

Abstract
Comparative studies of major histocompatibility complex (MHC) genes across vertebrate species can reveal the evolutionary processes that shape the structure and function of immune regulatory proteins. In this study, we characterized MHC class I sequences from six frog species representing three anuran families (Hylidae, Centrolenidae and Ranidae). Using cDNA from our focal species, we amplified a total of 79 unique sequences spanning exons 2–4 that encode the extracellular domains of the functional alpha chain protein. We compared intra- and interspecific nucleotide and amino-acid divergence, tested for recombination, and identified codon sites under selection by estimating the rate of non-synonymous to synonymous substitutions with multiple codon-based maximum likelihood methods. We determined that positive (diversifying) selection was acting on specific amino-acid sites located within the domains that bind pathogen-derived peptides. We also found significant signals of recombination across the physical distance of the genes. Finally, we determined that all the six species expressed two or three putative classical class I loci, in contrast to the single locus condition of Xenopus laevis. Our results suggest that MHC evolution in anurans is a dynamic process and that variation in numbers of loci and genetic diversity can exist among taxa. Thus, the accumulation of genetic data for more species will be useful in further characterizing the relative importance of processes such as selection, recombination and gene duplication in shaping MHC loci among amphibian lineages.
Keywords: anura, immunogenetics, major histocompatibility complex, molecular evolution
Introduction
The major histocompatibility complex (MHC) has been the subject of immunological and adaptive molecular evolution studies for over five decades (Clarke and Kirby, 1966; Doherty and Zinkernagel, 1975; Hedrick and Thompson, 1983; Hughes and Yeager, 1998). Many studies have characterized MHC genes within species (reviewed in Bernatchez and Landry (2003)) or investigated MHC evolution using model organisms at large phylogenetic scales (Flajnik et al., 1999b; Flajnik and Kasahara, 2001). Comparisons of intraspecific MHC diversity within lineages are less common, primarily due to the high variability in gene number, issues with identifying orthologous loci across species and the general difficulty of obtaining MHC data for species that lack whole genome sequences. Despite these problems, the number of comparative MHC studies in non-model organisms is slowly increasing due to the relevance of MHC variability in determining how wildlife populations respond to emerging infectious diseases (Acevedo-Whitehouse and Cunningham, 2006). Before pursuing such studies, however, researchers must have some basic knowledge about MHC genetic architecture, whether recovered sequences represent expressed gene copies and whether the loci being compared across populations or species originated from a single gene of the most recent common ancestor.
Current knowledge on MHC evolution in large, speciose vertebrate clades is often based on data from a limited number of taxa. This situation is especially true for frogs (anurans), where most MHC genetic data is predominately derived from two closely related species (Silurana tropicalis and Xenopus laevis) that belong to an ancient group of fully aquatic taxa (Pipidae; Figure 1). Understanding the differences in MHC variability among anuran species and between anuran MHC and that of other vertebrates has implications for evolutionary studies of acquired immunity, and increasingly, for amphibian conservation. Diseases are currently a major factor contributing to global amphibian declines and extinctions (for example, Berger et al., 1998; Lips et al., 2006). Given the growing evidence of associations between anuran MHC genotypes and susceptibility to bacterial, viral and fungal infections (for example, Barribeau et al., 2008; Teacher et al., 2009; May et al., 2011; Savage and Zamudio, 2011), understanding the evolution of MHC genes may hold important clues for determining which genetic differences may impact disease susceptibility among amphibian species.
Figure 1.
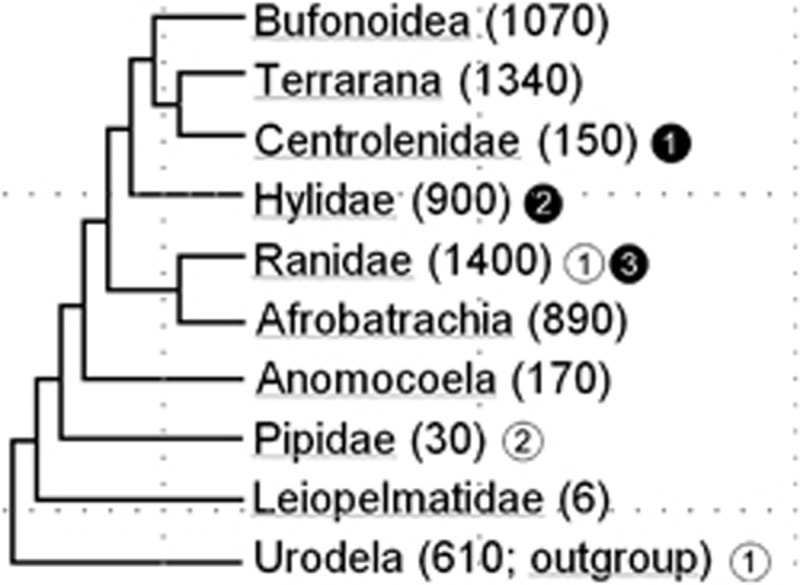
A simplified and abbreviated phylogeny of the major groups of anurans based on the relationships shown in Roelants et al. (2007) and Hedges et al. (2008). The approximate number of species in each group is given after the group name and is based on current numbers from AmphibiaWeb (accessed 20 February 2012). White circles contain the numbers of species from a particular group whose MHC class I alleles have been well-characterized while black circles contain the number of species characterized in the present study.
The MHC gene organization of the two well-characterized frogs, X. laevis (and S. tropicalis), is representative of the ‘primordial MHC' for jawed vertebrates because it is similar to that of other non-mammalian vertebrates in having separate but linked MHC class I and class II gene regions (Flajnik et al., 1999b; Ohta et al., 2006). Most Xenopus species express a single classical MHC class I gene (class Ia) that encodes receptor molecules that usually present intracellular pathogens—such as viruses—to cytotoxic T cells and is tightly linked to other genes (TAP1, TAP2 and LMP7) that assist in antigen processing (Flajnik et al., 1991; Flajnik et al., 1999b; Ohta et al., 2006). In addition, X. laevis express non-classical (class Ib) genes that encode molecules that are similar in sequence and structure to classical molecules (Flajnik et al., 1993). Class Ib genes belong to a separate MHC lineage that arose from an ancient duplication event and, although located on the same chromosome, are no longer linked to the MHC proper in frogs (Flajnik et al., 1993, 1999b; Ohta et al., 2006). Distinctions between the genes and gene products belonging to the two classes are not always clear, but class Ib genes generally show lower polymorphism, greater variation in sequence length, more restricted tissue distributions and lower expression levels relative to their classical counterparts (Flajnik et al., 1993; Braud et al., 1999).
The degree to which the X. laevis MHC is a good general model for the anuran MHC as a whole is largely unknown (but see Hauswaldt et al. (2007); Zeisset and Beebee (2009); Kiemnec-Tyburczy et al. (2010)). The anuran phylogeny includes 6000+ species of extant frogs, and their most recent common ancestor existed 250 million years ago (Roelants et al., 2007), permitting ample time for frog lineages to evolve differences within this gene complex. In addition, X. laevis and S. tropicalis are atypical frogs due to their fully aquatic life history, early divergence within the anuran phylogeny (Figure 1) and propensity to speciate via polyploidization; thus, data on the structure and diversity of MHC genes from a range of anuran taxa are needed to draw generalities for this clade.
