

Abstract
Kennedy's disease, an X-linked spinal and bulbar muscular atrophy, is characterized by loss of lower motor neurons. Mild sensory deficits, gynecomastia and infertility may be observed. Klinefelter's syndrome is a variation of sex chromosome disorder characterized by hypogonadism, gynecomastia and azoospermia, and the most frequent karyotype is XXY. A 55-year-old man who presented with slowly progressive and diffuse neurogenic muscle atrophy without bulbar or sensory symptoms. He also had Klinefelter's syndrome. Genetic study of Kennedy's disease was normal. Our patient differs from those with Kennedy's disease in the absence of bulbar and sensory symptoms. It is suggested that the X chromosome plays an important role in the biology of motor neurons.
Keywords: Electromyogram, gynecomastia, Klinefelter's syndrome, Kennedy's disease, muscle biopsy, progressive muscular atrophy
Introduction
Kennedy's disease, an X-linked spinal and bulbar muscular atrophy, is a neurodegenerative disorder characterized by loss of lower motor neurons (LMNs).[1] Patients exhibit progressive proximal and bulbar muscle atrophy, fasciculation of limb and facial muscles and mild sensory deficits. Gynecomastia and infertility may be observed. It is caused by the unstable expansion of CAG triplets in exon 1 of the androgenic receptor gene located on chromosome Xq11-12.
Klinefelter's syndrome is a variation of sex chromosome disorder characterized by hypogonadism, gynecomastia and azoospermia, and the most frequent karyotype is XXY. Neurological manifestations may include mental deficiency, dyslexia, ataxia and essential tremor. Peroneal muscular atrophy, familial spastic paraparesis,[2] myotonic dystrophy and benign neurogenic amyotrophy[3] associated with Klinefelter's syndrome have been occasionally described.
We describe a patient with Klinefelter's syndrome associated with progressive muscular atrophy (PMA) that mimics Kennedy's disease.
Case Report
A 55-year-old man without relevant medical history. Others members of his family had no neurological disease. He developed progressive and generalized weakness for over 20 years. Weakness began in the legs with difficulty in climbing stairs and has progressed very slowly during this time. For about 15 years, he also had proximal weakness of the arms. He had cramps, an unpleasant painful sensation caused by muscle contraction of the legs for over 10 years. He had mild gynecomastia and testicular atrophy.
On neurological examination, a slight bilateral shoulder girdle weakness was noticed, as follows: MRC scale was found to be 4/5 in deltoideus, triceps, biceps brachii and carpi and finger flexor/extension. Also, the MRC scale was 4/5 in iliopsoas, quadriceps, gluteus and gastrocnemius. Hand and foot muscles were normal. Distal and proximal limb muscles and trunk and facial muscles were atrophied. The cranial nerves showed facial weakness. Knee jerks were hypoactive; the other deep tendon reflexes were absent in all four limbs. Plantar responses were flexor bilaterally. His sensory system and coordination were normal.
Hemogram, vitamin B12, glucose, urea, creatinine, hepatic enzymes, creatine kinase, lactate, proteinogram and immunoelectrophoresis, phosphate, calcium, heavy metals, serology for syphilis and HIV, serum Ig M anti-GM1 and anti-HU antibodies were normal or negative. FSH 24.4 U/l (normal: 1–10.5), LH: 18.4 U/l (normal: 1–8.4), testosterone: 1.14 ng/ml (normal: 3–11) and estradiol: 72 pg/ml (normal: 0–54). Karyotype analysis revealed the XXY pattern.
Brain and spinal cord magnetic resonance imaging were normal. Electromyogram (EMG) of the anterior tibial, posterior tibial and biceps brachii [Figure 1] showed fibrillations at rest and neuromuscular units of high amplitude and long duration, indicating active neurogenic processes. Motor nerve conduction velocities of the median, ulnar, tibial and peroneal nerves were normal as were also the sensory conduction velocities of the median, ulnar and sural nerves. Absence of motor conduction blocks and F-wave study showed normal persistence and normal minimum latencies from the lower and upper limb nerves.
