPs evoked by repetitive nerve stimulation suggests that clinical use of riluzole does not affect the safety factor at the neuromuscular junction in treated patients. Incidentally, these findings indicate that careful consideration of in vitro results is necessary when extrapolating their clinical significance.
As previously seen in human tissue (Palma et al. 2011), we show here that preapplication of riluzole increases the block of IACh in cells expressing recombinant γ- or ɛ-AChRs. This is particularly evident when considering the IC50 of riluzole, which, for γ-AChRs, was as low as 8 μm after a preapplication of 120 s, whereas a value of about 22 μm was observed after a 20 s pretreatment in oocytes injected with membranes of denervated patients, containing almost exclusively γ-AChRs. The effect of pretreatment raises the possibility that intracellular signalling pathways mediate riluzole-induced block of AChRs. For instance, in neuronal membranes riluzole blocks PKC (Noh et al. 2000). However, we show here that in transfected HEK cells, riluzole's effect was preserved upon patch excision or blockade of G-proteins by the classical inhibitor GDPβS, indicating that soluble intracellular factors and G-proteins are not involved in the transduction of the riluzole effect. Moreover, the effect on IACh was observed in experimental systems as diverse as cDNA-injected oocytes (this work) and cultured human myotubes (Palma et al. 2011), which seems to be more consistent with a direct action of riluzole on AChR than with the transduction by a second messenger.
Being independent of ACh concentration, at least in oocytes, riluzole-induced block of IACh appears to be due to a non-competitive mechanism of action. Open-channel block is an unlikely candidate, as riluzole has a voltage-independent effect and does not influence the duration of ACh-evoked channel openings, the two hallmarks of open-channel blockade (Neher & Steinbach, 1978). In oocytes, riluzole did not change ACh potency at γ- or ɛ-AChRs, suggesting that ACh binding and channel gating are largely unaltered for both receptors (Colquhoun, 1998); therefore AChR desensitization was likely to be the functional parameter affected by riluzole. Single-channel recordings showed that riluzole had no effect on the conductance and open time distribution of ACh-evoked channels, but prolonged closed intervals, supporting the hypothesis that it acts by stabilizing AChR desensitized state(s), with no effect on the open state. Such an action has already been proposed for other substances, which have an enhanced effect on AChR channels following preapplication, for instance 3-(trifluoromethyl)-3-(m-iodophenyl)diazirine, some local anaesthetics and verapamil (Forman, 1999; Spitzmaul et al. 2009; Moriconi et al. 2010).
Both in oocytes and in HEK cells, riluzole blocked the γ-AChR more than the ɛ-AChR, thus adding to the list of compounds having differential actions on the two isoforms of muscle AChR. For instance, the γ-AChR is more sensitive than the ɛ-AChR to block by 5-hydroxytryptamine (Grassi, 1999) or hydrocortisone (Bouzat & Barrantes, 1996); fluoxetine alters channel kinetics of the γ-AChR (Garcia-Colunga et al. 1997) but not the ɛ-AChR (Harper et al. 2003). More striking, d-tubocurarine at low concentrations is a blocker of the ɛ-AChR, but a weak agonist of the γ-AChR (Steinbach & Chen, 1995).