The main goal of this study was to characterize MHC class I sequences in a number of phylogenetically distinct species, to gain a broader understanding of the evolutionary mechanisms that influence MHC diversity in this understudied vertebrate lineage. In total, we characterized six focal species that belong to three frog families, two of which—Hylidae and Centrolenidae—have not been examined in any MHC studies (Figure 1). We focused on characterizing MHC class I sequences from frogs for two general reasons. First, the effects of selection and other processes on MHC are so complex and diverse among species studied to date that we wanted to concentrate on a single class of genes. Second, the single class I locus condition of several characterized frogs suggests that studies of class I evolution may be more tractable than those of other multi-locus immune genes. A single classical class I locus is expressed in S. tropicalis, X. laevis and R. temporaria (a member of a more recently derived clade, Ranidae; Figure 1). Thus, it has been hypothesized that this single locus condition has been conserved throughout the anuran lineage (Teacher et al., 2009). In other vertebrate lineages, however, ancestral linkage patterns and gene arrangements have been greatly modified over evolutionary history. Mammalian MHC class I genes, in particular, are seemingly less evolutionarily constrained and have duplicated and diverged numerous times (Flajnik and Kasahara, 2001). The accumulation of sequence data for distantly related frog species will allow us to examine whether some of the generalities that apply to MHC evolution in other, more broadly surveyed clades such as mammals also apply to frogs. Our primary objectives were to (i) characterize the genetic diversity of alleles and infer putative numbers of loci for each species; (ii) test for the presence of recombination over physical distances along the gene sequences; and (iii) determine whether selection has acted on anuran MHC genes (including what codons may have experienced positive selection).
Materials and methods
Tissue sampling and nucleic acid extraction
We sampled six species, three from Central America: Agalychnis callidryas (red-eyed tree frog), Espadarana prosoblepon (emerald glass frog) and Smilisca phaeota (masked tree frog); and three from North America: Lithobates catesbeianus (bullfrog), L. clamitans (green frog) and L. yavapaiensis (lowland leopard frog). Agalychnis callidryas and S. phaeota belong to the family Hylidae; E. prosoblepon belongs to the family Centrolenidae, and the other three species belong to the family Ranidae (Figure 1). We collected five individuals of each species from a single locality within each species' range (Supplementary Table S1) and euthanized them in accordance with an approved Cornell University Animal Care and Use Protocol. We extracted RNA from intestinal tissue preserved in RNA later (Invitrogen, Carlsbad, CA, USA) using TRIzol, following the manufacturer's instructions (Invitrogen).
DNA amplification and cloning
We used complementary DNA (hereafter cDNA) as a template for PCR amplification of the coding regions of MHC class I from all species. We generated cDNA using an ImProm-II reverse transcription system (Promega, Madison, WI, USA) with an oligo(dT)15 primer under standard conditions. The resulting cDNA templates were diluted 1:10 for use in PCR.
The targeted gene regions included exons 2–4 that encode three extracellular domains of the class I alpha chain protein. We initially experimented with two types of DNA polymerase (Thermus (Taq: Roche Diagnostics, Indianapolis, IN, USA) versus Pyrococcus (Pyr; Phusion High-Fidelity DNA Polymerase: Finnzymes, Lafayette, CO, USA)) to assess whether the type of polymerase had an effect on the generation of spurious polymorphisms. The use of different polymerases did not have a significant effect on the genetic diversity between pools of Taq versus Pyr MHC alleles; thus both polymerases produced similar results (data not shown) and Taq was used for all subsequent amplifications. We amplified across the three exons using a single primer pair designed from L. pipiens and X. laevis class I sequences (Flajnik et al., 1999a). The forward (5′-AGTCAYWCYCTGCGBWATTAT-3′) and reverse (5′-GHWYYTYMTYCAGRCTGCTGT-3′) primers anneal at the 5′ end of exon 2 and 3′ end of exon 4, respectively. PCR cycling conditions were as follows: denaturation at 95 °C for 3 min, 35 cycles of 94 °C for 30 s, 58 °C for 1 min (touchdown −1–54 °C, with 5 cycles at each degree interval, then 15 cycles at 54 °C) and 72 °C for 1 min 30 s, and a final elongation step at 72 °C for 8 min. The resulting products were cloned using the pGEM-Teasy vector system (Promega). We amplified 20 (±2) positive clones per individual using universal M13 primers. PCR products were purified using an alkaline phosphatase-exonuclease I reaction and sequenced on an ABI 3100 automated sequencer (Applied Biosystems, Carlsbad, CA, USA) using Big Dye v3.1 chemistry (Kiemnec-Tyburczy et al., 2010).
Identifying genetic diversity
We initially used translated nucleotide BLAST queries to verify MHC class I homology of our amplicons to frog (R. temporaria and X. laevis), human and chicken class I amino-acid sequences. We edited the data using Sequencher v.4.7 and screened all sequences for disruptions of the amino-acid reading frame in DnaSP v4.5 (Rozas et al., 2003). We also performed data screening to minimize the inclusion of PCR and cloning artifacts (spurious errors) in our data set. Our analyses only included sequences that were (i) obtained from three or more independent clones (as in Hauswaldt et al. (2007)) and (ii) that differed from each other by more than three substitutions (van Oosterhout et al., 2006).
For all measures of nucleotide evolutionary divergence for MHC class I sequences, we calculated the number of nucleotide base substitutions per site by averaging over sequences using the Tajima–Nei distance implemented in MEGA 5.05 (Tamura et al., 2011). All amino-acid distances were calculated with the Poisson correction in MEGA. Gaps and missing data were excluded from downstream calculations.
Identifying recombination
We screened our data sets for recombination by calculating the correlation between physical distance along the gene sequence and three indices of linkage disequilibrium (LD): r2 (Hill and Robertson, 1968), D' (Lewontin, 1964) and G4 (McVean et al., 2002). In the absence of recombination, adjacent sites should be as tightly linked to one another as distant sites (Meunier and Eyre-Walker, 2001); thus, the null hypothesis predicts no correlation between LD and distance between sites. The program Permute (www.danielwilson.me.uk/omegaMap/permute.html) was used to determine statistical significance by randomly permuting the sites 999 times and recalculating the correlation coefficient for each simulated data set. The use of multiple LD measures accounted for the confounding effects of repeat mutations that can produce false-positives for recombination. Sites with more than two variants were excluded from these analyses because LD statistics are not defined in those cases.
Tests of selection acting on MHC class I loci
We tested whether selection was acting at two levels: within each frog species and across all the six species. For the intraspecific comparisons, we tested whether there was a signal of positive selection (1) in each of the three domains as a whole and (2) at peptide-binding region (PBR) sites vs non-PBR sites. The rationale for partitioning PBR and non-PBR sites was that the residues that compose the PBR are those that bind the pathogenic peptides and for this reason, they often show signatures of positive (diversifying) selection, whereas other regions of the protein do not. PBR sites were inferred as the sites that contact pathogenic peptides (A pocket, F pocket and other peptide contacts) in HLA-A2 as defined by Wallny et al. (2006) and references therein. Although the eight docking residues listed Supplementary Table S3 do contact the peptide, we excluded them from the analysis because they are known to be conserved across vertebrates (Flajnik et al., 1993) and thus are not likely to be under diversifying selection. The two tests of selection were performed using an one-tailed Z-test of selection in MEGA. The Z-test compared the relative rates of non-synonymous substitutions per non-synonymous site (dN) to number of synonymous substitutions per synonymous site (dS) across each domain for each species. When dN to dS are equal (dN−dS=0), a site is evolving neutrally, whereas an excess of dN relative to dS (dN−dS>0) is a signal of positive (diversifying) selection, and a deficiency is indicative of purifying (or negative) selection (dN−dS<0). Standard errors were determined using 500 bootstrap replicates.