Figure 1.
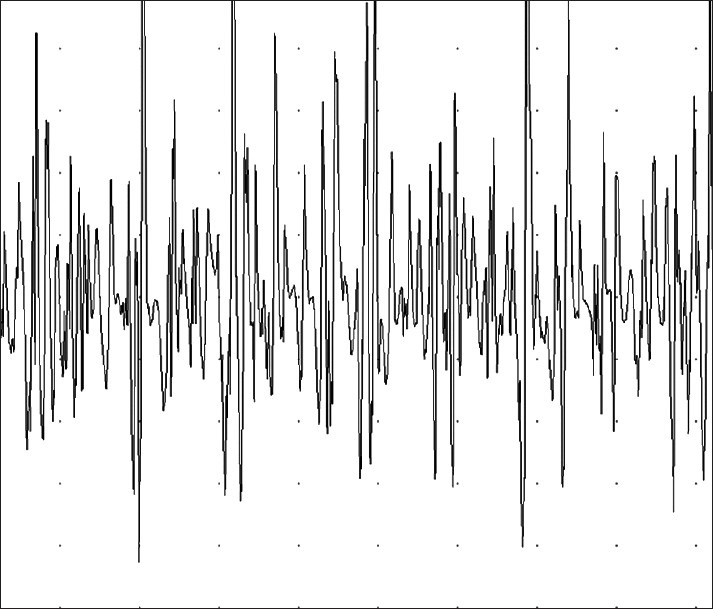
EMG of biceps brachii shows delayed recruitment and fast firing of high amplitude as well as long duration motor unit potentials. This pattern suggests chronic partial denervation with evidence of reinervation
Sural nerve biopsy showed no abnormality. Gastrocnemius muscle biopsy was stained with hematoxylin–eosin, Gomori trichorme, NADH-TR, periodic acid-Schiff reaction and myosin ATPase. There were groups of small angulated fibers and fiber-type grouping and a predominance of type 1 fibers. Biochemical analysis was negative.
Survival motor neuron (SMN) gene implicated in hereditary spinal muscular atrophy (SMA) showed no deletions in exons 7 and 8. Androgen receptor gene revealed 26 and 28 CAG repeats on X chromosomes (pathological more than 40).
Discussion
Clinical presentation with degeneration of the LMNs of the spinal cord in the absence of upper motor neuron (UMN) and bulbar features, often with a very slowly progressive course, acting up to 20–30 years is suggestive of PMA.[4] PMA is an adult-onset, nonhereditary progressive disease of the LMNs. Inclusion criteria are age of onset older than 18 years, disease duration of less than 4 years from the time of onset of weakness and clinical and electrophysiological evidence of progressive LMN involvement in one or more of four regions. Exclusion criteria include motor conduction blocks, definite clinical UMN signs (pseudobulbar symptoms, extensor plantar response and spasticity), objective sensory signs, history of diseases that may mimic motor neuron disease (NMD) as poliomyelitis, family history of inherited SMA atrophy and delection in the SMN gene or an expansion of CAG repeats more than 40 in the androgen receptor gene.
In a prospective study of amyotrophic lateral sclerosis (ALS), 70% of the patients with LMN signs had developed UMN and bulbar signs characteristic of ALS after 6 years.[5] Therefore, probably fewer than 10% of the patients with NMD will continue to show LMN signs only. We can rule out ALS in our patient because he has had LMN signs only for more than 20 years.
SMA type IV is an autosomal-recessive disorder of the LMNs. The gene involved (SMN gene) is located on chromosome 5q13. Absence of family history or deletion in this gene reasonably rule out this disease.
Sporadic cases of LMN disorders may be attributable to heavy metal intoxication, remote effects of carcinoma, syphilis and, at times, subjects who have stabilised or recovered from an episode of poliomyelitis at a young age subsequently develop progressive LMN dysfunction.[6]
Our patient resembled Kennedy's disease in regard to feminisation and neurogenic muscular atrophy. Bulbar symptoms, which are one of the hallmarks of Kennedy's disease, were, however, absent and neither had sensory symptoms or sensory nerve conduction study disturbances.[7] Muscular biopsy ruled out a myopathy. Fasciculations are not unique to LMNs disease; they may also occur in polyneuropathy or muscle diseases.