While we show that riluzole-induced block of ɛ-AChRs is unlikely to occur during neuromuscular transmission, blockade of γ-AChRs may play some role in disease history. Since muscle fibres of ALS patients undergo denervation and subsequent reinnervation by collateral sprouting of healthy motor neurones (Eisen & Swash, 2001), the γ-AChR isoform may transiently coexist with ɛ-AChRs at reinnervated endplates, as occurs in rodents (Yampolsky et al. 2010). However, also for γ-AChRs, brief ACh-evoked currents were little affected by clinical concentrations of riluzole, again suggesting that synaptic transmission is unlikely to be impaired. In non-innervated muscles, activation of extrasynaptic γ-AChRs might be induced by the tonic release of ACh or an ACh-like compound, well documented in cultured myotubes and denervated muscle in vivo (Krnjevic & Straughan, 1964; Krause et al. 1995; Bandi et al. 2005). Whether or not γ-AChRs influence muscle reinnervation in humans is unknown; in rodents reinnervation of partially denervated muscles is impaired by α-bungarotoxin (Connold & Vrbova, 1991). Other studies, however, point out that full chronic AChR blockade is required to prevent collateral sprouting of motor axons (Pestronk & Drachmann, 1985), so that the partial riluzole-induced reduction of AChR function might be non-detrimental to the reinnervation process, a hypothesis that deserves analysis in adequate experimental systems. On the other hand, it has long been known that, during chick and mouse development, blockade or reduction of γ-AChR favours motor neuron survival (Oppenheim et al. 2000; Terrado et al. 2001; Liu et al. 2008; Yampolsky et al. 2008), thus allowing the suggestion that riluzole action might be beneficial against ongoing motor neuron degeneration through this route, too.
At present, the idea that muscle denervation may precede – or even contribute to – motor neuron death is gaining support (as reviewed by Musarò, 2010; Dadon-Nachum et al. 2011), so that investigation of the physiological consequences of riluzole-induce blockade of γ-AChRs warrants further studies to understand whether it contributes, positively or negatively, to disease progression.
Acknowledgments
The authors thank all the patients who made this study possible by donating muscle tissue. This work was supported by grants from Association Française contre le Myopathies (F.G.), Fondazione Viva la Vita (to M.I. and C.L.), Ministero della Salute Antidoping project (to C.L. and E.P.), Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR-PRIN grants to E.P. and C.L.). C.D. and L.C. were supported by the PhD Programme in Neurophysiology at Sapienza University, Rome. The authors declare no conflict of interests.
Glossary
Abbreviations
- AChR
acetylcholine receptor
- ALS
amyotrophic lateral sclerosis
- CMAP
compound muscle action potential
- IACh
ACh-evoked whole-cell current
- QACh
integral of ACh-evoked whole-cell current
- PKC
protein kinase C
Author contributions
F.G., E.P., M.I. and C.L. planned and discussed experiments; C.D., L.C., C.R., M.C. and A.M. performed experiments and critically analysed the data; E.G. and M.I. examined patients; M.I. performed biopsies; F.G., E.P. and M.I. wrote the paper; all authors approved the manuscript.
Supplementary material
Supplemental Table 1
Supplemental Table 2
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Bandi E, Bernareggi A, Grandolfo M, Mozzetta C, Augusti-Tocco G, Ruzzier F, Lorenzon P. Autocrine activation of nicotinic acetylcholine receptors contributes to Ca2+ spikes in mouse myotubes during myogenesis. J Physiol. 2005;568:171–180. doi: 10.1113/jphysiol.2005.091439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham MC. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci Ther. 2011;17:4–31. doi: 10.1111/j.1755-5949.2009.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzat C, Barrantes FJ. Modulation of muscle nicotinic acetylcholine receptors by the glucocorticoid hydrocortisone. Possible allosteric mechanism of channel blockade. J Biol Chem. 1996;271:25835–25841. doi: 10.1074/jbc.271.42.25835. [DOI] [PubMed] [Google Scholar]