To compare the strength of selection occurring across the sequences from multiple anuran species, we combined the sequences from all our focal species into a single data set, partitioned into the three exons that encode the protein domains. We first examined whether selection was acting across each domain alignment as a whole using three tests: (1) an one-tailed Z-test, (2) PARRIS (hosted at the Datamonkey server (http://www.datamonkey.org)) and (3) Codeml (implemented in PAML). We used likelihood ratio tests to assess the fit of the data to two pairs of nested codon-substitution models in Codeml: (1) the M1a model of nearly neutral evolution vs M2a model of selection and (2) the M7 model (which fits ω into 10 site classes in the interval 0–1) vs M8 model (which adds an extra site class to M7 that allows ω to be>1) (Yang, 2007). PARRIS also uses a likelihood ratio test to select between two nested models, while allowing synonymous rate variation, topology and branch lengths to vary across recombination breakpoints (Scheffler et al., 2006).
We then tested whether selection had acted on each codon site across the multi-species alignment. We used four methods to assess whether selection had acted on a site: single likelihood ancestral counting (SLAC), fixed effects likelihood (FEL), random effects likelihood (REL) and maximum likelihood models of codon evolution in Codeml (implemented in PAML; Yang, 2007). SLAC, FEL and REL are included in the HyPhy software package (hosted at the Datamonkey server). SLAC is the most conservative of the three HyPhy methods, while REL is the most powerful (Pond and Frost, 2005). Bayes empirical Bayes (in Codeml) was used to identify codon sites under positive selection by identifying sites whose posterior probabilities were ⩾0.95 under both the M2a and M8 models. By comparing all methods, we could better assess which sites showed strong evidence of selection.
To build the input tree used for all four methods, we first tested each domain alignment for the best-fit model of nucleotide evolution using the Datamonkey server model selection tool. This analysis compared the fit of over 200 nucleotide substitution models to the observed data using the Akaike information criterion. We used MrBayes 3.1 (Ronquist and Huelsenbeck, 2003) to generate a 95% credible set of rooted MHC genealogies using the appropriate nucleotide model (HKY+ Γ for exon 2, GTR+Γ for exon 3, HKY+Γ for exon 4). We ran two Markov chains for 1 × 106 iterations, retaining every 500th sample from the posterior distribution and starting values for each chain chosen randomly. We tested for convergence, effective sample sizes and the appropriate amount of burn-in by examining the posterior distributions in Tℛ𝒜𝒞ℰℛ v1.4 (Drummond and Rambaut, 2007). We performed each of the four methods on the individual exon alignments to avoid biases introduced by recombination, which we detected only across larger physical distances than those covered by each exon (see Results).
Phylogenetic tree reconstruction
To ascertain the genealogical relationships among the frog MHC class I sequences, we compiled an amino-acid data set consisting of: (1) the sequences from each of our focal species, (2) X. laevis Ia and Ib sequences and (3) two urodele amphibian sequences as outgroups. We then partitioned these sequences into domains and tested each domain for signals of recombination using the three measures calculated by Pℰℛℳ𝒰𝒯ℰ (Wilson and McVean, 2006) to ensure that recombination would not bias the accuracy of our phylogenetic inference (Schierup and Hein, 2000). We used ProtTest 3 (Darriba et al., 2011) to determine which amino-acid replacement matrix best fits the amino-acid alignment of each domain, based on the Akaike Information Criterion (JTT+ Γ for α1, α2 and α3). Genealogical trees for each exon were constructed separately using maximum likelihood methods in PhyML 3.0 (Guindon et al., 2010) with the proportion of invariable sites and gamma shape parameter estimated from the data. We calculated branch support using 1000 bootstrap replicates. In some cases, sequences were 100% identical in one or two domains and in these cases, only one representative was included in the alignment, but the names of both sequences were noted on the tree (for example, Lica-UA*01/08).
Results
We recovered 1–5 unique cDNA sequences per frog that encoded proteins with high similarity to other vertebrate MHC class I genes. The strongest BLAST hits recovered were the MHC class I genes of other vertebrates, with the highest similarities to published anuran sequences from L. pipiens, X. laevis and S. tropicalis. We isolated a total of 79 putative class I sequences (see Supplementary Table S2 for accession nos.). The total length of the fragments ranged from 750 to 759 nucleotides, excluding primer sequences and spanned exons 2–4. These sequences encoded most of the α1, the complete α2 and part of the α3 domain. Generally, the MHC class I sequences we amplified encoded 80 codons of the α1 domain, 91 codons of the α2 domain and 80 codons of the α3 domain, although we did identify length variants both within and among species. Compared to human HLA-A*0201V, frogs had 1–4 fewer amino acids (aa) in their overall sequences (Figure 2). The shortest fragment was amplified from E. prosoblepon (250 aa) and the longest from L. clamitans and L. catesbeianus (253 aa). Most of the deletions that differed within frogs occurred in the α1 domain. All frog sequences had a two-aa insertion in the α2 domain, relative to human HLA. Even though we isolated length variants, we did not sequence expressed pseudogenes, as identified by frameshift mutations or premature stop codons.
Figure 2.

Alignment of representative amino-acid sequences of MHC class I sequences isolated from A. callidryas (Agca), E. prosoblepon (Espr), L. catesbeianus (Lica), L. clamitans (Licl), L. yavapaiensis (Liya) and S. phaeota (Smph). Sequences are aligned to classical MHC class I sequences from A. mexicanum (Amme; U83137), X. laevis (Xela; DQ149606) and human (HLA-A*0201V; AJ621243). Domains are separated according to Flajnik et al. (1991). Numbering is in reference to the consensus amino acids in the alignment. The dots represent amino-acid residues that are identical to the top sequence; dashes represent gaps in the sequences; ▪ denotes disulfide bridge-forming cysteines; ♦ denotes salt bridge-forming sites; ▴ denotes an N-glycosylation site; CD8 box denotes the predicted CD8-binding site; and * denotes the antigen N- and C-termini peptide-docking sites.
The frog sequences we isolated generally showed high levels of amino-acid identity at residues known to have functional significance in class Ia proteins, such as sites of glycosylation and disulfide bridge formation (Figure 2). Conservative substitutions were common across some of the functional residues in the six frogs, including Y to F/H, types of conservative substitutions that are common in the classical class I sequences of some non-mammalian vertebrates (for example, fish; Lukacs et al., 2010). We did note, however, the presence of non-conservative substitutions in peptide-docking residues that are usually conserved between divergent groups of vertebrates (for example, Xenopus and humans; Flajnik et al., 1993). This phenomenon was especially illustrated by the amino-acid variants found at sites 116 and 166. These two sites—predominately Y in other vertebrates—were substituted to F, L, R, H or C in various frog sequences (Supplementary Table S3). In another case, multiple frog species had non-conservative substitutions that dramatically changed the biochemical properties of that site; W140 was changed to a hydrophobic amino acid (L, I, V) in five of the six species. Several of these types of non-conservative substitutions were also present in the class I sequences isolated from R. temporaria (Teacher et al., 2009).