Review of the literature revealed a patient with Klinefelter's syndrome associated with infantile LMN disease caused by mutation of the SMN gene (Werdning-Hoffmann disease).[8] Two cases of benign neurogenic amyotrophic have been described in patients with Klinefelter's syndrome,[3] similar to our patient, but one had UMN disorder associated and the other had serum creatine kinase raised and essential tremor.
It may be a fortuitous association of Klinefelter's syndrome and LMNs disease. The clinical course of the present case and others[3] resembled that of hereditary NMD rather than sporadic ALS, which is far more common, yet none of these cases had a family history of NMD. It is suggested that the X chromosome plays an important role in the biology of motor neurons. Sex chromosome disorder (47, XXY) arises through a phenomenon such as lionisation[9] upon the emergence of this lesive mechanism. In the same way as do the clinical consequences seen in women heterozygous for the CAG repeats in Kennedy's disease.[10]
Footnotes
Source of Support: Nil
Conflict of Interest: Nil
References
- 1.Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset.A sex-linked recessive trait. Neurology. 1968;18:671–80. doi: 10.1212/wnl.18.7.671. [DOI] [PubMed] [Google Scholar]
- 2.Uzicanin S, Catibusic F, Terzic S, Zubcevic S. Familiar spastic paraplegia presenting in a boy with Klinefelter syndrome. Case report. Med Arh. 2007;61:52–3. [PubMed] [Google Scholar]
- 3.Matsubara S, Yoshino M, Takamori M. Benign neurogenic amyotrophy in Klinefelter's syndrome. J Neurol Neurosurg Psychiatry. 1994;57:640–2. doi: 10.1136/jnnp.57.5.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Visser J, de Jong JM, de Visser M. The history of progressive muscular atrophy: Syndrome or disease? Neurology. 2008;70:723–7. doi: 10.1212/01.wnl.0000302187.20239.93. [DOI] [PubMed] [Google Scholar]
- 5.Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardinam O. Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie House diagnostic criteria. Arch Neurol. 2000;57:1171–6. doi: 10.1001/archneur.57.8.1171. [DOI] [PubMed] [Google Scholar]
- 6.van den Berg-Vos RM, Visser J, Franssen H, de Visser M, de Jong JM, Kalmijn S, et al. Sporadic lower motor neuron disease with adult onset: Classification of subtypes. Brain. 2003;126:1036–47. doi: 10.1093/brain/awg117. [DOI] [PubMed] [Google Scholar]
- 7.Meriggioli MN, Rowin J, Sanders DB. Distinguishing clinical and electrodiagnostic features of X-linked bulbospinal neuronopathy. Muscle Nerve. 1999;22:1693–7. doi: 10.1002/(sici)1097-4598(199912)22:12<1693::aid-mus11>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 8.Migita M, Uchikoba Y, Orimo H, Shimada T, Matsumoto T, Hayakawa J, et al. Genetic diagnosis of Werdning-Hoffmann disease: A problem for application to prenatal diagnosis. J Nippon Med Sch. 2003;70:45–8. doi: 10.1272/jnms.70.45. [DOI] [PubMed] [Google Scholar]
- 9.Heard E, Clerc P, Avner P. X-chromosome inactivation in mammals. Annu Rev Genet. 1997;31:571–610. doi: 10.1146/annurev.genet.31.1.571. [DOI] [PubMed] [Google Scholar]
- 10.Greenland KJ, Beilin J, Castro J, Varghese PN, Zajac JD. Polymorphic CAG repeat length in the androgen receptor gene and association with neurodegeneration in a heterozygous female carrier of Kennedy's disease. J Neurol. 2004;251:35–41. doi: 10.1007/s00415-004-0266-x. [DOI] [PubMed] [Google Scholar]