- Chandu BR, Nama S, Kanala K, Challa BR, Shaik RP, Khagga M. Quantitative estimation of riluzole in human plasma by LC-ESI-MS/MS and its application to a bioequivalence study. Anal Bioanal Chem. 2010;398:1367–1374. doi: 10.1007/s00216-010-4034-8. [DOI] [PubMed] [Google Scholar]
- Connold AL, Vrbova G. Temporary loss of activity prevents the increase of motor unit size in partially denervated rat soleus muscles. J Physiol. 1991;434:107–119. doi: 10.1113/jphysiol.1991.sp018461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D. Binding, gating, affinity and efficacy: The interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadon-Nachum M, Melamed E, Offen D. The ‘dying-back’ phenomenon of motor neurons in ALS. J Mol Neurosci. 2011;43:470–477. doi: 10.1007/s12031-010-9467-1. [DOI] [PubMed] [Google Scholar]
- Di Angelantonio S, Piccioni A, Moriconi C, Trettel F, Cristalli G, Grassi F, Limatola C. Adenosine A2A receptor induces protein kinase A-dependent functional modulation of human α3β4 nicotinic receptor. J Physiol. 2011;589:2755–2766. doi: 10.1113/jphysiol.2011.207282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Castro A, Martinello K, Grassi F, Eusebi F, Engel AG. Pathogenic point mutations in a transmembrane domain of the epsilon subunit increase the Ca2+ permeability of the human endplate ACh receptor. J Physiol. 2007;579:671–677. doi: 10.1113/jphysiol.2007.127977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen A, Swash M. Clinical neurophysiology of ALS. Clin Neurophysiol. 2001;112:2190–2201. doi: 10.1016/s1388-2457(01)00692-7. [DOI] [PubMed] [Google Scholar]
- Forman SA. A hydrophobic photolabel inhibits nicotinic acetylcholine receptors via open-channel block following a slow step. Biochemistry. 1999;38:14559–14564. doi: 10.1021/bi9914457. [DOI] [PubMed] [Google Scholar]
- Fucile S, Sucapane A, Grassi F, Eusebi F, Engel AG. The human adult subtype ACh receptor channel has high Ca2+ permeability and predisposes to endplate Ca2+ overloading. J Physiol. 2006;573:35–43. doi: 10.1113/jphysiol.2006.108092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Colunga J, Awad JN, Miledi R. Blockage of muscle and neuronal nicotinic acetylcholine receptors by fluoxetine (Prozac) Proc Natl Acad Sci U S A. 1997;94:2041–2044. doi: 10.1073/pnas.94.5.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi F. 5-Hydroxytryptamine blocks the fetal more potently than the adult mouse muscle acetylcholine receptor. Pflugers Arch. 1999;437:903–909. doi: 10.1007/s004240050861. [DOI] [PubMed] [Google Scholar]
- Harper CM, Fukodome T, Engel AG. Treatment of slow-channel congenital myasthenic syndrome with fluoxetine. Neurology. 2003;60:1710–1713. doi: 10.1212/01.wnl.0000061483.11417.1b. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. Further observations on the distribution of actylcholine-reactive sites in skeletal muscle. J Physiol. 1964;170:379–388. doi: 10.1113/jphysiol.1964.sp007338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause RM, Hamann M, Bader CR, Liu JH, Baroffio A, Bernheim L. Activation of nicotinic acetylcholine receptors increases the rate of fusion of cultured human myoblasts. J Physiol. 1995;489:779–790. doi: 10.1113/jphysiol.1995.sp021091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krnjevic K, Straughan DW. The release of acetylcholine from the denervated rat diaphragm. J Physiol. 1964;170:371–378. doi: 10.1113/jphysiol.1964.sp007337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Liboux A, Cachia JP, Kirkesseli S, Gautier JY, Guimart C, Montay G, Peeters PA, Groen E, Jonkman JH, Wemer J. A comparison of the pharmacokinetics and tolerability of riluzole after repeat dose administration in healthy elderly and young volunteers. J Clin Pharmacol. 1999;39:480–486. [PubMed] [Google Scholar]
- Liu Y, Padgett D, Takahashi M, Li H, Sayeed A, Teichert RW, Olivera BM, McArdle JJ, Green WN, Lin W. Essential roles of the acetylcholine receptor γ-subunit in neuromuscular synaptic patterning. Development. 2008;135:1957–1967. doi: 10.1242/dev.018119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miledi R, Palma E, Eusebi F. Microtransplantation of neurotransmitter receptors from cells to Xenopus oocyte membranes: new procedure for ion channel studies. Methods Mol Biol. 2006;322:347–355. doi: 10.1007/978-1-59745-000-3_24. [DOI] [PubMed] [Google Scholar]