Number of expressed MHC class Ia loci
We recovered sequences from multiple loci from all six species, based on the number of unique sequences cloned from single individuals. The number of putative loci varied from 2 to 3 among the three families (Centrolenidae, Hylidae and Ranidae). Although we could not determine the total number of loci in each frog without a reference genome, we conservatively estimated the minimum number of loci for each species by dividing the maximum number of alleles found in any one individual by two (Table 1). Our results indicated the presence of 1–2 additional loci not found in S. tropicalis or R. temporaria. We also recovered 2–5 additional unique sequences from single clones, which were excluded from further analyses because we could not verify their repeated presence among multiple sequenced clones. The sequences reported here therefore represent a conservative subset of the unique sequences we amplified from the cDNA of our focal frogs.
Table 1. MHC class I genetic divergence within six species of frogs.
| Species | No. of unique sequences | Ave. nucleotide divergence | Ave. amino-acid divergence | ||||||
|---|---|---|---|---|---|---|---|---|---|
| |
|
Exon 2 |
Exon 3 |
Exon 4 |
Full-length |
Exon 2 |
Exon 3 |
Exon 4 |
Full-length |
| A. callidryas | 19 | 0.304 | 0.212 | 0.102 | 0.202 | 0.320 | 0.349 | 0.132 | 0.287 |
| E. prosoblepon | 12 | 0.233 | 0.145 | 0.066 | 0.153 | 0.329 | 0.230 | 0.094 | 0.225 |
| L. catesbeianus | 12 | 0.287 | 0.115 | 0.042 | 0.132 | 0.377 | 0.201 | 0.066 | 0.197 |
| L. clamitans | 16 | 0.292 | 0.113 | 0.031 | 0.130 | 0.367 | 0.196 | 0.050 | 0.193 |
| L. yavapaiensis | 9 | 0.115 | 0.080 | 0.038 | 0.076 | 0.219 | 0.129 | 0.064 | 0.134 |
| S. phaeota | 11 | 0.141 | 0.122 | 0.054 | 0.102 | 0.218 | 0.182 | 0.087 | 0.160 |
Genetic variability across frog MHC class I sequences
The MHC class I sequences we isolated were extremely polymorphic at both the nucleotide and amino-acid levels, a pattern consistent with observed variation in MHC class Ia sequences in other vertebrates (Flajnik and Kasahara, 2001). The low frequency of identical sequences recovered from conspecific individuals is a second illustration of the polymorphic nature of the MHC class I alleles in the populations of frogs we sampled. Most sequences were isolated from single individuals within each species (Supplementary Table S2). For example, of the 19 sequences recovered from A. callidryas, only one sequence was found in more than one individual. In total, 69 sequences were found solely in a single individual over all species combined. Within species, the number of alleles found in a single individual ranged from 7 (in L. yavapaiensis) to 18 (in A. callidryas). In addition, the magnitude of nucleotide and amino-acid divergence differed greatly across all species, as well as among sequences within individuals of the same species. At the level of species, A. callidryas displayed the highest nucleotide and amino-acid divergence across all three domains/exons (Table 1), whereas L. yavapaiensis showed the lowest. At the level of individuals, we again found that some A. callidryas expressed highly divergent classical class I sequences within individuals, while L. yavapaiensis, we detected less intraindividual variation (Table 2).
Table 2. Variation in the sequence divergence of MHC class I sequences among individuals.
| Species | Mean no. of sequences/individuals | Range of no. of sequences/individuals |
Range of divergence within an individuala |
|
| |
|
|
Nucleotide |
Amino acid |
| A. callidryas | 3.8 | 3–5 | 0.075–0.336 | 0.120–0.451 |
| E. prosoblepon | 2.4 | 1–4 | 0–0.230 | 0–0.336 |
| L. catesbeianus | 2.4 | 1–5 | 0–0.137 | 0–0.206 |
| L. clamitans | 3.2 | 2–5 | 0.039–0.163 | 0.070–0.216 |
| L. yavapaiensis | 1.8 | 1–4 | 0–0.085 | 0–0.158 |
| S. phaeota | 2.2 | 2–5 | 0–0.154 | 0–0.242 |
A ‘0' starting range indicates that this species had one or more individuals from which a single sequence was amplified
Sequence divergence also revealed patterns consistent with the functional roles of each extracellular domain. In all frog species, the exons that encode the two domains that putatively interact with pathogen-derived peptides—α1 and α2—had higher levels of divergence than α3 (Table 1), which interacts with the β2-microglobulin and functions primarily in maintaining protein structure. Comparing divergence of α1 and α2, we found in almost all cases that the α1 domain had higher levels (both nucleotides and amino acids) of divergence than α2. This finding is similar to patterns of variation documented for X. laevis class Ia domains (divergence of α1>α2>α3 domains; Flajnik et al., 1999a).
Presence of recombination
We tested two full-length cDNA (exon 2–4) class I data sets for signals of recombination: one used to estimate selection on specific sites and one used to generate the phylogenetic trees for each protein domain (Figure 3). In both cases, we detected recombination in the data sets (P<0.01 in all cases), as evidenced by the significant negative correlations between the three LD statistics and physical distance between sites. When we partitioned each data set into the three exons, however, none of the three measures detected a significant signal of recombination (all P>0.05). This result indicates that the physical distance over which recombination typically occurs in these frogs exceeds the length of individual exons.
Figure 3.

Evolutionary relationships of anuran MHC class Ia and Ib sequences. Consensus trees were constructed with PhyML on amino-acid alignments of either the α1 (a), α2 (b) or α3 (c) domains (see text for details). Bootstrap values above 70% are denoted by asterisks below branches (***=90–100% **=80–89% *=70–79%). Scales indicate the number of amino-acid substitutions per site. Some highly supported groups of sequences were compressed into blocks (the thickness of triangle was roughly proportional to number of sequences). GenBank Accession Nos. for X. laevis class Ia (Xela r/r: AF185582; Xela f/f: AF185580; Xela a/c-1: AF185583; Xela j/j: AF185586), X. laevis class Ib (XNC1: M58019; XNC2: L20725; XNC3: L20726; XNC4: L20727; XNC6: L20729; XNC5: L20728; XNC7: L20730; XNC8: L20731; XNC10: FJ589642; XNC11: FJ589643) and A. mexicanum class Ia (1: U83137; 2: U83138).
Tests of selection within and across species
We first tested each domain of each species separately for evidence of positive selection using the one-tailed Z-test. All of these tests showed no evidence of selection in any species when considering all amino acids of a single exon (all P>0.05). This result is not surprising given that most of the MHC class I sites are expected to be neutrally evolving, with a smaller subset (especially those that are functionally constrained) expected to be under purifying selection. Thus, the signal of positive selection on a small number of residues might be overwhelmed by a greater number of sites evolving neutrally or under purifying selection. When we tested the PBR and non-PBR sites separately, an excess of dN was detected in the PBR sites of the α1 and α2 domain of five of the six species (Table 3). In contrast, no evidence of positive selection was detected in the α3 domain or the combination of the non-PBR sites remaining in the α1 and α2 domains.
Table 3. Comparisons of non-synonymous (dN) and synonymous (dS) substitutions per site of the MHC class I domains from six frog species.
| Species | dN−dS | ||
|---|---|---|---|
| |
α1+α2 PBR |
α1+α2 non-PBR |
α3 |
| A. callidryas | 0.476±0.126a | −0.069±0.018 | −0.037±0.027 |
| E. prosoblepon | 0.498±0.113a | −0.043±0.019 | −0.038±0.020 |
| L. catesbeianus | 0.343±0.133 | −0.035±0.021 | −0.039±0.020 |
| L. clamitans | 0.399±0.123a | −0.046±0.024 | −0.021±0.014 |
| L. yavapaiensis | 0.375±0.096a | 0.014±0.016 | −0.006±0.014 |
| S. phaeota | 0.295±0.089a | −0.011±0.018 | −0.004±0.020 |
Mean dN is significantly higher than mean dS (P<0.05).
We also tested the larger data set of anuran sequences for evidence of positive selection over the alignments of each protein domain. For α1 and α2, PARRIS detected significant evidence of positive selection (P<0.1), as did Codeml (M2a model was a better fit to the data than the model of neutrality M1a, and M8 was a significantly better fit than M7). Codeml, however, detected positive selection acting on α3, whereas PARRIS did not. The Z-test of selection did not show significant excess of dN over dS in any of the three domains in this larger data set.
Patterns of selection across codons
We used four codon-based maximum likelihood methods to assess whether selection had acted on each codon in an alignment of MHC class I sequences from six frog species. The methods differed in which sites they identified as under positive selection (Table 4). Most methods found positive selection acting on subset of codon sites within exon 2 and exon 3. REL and Codeml, however, found a total of three sites under selection in exon 4. About half of the sites identified by any method were predicted to be PBR sites in the alpha chain (comprised of the α1 and α2 domains). In contrast, 80% of the sites identified as under selection by all methods were predicted to be PBR sites. The remaining sites in exons 2 and 3 were often only predicted by one or two methods to be under selection (with the exception of sites 3 and 147). We found approximately equal numbers of positively selected sites in exon 2 and exon 3, and approximately one third of the sites were identified by all four methods (Table 4). REL predicted the most sites under selection, while SLAC predicted the fewest. Given the effects of diversifying selection on the α1 and α2 domains and the functional constraints imposed on the α3 domain (Kaufman et al., 1992), exons 2 and 3 are expected to have higher dN−dS values relative to exon 4. Our results are consistent with this prediction; far fewer sites were under positive selection in exon 4 (Table 4) and, generally, the values of dN−dS were lowest for this exon (Table 3).
Table 4. Codon sites predicted to be under positive selection in six species of frogs.
| Method | Sites under positive selection | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Exon 2 |
Exon 3 |
Exon 4 |
|||||||||||||||||||||||||||||||
| |
1 |
3 |
16 |
26 |
37 |
38 |
47 |
55 |
56 |
60 |
62 |
63 |
66 |
67 |
74 |
83 |
85 |
90 |
97 |
107 |
109 |
110 |
114 |
126 |
136 |
137 |
147 |
151 |
158 |
165 |
179 |
216 |
240 |
| SLAC | x | x | x | x | x | x | x | x | x | x | x | x | x | ||||||||||||||||||||
| FEL | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | |||||||||||||
| REL | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | ||||||||
| Codeml | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | |||||||||
Bold sites were identified as putative PBR sites based on those in Wallny et al. (2006). Numbering is based on alignment in Figure 2.
Genealogical relationships of MHC class I exons
To gain insight into the evolutionary history of the amphibian MHC class I domains, we constructed a phylogenetic tree of class I sequences from the six focal species and other amphibians for each protein domain. The high amino-acid diversity in the α1 and α2 domains within and across species likely contributed to the low branch support in the trees (Figures 3a and b). The trees showed that the frog sequences we isolated were distinct from the class Ib sequences of X. laevis; in all three trees, the X. laevis Ib sequences formed a well-supported monophyletic group (Figure 3). The class Ia sequences of X. laevis also comprised a separate clade in all three trees. The expected phylogenetic relationships among frogs were reflected on a broad level in the MHC α3 domain tree. In the α3 tree, we inferred four major groups: the X. laevis Ia sequences, X. laevis Ib sequences, central American sequences and north American sequences (Figure 3c). The three central American species belong to two closely related anuran families (Hylidae and Centrolenidae), while Lithobates is a distantly related genus in the family Ranidae (Figure 1). The north and central American frogs formed two distinct groups in all trees; however, in the α1 and α2 trees they were nested within central American species. A subset of sequences isolated from E. prosoblepon and A. callidryas clustered into extremely divergent groups of sequences that did not group with other sequences from the same species (Figure 3c). Instead, these sequences grouped with sequences from another species (for example, Espr-UA*01, Agca-UA*01 group). This result was indicative of trans-species polymorphism, where similar alleles were passed from the common ancestor of multiple species to the descendents (Klein et al., 1998). A similar pattern of allele sharing was also evident in the MHC sequences from the more closely related members of the genus Lithobates (Figure 3). Sequences from multiple Lithobates species grouped together within a single clade in the α1, α2 and α3 trees.
Discussion
MHC class I transcripts from six phylogenetically disparate frog species show high sequence polymorphism and signatures of positive selection characteristic of functional MHC class Ia loci. We found that putative MHC class I genes were most diverse in the central American species A. callidryas, but genetic divergence was also high in some north American ranids. We documented patterns in the class I evolutionary trees that indicated trans-species polymorphism had occurred (some allelic lineages on the tree contained sequences from multiple species). The genus Lithobates may be as old as 36–39 million years, while the divergence between A. callidryas and E. prosoblepon dates back to 68–91 million years ago (Roelants et al., 2011). In comparison, the oldest DRB1 allelic lineages in primates are estimated to be ∼55 million years old (Klein et al., 1998) and allelic lineages are shared between Ruminanta genera that diverged over 30 million years ago (Ballingall et al., 2010).
Although our methods and results do not characterize the complete genomic architecture of the MHC of our six focal species, they provide a conservative estimate of the numbers of classical MHC class I loci for these frog species (Table 5). We used the three criteria to define our sequences as class Ia loci: (i) the alleles do not show the typical pattern of additional insertions and deletions that are present in X. laevis 1b alleles; (ii) sequences in our data set do not group with the non-classical X. laevis sequences phylogenetically; and (iii) our sequences contain conserved amino acids at sites that are regarded as necessary for classical class I proteins to function. Although it is possible some of the sequences we identified were class Ib loci, our methodology provides evidence that the sequences are functional Ia alleles. Three other anuran species, R. temporaria, X. laevis and S. tropicalis express a single MHC class Ia locus (Flajnik et al., 1991, 1993; Teacher et al., 2009), but our study shows that numbers expressed of class Ia loci vary across major frog lineages (Table 5). We documented the presence of 2–3 expressed class Ia loci in other frog species, including three species within the genus Lithobates.