- Mohammadi B, Lang N, Dengler R, Bufler J. Interaction of high concentrations of riluzole with recombinant skeletal muscle sodium channels and adult-type nicotinic receptor channels. Muscle Nerve. 2002;26:539–545. doi: 10.1002/mus.10230. [DOI] [PubMed] [Google Scholar]
- Moriconi C, Di Castro MA, Fucile S, Eusebi F, Grassi F. Mechanism of verapamil action on wild-type and slow-channel mutant human muscle acetylcholine receptor. J Neurochem. 2010;114:1231–1240. doi: 10.1111/j.1471-4159.2010.06842.x. [DOI] [PubMed] [Google Scholar]
- Musarò A. State of the art and the dark side of amyotrophic lateral sclerosis. World J Biol Chem. 2010;1:62–68. doi: 10.4331/wjbc.v1.i5.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Steinbach JH. Local anaesthetics transiently block currents through single acetylcholine-receptor channels. J Physiol. 1978;277:153–176. doi: 10.1113/jphysiol.1978.sp012267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh KM, Hwang JY, Shin HC, Koh JY. A novel neuroprotective mechanism of riluzole: direct inhibition of protein kinase C. Neurobiol Dis. 2000;7:375–383. doi: 10.1006/nbdi.2000.0297. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Prevette D, D’Costa A, Wang S, Houenou LJ, McIntosh JM. Reduction of neuromuscular activity is required for the rescue of motoneurons from naturally occurring cell death by nicotinic-blocking agents. J Neurosci. 2000;20:6117–6124. doi: 10.1523/JNEUROSCI.20-16-06117.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma E, Esposito V, Mileo AM, Di Gennaro G, Quarato P, Giangaspero F, Scoppetta C, Onorati P, Trettel F, Miledi R, Eusebi F. Expression of human epileptic temporal lobe neurotransmitter receptors in Xenopus oocytes: An innovative approach to study epilepsy. Proc Natl Acad Sci U S A. 2002;99:15078–15083. doi: 10.1073/pnas.232574499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma E, Inghilleri M, Conti L, Deflorio A, Frasca V, Manteca A, Pichiorri F, Roseti C, Torchia G, Limatola C, Grassi F, Miledi R. Physiological characterization of human muscle acetylcholine receptors from ALS patients. Proc Natl Acad Sci U S A. 2011;108:20184–20188. doi: 10.1073/pnas.1117975108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestronk A, Drachman DB. Motor nerve terminal outgrowth and acetylcholine receptors: inhibition of terminal outgrowth by α-bungarotoxin and anti-acetylcholine receptor antibody. J Neurosci. 1985;5:751–758. doi: 10.1523/JNEUROSCI.05-03-00751.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzmaul G, Gumilar F, Dilger JP, Bouzat C. The local anaesthetics proadifen and adiphenine inhibit nicotinic receptors by different molecular mechanisms. Br J Pharmacol. 2009;157:804–817. doi: 10.1111/j.1476-5381.2009.00214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach JH, Chen Q. Antagonist and partial agonist actions of d-tubocurarine at mammalian muscle acetylcholine receptors. J Neurosci. 1995;15:230–240. doi: 10.1523/JNEUROSCI.15-01-00230.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrado J, Burgess RW, DeChiara T, Yancopoulos G, Sanes JR, Kato AC. Motoneuron survival is enhanced in the absence of neuromuscular junction formation in embryos. J Neurosci. 2001;21:3144–3150. doi: 10.1523/JNEUROSCI.21-09-03144.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzemann V, Brenner HR, Sakmann B. Neural factors regulate AChR subunit mRNAs at rat neuromuscular synapses. J Cell Biol. 1991;114:125–141. doi: 10.1083/jcb.114.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yampolsky P, Gensler S, McArdle J, Witzemann V. AChR channel conversion and AChR-adjusted neuronal survival during embryonic development. Mol Cell Neurosci. 2008;37:634–645. doi: 10.1016/j.mcn.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Yampolsky P, Pacifici PG, Witzemann V. Differential muscle-driven synaptic remodeling in the neuromuscular junction after denervation. Eur J Neurosci. 2010;31:646–658. doi: 10.1111/j.1460-9568.2010.07096.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.