Table 5. Minimum numbers of putative MHC class Ia loci expressed in amphibian species.
| Family | Species | No. of loci | Reference |
|---|---|---|---|
| Hylidae | A. callidryas | 3 | This study |
| Hylidae | S. phaeota | 3 | This study |
| Centrolenidae | E. prosoblepon | 2 | This study |
| Ranidae | L. catesbeianus | 3 | This study |
| Ranidae | L. clamitans | 3 | This study |
| Ranidae | L. pipiens | ⩽1 | Flajnik et al. (1999a, 1999b) |
| Ranidae | L. yavapaiensis | 2 | This study |
| Ranidae | R. temporaria | 1 | Teacher et al. (2009) |
| Pipidae | S. tropicalis | 1 | Ohta et al. (2006) |
| Pipidae | X. laevis | 1 | Goyos et al. (2011) |
| (Urodela) Ambystomatidae | A. mexicanum | ⩽2 | Sammut et al. (1999) |
The number of sequences obtained from our focal species of frogs is similar to those obtained in an initial characterization of MHC class I genes of the axolotl (Ambystoma mexicanum), a urodele amphibian (Sammut et al., 1999). The axolotl expresses 11–21 unique amino-acid sequences per individual, indicative of multiple class I loci. This multi-locus condition is more similar to the genomic architecture of mammals, birds and fish, which have widely differing numbers of class I loci and high levels of intraspecific polymorphism (Nonaka et al., 1997; Flajnik and Kasahara, 2001). For example, fish express anywhere from three class I loci in salmonids to 17 in some cichlids (Sato et al., 1997; Miller and Withler, 1998). The chicken genome contains two classical class I loci (Kaufman et al., 1999) while great reed warblers express up to 6 loci (Westerdahl et al., 2004). It is therefore possible that gene duplications or genome polyploidization have given rise to MHC class I paralogs in species nested within the anuran tree. We hypothesize that these differences, even potentially within genera, may be due to species-specific gene duplication events that may have occurred in frogs as they have in other vertebrate groups. A number of Xenopus and Silurana species have evolved by allopolyploidization, resulting in tetra-, octo- and dodecaploid species over the past 10–81 million years (Evans et al., 2004). While most of these polyploids revert to disomic inheritance of the MHC, the dodecaploid X. ruwenzoriensis displays polysomic inheritance and expresses multiple class I paralogs (Sammut et al., 2002). Frogs, in general, show disproportionately high rates of polyploid speciation relative to all vertebrates, with some of the best-known examples occurring in the Hylidae (Haddad et al., 1994; Ptacek et al., 1994), the family to which A. callidryas and S. phaeota belong. In some species, geographic variation in ploidy level has even been described (Haddad et al., 1994). Karyotypic data from our focal species, however, show that A. callidryas (Duellman and Cole, 1965), L. catesbeianus (Elinson and Briedis, 1981), L. clamitans (Elinson and Briedis, 1981), S. phaeota (Duellman and Trueb, 1966) and L. yavapaiensis (Hillis, 1988) are diploid. Overall, while the number of unique MHC class I sequences within individuals in this study is striking when compared with other frogs examined to date, our results are not entirely unexpected, especially given the small percentage of frog species whose MHC genes have been characterized (Figure 1) and the diversity of ploidy levels within anurans.
MHC variability is predominantly caused by the accumulation of point mutations over millions of years, with recombination, gene duplication and gene convergence generating additional allelic diversity (Parham and Ohta, 1996; Little and Parham, 1999; Bos and Waldman, 2006a). The large number of sequences recovered within individual frogs in this study may also reflect the joint effects of intra- and interlocus recombination acting across several MHC class I paralogs. Conversion between class Ia and class Ib is an expected outcome of their common ancestry. Non-classical MHC loci may generate additional genetic diversity by serving as donors that can be partially transferred to Ia genes via recombination or serve as templates from the formation of new class Ia genes (Watkins et al., 1990). In addition, we detected significant correlations between LD and physical distance between sites across all three MHC class I protein domains, indicating that recombination is central to increasing genetic diversity in these species. The signal was lost, however, when we partitioned each domain into a separate data set. One possible mechanism underlying these results is the exchange of entire exons during recombination, as opposed to shorter motifs within exons. In X. laevis, at least two recombinant alleles (Xela-UAA*06 and UAA*11) have identical α1 domain sequences, but divergent α2 and α3 domains (Bos and Waldman, 2006a). Our results are consistent with this pattern, especially given that we amplified sequences from five of the six species that were identical in one domain, but differed in others. Our preliminary analyses suggest that breakpoints may be located at around exon junctions (data not shown) but further population sampling within species is needed to identify breakpoint hotspots that exhibit higher rates of recombination relative to surrounding sites. As the sequence database for amphibian MHC genes continues to grow, it will hopefully become possible to distinguish the relative contributions of recombination and selection in generating MHC diversity in frogs.
Natural selection is a key process that acts on MHC genes in most vertebrate taxa (Hughes and Yeager, 1998; Bernatchez and Landry, 2003). Our tests for selection clearly showed that a proportion of codon sites putatively located in the PBR of the MHC class I sequences from central and north American frogs have elevated levels of dN over dS indicative of positive selection. When we examined the alignment of six species, the majority of positively selected sites were located in the α1 and α2 domains, the two domains that bind foreign peptides derived from pathogens. This result is concordant with that found in the alleles of X. laevis (Bos and Waldman, 2006b). Although the majority of sites under selection were predicted to be PBR based on the crystal structure of human class I, there are others that showed significant signatures of positive selection that were not located in the human PBR. Given the differences between the crystal structures of chicken and human class I molecules (Koch et al., 2007), the structure of the peptide-binding groove in frogs is necessary to decode the functional role of the sites under positive selection. Regardless, it is clear that over the evolutionary history of anurans diversifying selection has shaped MHC class I genes. Elevated amino-acid diversity, especially in gene regions encoding the antigen-binding sites, intra- and intergenic recombination and gene duplication are possible evolutionary mechanisms that enhance an individual's ability to combat disease (Hedrick, 1994; Parham and Ohta, 1996; Hughes and Yeager, 1998). The evolutionary history of a population may therefore be important when individuals are faced with an epizootic factor, as selection may favor individuals with elevated MHC polymorphism or specific resistance-conferring alleles (Hughes and Yeager, 1998; Bernatchez and Landry, 2003; Richmond et al., 2009). In fact, recent studies on ecological time scales have shown that natural amphibian populations recently exposed to infectious diseases have short-term changes in allele frequency that may be correlated with differential survival (for example, Teacher et al., 2009; May et al., 2011). Thus, anurans represent tractable systems for studying MHC evolution over both short and long timescales.
Data archiving
Sequence data have been deposited at GenBank Accession No.JQ679312–JQ679390.
Acknowledgments
We thank Daniel Wilson, Gil McVean, José Andrés, Adam Richman and Rayna Bell for helpful discussions about analytical methods, and Martin Flajnik for advice on MHC evolution. Steve Bogdanowicz and José Andrés provided guidance with molecular techniques. James Austin, Joseph Mendelson III, Jeanne Robertson and Bunk Moreland graciously contributed samples to this study. Cora Johnston, Stephanie Wein and Rebecca McCaffery provided assistance with specimen collection. We thank the Smithsonian Tropical Research Institute staff for assistance with permits. Funding for this study was provided by the Cornell University ADVANCE Research Initiation Grant, National Science Foundation Grants (DEB-0815315 and DEB-0717741), and the Biogeochemistry & Biocomplexity Initiative at Cornell University.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Supplementary Material
References
- Acevedo-Whitehouse K, Cunningham AA. Is MHC enough for understanding wildlife immunogenetics. Trends Ecol Evol. 2006;21:433–438. doi: 10.1016/j.tree.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Ballingall KT, Rocchi MS, McKeever DJ, Wright F. Trans-species polymorphism and selection in the MHC class II DRA genes of domestic sheep. PLoS ONE. 2010;5:e11402. doi: 10.1371/journal.pone.0011402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barribeau SM, Villinger J, Waldman B. Major histocompatibility complex based resistance to a common bacterial pathogen of amphibians. PLoS One. 2008;3:e2692. doi: 10.1371/journal.pone.0002692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger L, Speare R, Daszak P, Green DE, Cunningham AA, Goggin CL, et al. Chytridiomycosis causes amphibian mortality associated with population declines in the rain forests of Australia and central America. Proc Natl Acad Sci USA. 1998;95:9031–9036. doi: 10.1073/pnas.95.15.9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernatchez L, Landry C. MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years. J Evol Biol. 2003;16:363–377. doi: 10.1046/j.1420-9101.2003.00531.x. [DOI] [PubMed] [Google Scholar]
- Bos DH, Waldman B. Evolution by recombination and transspecies polymorphism in the MHC class I gene of Xenopus laevis. Mol Biol Evol. 2006a;23:137–143. doi: 10.1093/molbev/msj016. [DOI] [PubMed] [Google Scholar]
- Bos DH, Waldman B. Polymorphism, natural selection, and structural modeling of class Ia MHC in the African clawed frog (Xenopus laevis) Immunogenetics. 2006b;58:433–442. doi: 10.1007/s00251-006-0114-5. [DOI] [PubMed] [Google Scholar]
- Braud VM, Allan DSJ, McMichael AJ. Functions of nonclassical MHC and non-MHC-encoded class I molecules. Curr Opin Immunol. 1999;11:100–108. doi: 10.1016/s0952-7915(99)80018-1. [DOI] [PubMed] [Google Scholar]
- Clarke B, Kirby D. Maintenance of histocompatibility polymorphisms. Nature. 1966;211:999–1000. doi: 10.1038/211999a0. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011. pp. 1164–1165. [DOI] [PMC free article] [PubMed]
- Doherty P, Zinkernagel R. Enhanced immunological surveillance in mice heterozygous at the H-2 gene complex. Nature. 1975;256:50–52. doi: 10.1038/256050a0. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duellman WE, Cole CJ. Studies of chromosomes of some anuran amphibians. Syst Zool. 1965;14:139–143. [PubMed] [Google Scholar]
- Duellman WE, Trueb L. Neotropical hylid frogs, genus Smilisca. Museum Nat History (Univ Kansas Publ) 1966;17:281–375. [Google Scholar]
- Elinson RP, Briedis A. Triploidy permits survival of an inviable amphibian hybrid. Dev Genet. 1981;2:357–367. [Google Scholar]
- Evans BJ, Kelley DB, Tinsley RC, Melnick DJ, Cannatella DC. A mitochondrial DNA phylogeny of African clawed frogs: phylogeography and implications for polyploid evolution. Mol Phylogenet Evol. 2004;33:197–213. doi: 10.1016/j.ympev.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Flajnik MF, Kasahara M. Comparative genomics of the MHC: glimpses into the evolution of the adaptive immune system. Immunity. 2001;15:351–362. doi: 10.1016/s1074-7613(01)00198-4. [DOI] [PubMed] [Google Scholar]
- Flajnik MF, Kasahara M, Shum B, Salter-Cid L, Taylor E, Du Pasquier L. A novel type of class I gene organization in vertebrates: a large family of non-MHC-linked class I genes is expressed at the RNA level in the amphibian Xenopus. EMBO J. 1993;12:4385–4396. doi: 10.1002/j.1460-2075.1993.tb06123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flajnik MF, Ohta Y, Greenberg AS, Salter-Cid L, Carrizosa A, Du Pasquier L, et al. Two ancient allelic lineages at the single classical class I locus in the Xenopus MHC. J Immunol. 1999a;163:3826–3833. [PubMed] [Google Scholar]
- Flajnik MF, Ohta Y, Namikawa-Yamada C, Nonaka M. Insight into the primordial MHC from studies in ectothermic vertebrates. Immunol Rev. 1999b;167:59–67. doi: 10.1111/j.1600-065x.1999.tb01382.x. [DOI] [PubMed] [Google Scholar]
- Flajnik MJ, Canel C, Kramer J, Kasahara M. Evolution of the major histocompatibility complex: molecular cloning of major histocompatibility complex class I from the amphibian Xenopus. Proc Natl Acad Sci USA. 1991;88:537–541. doi: 10.1073/pnas.88.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyos A, Sowa J, Ohta Y, Robert J. Remarkable conservation of distinct nonclassical MHC class I lineages in divergent amphibian species. J Immunol. 2011;186:372–381. doi: 10.4049/jimmunol.1001467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Haddad CFB, Pombal JPJ, Batistic RF. Natural hybridization between diploid and tetraploid species of leaf-frogs, genus Phyllomedusa (Amphibia) J Herpetol. 1994;28:425–430. [Google Scholar]
- Hauswaldt JS, Stuckas H, Pfautsch S, Tiedemann R. Molecular characterization of MHC class II in a nonmodel anuran species, the fire-bellied toad Bombina bombina. Immunogenetics. 2007;59:479–491. doi: 10.1007/s00251-007-0210-1. [DOI] [PubMed] [Google Scholar]
- Hedges SB, Duellman WE, Heinicke MP. New World direct-developing frogs (Anura: Terrarana): molecular phylogeny, classification, biogeography, and conservation. Zootaxa. 2008;1737:1–182. [Google Scholar]
- Hedrick P. Evolutionary genetics at the major histocompatibility complex. Am Nat. 1994;143:945–964. [Google Scholar]
- Hedrick PW, Thompson G. Evidence for balancing selection at HLA. Genetics. 1983;104:449–456. doi: 10.1093/genetics/104.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG, Robertson A. Linkage disequilibrium in finite populations. Theor Appl Genet. 1968;38:226–231. doi: 10.1007/BF01245622. [DOI] [PubMed] [Google Scholar]
- Hillis DM. Systematics of the Rana pipiens complex: puzzle and paradigms. Ann Rev Ecol Syst. 1988;19:39–63. [Google Scholar]
- Hughes A, Yeager M. Natural selection at major histocompatibility complex loci of vertebrates. Annu Rev Genet. 1998;32:415–434. doi: 10.1146/annurev.genet.32.1.415. [DOI] [PubMed] [Google Scholar]
- Kaufman J, Andersen R, Avila D, Engberg J, Lambris J, Salomonsen J, et al. Different features of the MHC class I heterodimer have evolved at different rates. Chicken B-F and beta 2-microglobulin sequences reveal invariant surface residues. J Immunol. 1992;148:1532–1546. [PubMed] [Google Scholar]
- Kaufman J, Milne S, Gobel TW, Walker BA, Jacob JP, Auffray C, et al. The chicken B locus is a minimal essential major histocompatibility complex. Nature. 1999;401:923–925. doi: 10.1038/44856. [DOI] [PubMed] [Google Scholar]
- Kiemnec-Tyburczy KM, Richmond JQ, Savage AE, Zamudio KR. Selection, trans-species polymorphism and locus identification of major histocompatibility complex class IIβ alleles of New World ranid frogs. Immunogenetics. 2010;62:741–752. doi: 10.1007/s00251-010-0476-6. [DOI] [PubMed] [Google Scholar]
- Klein J, Sato A, Nagl S, O'hUigin C. Molecular trans-species polymorphism. Annu Rev Ecol Syst. 1998;29:1–C1. [Google Scholar]
- Koch M, Camp S, Collen T, Avila D, Salomonsen J, Wallny HJ, et al. Structures of an MHC class I molecule from B21 chickens illustrate promiscuous peptide binding. Immunity. 2007;27:885–899. doi: 10.1016/j.immuni.2007.11.007. [DOI] [PubMed] [Google Scholar]
- Lewontin RC. The interaction of selection and linkage. I. Genetic considerations; heterotic models. Genetics. 1964;49:49–67. doi: 10.1093/genetics/49.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lips KR, Brem F, Brenes R, Reeve JD, Alford RA, Voyles J, et al. Emerging infectious disease and the loss of biodiversity in a Neotropical amphibian community. Proc Natl Acad Sci USA. 2006;103:3165–3170. doi: 10.1073/pnas.0506889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little AM, Parham P. Polymorphism and evolution of HLA class I and II genes and molecules. Rev Immunogenet. 1999;1:105–123. [PubMed] [Google Scholar]
- Lukacs MF, Harstad H, Bakke HG, Beetz-Sargent M, McKinnel L, Lubieniecki KP, et al. Comprehensive analysis of MHC class I genes from the U-, S-, and Z-lineages in Atlantic Salmon. BMC Genomics. 2010;11:154. doi: 10.1186/1471-2164-11-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May S, Zeisset I, Beebee TJC. Larval fitness and immunogenetic diversity in chytrid-infected and uninfected natterjack toad (Bufo calamita) populations. Conserv Genet. 2011;12:805–811. [Google Scholar]
- McVean G, Awadalla P, Fearnhead P. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics. 2002;160:1231–1241. doi: 10.1093/genetics/160.3.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J, Eyre-Walker A. The correlation between linkage disequilibrium and distance: implications for recombination in Hominid mitochondria. Mol Biol Evol. 2001;18:2132–2135. doi: 10.1093/oxfordjournals.molbev.a003756. [DOI] [PubMed] [Google Scholar]
- Miller KM, Withler RE. The salmonids class I MHC: limited diversity in a primitive teleost. Immunol Rev. 1998;166:279–293. doi: 10.1111/j.1600-065x.1998.tb01269.x. [DOI] [PubMed] [Google Scholar]
- Nonaka M, Namikawa C, Kato Y, Sasaki M, Salter-Cid L, Flajnik MF. Major histocompatibility complex gene mapping in the amphibian Xenopus implies a primordial organization. Proc Natl Acad Sci USA. 1997;94:5789–5791. doi: 10.1073/pnas.94.11.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta Y, Goetz W, Hossain MZ, Nonaka M, Flajnik MF. Ancestral organization of the MHC revealed in the amphibian Xenopus. J Immunol. 2006;176:3674–3685. doi: 10.4049/jimmunol.176.6.3674. [DOI] [PubMed] [Google Scholar]
- Parham P, Ohta T. Population biology of antigen presentation by MHC class I molecules. Science. 1996;272:67–74. doi: 10.1126/science.272.5258.67. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Ptacek MB, Gerhardt HC, Sage RD. Speciation by polyploidy in treefrogs: multiple origins of the tetraploid, Hyla versicolor. Evolution. 1994;48:898–908. doi: 10.1111/j.1558-5646.1994.tb01370.x. [DOI] [PubMed] [Google Scholar]
- Richmond JQ, Savage AE, Zamudio KR, Rosenblum EB. Toward immunogenetic studies of amphibian chytridiomycosis: linking innate and acquired immunity. BioScience. 2009;59:311–320. [Google Scholar]
- Roelants K, Gower DJ, Wilkinson M, Loader SP, Biju SD, Guillaume K, et al. Global patterns of diversification in the history of modern amphibians. Proc Natl Acad Sci USA. 2007;104:887–892. doi: 10.1073/pnas.0608378104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelants K, Haas A, Bossuyt F. Anuran radiations and the evolution of tadpole morphospace. Proc Natl Acad Sci USA. 2011;108:8731–8736. doi: 10.1073/pnas.1100633108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Sammut B, Du Pasquier L, Ducoroy P, Laurens V, Marcuz A, Tournefier A. Axolotl MHC architecture and polymorphism. Eur J Immunol. 1999;29:2897–2907. doi: 10.1002/(SICI)1521-4141(199909)29:09<2897::AID-IMMU2897>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Sammut B, Marcuz A, Du Pasquier L. The fate of duplicated major histocompatibility complex class Ia genes in a dodecaploid amphibian, Xenopus ruwenzoriensis. Eur J Immunol. 2002;32:1593–1604. doi: 10.1002/1521-4141(200206)32:6<1593::AID-IMMU1593>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Sato A, Klein D, Sültmann H, Figueroa F, O'hUigin C, Klein J. Class I MHC genes of cichlid fishes: identification, expression, and polymorphism. Immunogenetics. 1997;46:63–72. doi: 10.1007/s002510050243. [DOI] [PubMed] [Google Scholar]
- Savage AE, Zamudio KR. MHC genotypes associate with resistance to a frog-killing fungus. Proc Natl Acad Sci USA. 2011;108:16705–16710. doi: 10.1073/pnas.1106893108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffler K, Martin DP, Seoighe C. Robust inference of positive selection from recombining coding sequences. Bioinformatics. 2006;22:2493–2499. doi: 10.1093/bioinformatics/btl427. [DOI] [PubMed] [Google Scholar]
- Schierup MH, Hein J. Consequences of recombination on traditional phylogenetic analysis. Genetics. 2000;156:879–891. doi: 10.1093/genetics/156.2.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teacher AGF, Garner TWJ, Nichols RA. Evidence for directional selection at a novel major histocompatibility class I marker in wild common frogs (Rana temporaria) exposed to a viral pathogen (Ranavirus) PLoS ONE. 2009;4:e4616. doi: 10.1371/journal.pone.0004616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oosterhout C, Joyce DA, Cummings SM, Blais J, Barson NJ, Ramnarine IW, et al. Balancing selection, random genetic drift, and genetic variation at the major histocompatibility complex in two wild populations of guppies (Poecilia Reticulata) Evolution. 2006;60:2562–2574. [PubMed] [Google Scholar]
- Wallny H-J, Avila D, Hunt LG, Powell TJ, Riegert P, Salomonsen J, et al. Peptide motifs of the single dominantly expressed class I molecule explain the striking MHC-determined response to rous sarcoma virus in chickens. Proc Natl Acad Sci USA. 2006;103:1434–1439. doi: 10.1073/pnas.0507386103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins D, Chen Z, Hughes AL, Evans M, Tedder T, Letvin N. Evolution of the MHC class I genes of a new world primate from ancestral homologues of human non-classical genes. Nature. 1990;346:60–63. doi: 10.1038/346060a0. [DOI] [PubMed] [Google Scholar]
- Westerdahl H, Wittzell H, von Schantz T, Bensch S. MHC class I typing in a songbird with numerous loci and high polymorphism using motif-specific PCR and DGGE. Heredity. 2004;92:534–542. doi: 10.1038/sj.hdy.6800450. [DOI] [PubMed] [Google Scholar]
- Wilson DJ, McVean G. Estimating diversifying selection and functional constraint in the presence of recombination. Genetics. 2006;172:1411–1425. doi: 10.1534/genetics.105.044917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol Biol Evol. 2007;8:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zeisset I, Beebee TJC. Molecular characterization of major histocompatibility complex class II alleles in the common frog, Rana temporaria. Mol Ecol. 2009;9:738–745. doi: 10.1111/j.1755-0998.2009.02535.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.