

In this issue of PNAS, Chen et al. (1) show feasibility for producing a highly sensitive biosensor, capable of detecting nanomolar quantities of biologically interesting molecules in “real time.” If the phenomenon can be generalized to other systems, it could have significant implications for a wide variety of fields, including toxicology and medical diagnostics.
The last two decades have witnessed the emergence of conjugated polymers as an intriguing class of organic macromolecules that have the electrical and optical properties of metals and semiconductors and, in addition, have the processing advantages and mechanical properties of polymers (2). The study of conjugated (semiconducting) polymers has resulted in fundamental insights into the understanding of the chemistry and physics of this novel class of materials (3, 4), and it has stimulated the development of a number of applications (5, 6). Included among these are all-polymer integrated electronic circuits (7), photodetectors and solar cells (8), and flat-panel emissive displays fabricated from polymer light emitting diodes (9). Chen et al. (1) have now demonstrated that the luminescent properties of such polymers can be further exploited to develop a new form of biosensor.
Conjugated polymers such as poly(phenylene vinylene) (PPV) and its soluble derivatives are known to exhibit photoluminescence with high quantum efficiency (6). This luminescence can be described in terms of semiconductor band theory. On photoexcitation, an electron is excited from the highest occupied energy band (the π-band) to the lowest unoccupied energy band (the π*-band). The excited electron and the oppositely charged “hole” (the empty state in the π-band) attract one another. When the excited electron recombines with the hole, a photon is emitted (luminescence or fluorescence). The wavelength of the absorbed light is determined by the π-π* energy gap and can be manipulated by altering the molecular structure of the polymer. Conjugated polymers have been demonstrated with emission colors that span the full range of the visible spectrum (6).
By functionalizing the conjugated backbone with suitable side-chains, these macromolecules can be made soluble in common organic solvents and in water. Typical molecular weights are on the order of 106 Da, corresponding to ≈1,000 monomer repeat units per macromolecule.
The fundamental scientific discovery addressed in the paper by Chen et al. is that the luminescence of a semiconducting polymer in aqueous solution can be quenched by using extremely low concentrations of a cationic electron acceptor (1). The mechanism for the quenching is ultrafast photoinduced electron transfer (10); the excited electron transfers to a nearby acceptor within a few hundred femtoseconds (11), more than four orders of magnitude faster than the luminescence decay time. Because the electron and the hole are separated after the electron transfer step (the electron is on the molecular acceptor and the hole is left behind on the polymer chain), the luminescence is quenched. The photoinduced electron transfer step is exponentially sensitive to the distance separating the electron on the polymer chain from the acceptor. If the acceptor is removed from the vicinity of the polymer chain by ≈1 nm, the electron transfer rate will be so slow that the radiative recombination channel (luminescence) will again dominate.
The Stern-Volmer constant, Ksv, provides a quantitative measure of the luminescence quenching:
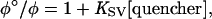 |
where φ° is the intensity of fluorescence in the absence of the quencher and φ is the intensity of fluorescence in the presence of the quencher. The equation reveals that φ°/φ increases in direct proportion to the concentration of the quenching moiety, and the constant Ksv defines the efficiency of quenching. When all other variables are held constant, the higher the Ksv, the lower the concentration of quencher required to quench the luminescence.
Chen et al. demonstrate that the luminescence of the sulfonated (anionic) and water soluble conjugated polymer, MPS-PPV (Fig. 1), is quenched by the cationic molecule methylviologen (MV2+) with a Ksv of 107 (1). By contrast, quenching of the fluorescent molecule, stilbene, (the equivalent of 1.5 repeat units of PPV) has a Ksv of 15. Moreover, the largest amplification of fluorescence quenching reported earlier in the literature corresponds to Ksv = 65 (12).
Figure 1.

Chemical structure of a single repeat unit of the luminescent polymer MPS-PPV.
The huge enhancement of fluorescence quenching observed for the MV2+/2,5-methoxy propyloxysulfonate (MPS)–PPV system arises from a combination of two effects. First, in aqueous solution, there is an equilibrium:
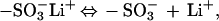 |
such that the luminescent polymer is negatively charged (anionic). As a result, the positively charged acceptor (MV2+) and the anionic polymer form a weak complex, thereby significantly enhancing the local concentration. Second, from the concentrations used in the experiments, it appears that a single MV2+ molecule can quench the fluorescence derived from an entire ≈1,000 repeat unit polymer chain. The mechanism of this remarkable extended quenching is not yet understood and will need to be more carefully addressed. Nonetheless, the combination of this effect with the enhanced local concentration resulting from the ionic charge interaction leads to the million-fold amplification factor.
Because MV2+ quenches quantitatively at extremely low concentrations, the resultant detectable fluorescence could potentially be harnessed as a biosensor; specifically, removing minute amounts of MV2+ from the polymer could theoretically increase the fluorescence in proportion to the amount of MV2+ removed. By connecting the quencher to a biologically interesting ligand through a flexible tether, Chen and colleagues were in fact able to demonstrate that such an approach is feasible (Fig. 2) (1). First, they constructed the quencher/ligand moiety by covalently linking the MV2+ to biotin, a small ligand molecule (molecular mass of ≈250 Da) that specifically binds with high affinity (binding constant, Kd ≈ 10−15) to the much larger protein, avidin (molecular mass of ≈64,000 Da). The MV2+–biotin (quencher/ligand) was able to effectively quench the MPS-PPV, albeit not as efficiently as the MV2+ alone. The investigators then showed that as little as 1 × 10−8 M avidin added to a mixture of MV2+–biotin plus MPS-PPV unquenched the fluorescence. Importantly, avidin did not have any effect on the quenched state of MPS-PPV plus MV2+ alone (the avidin effect only occurred in the presence of MV2+–biotin). Moreover, the specificity of the biotin–avidin interaction was demonstrated by showing that addition of another protein of similar size to avidin, the cholera toxin receptor, did not unquench the fluorescence.
Figure 2.

Schematic representation of a polymer biosensor. (A) Quencher-Biotin is removed by avidin and results in fluorescence as performed by Chen et al. (1). (B) Quencher linked to an antibody may be removed from the polymer by its specific antigen and result in fluorescence. (C) Quencher linked to an oligonucleotide may be removed from the polymer by a specific complementary sequence of DNA or RNA and result in fluorescence.
As noted earlier, when the acceptor is removed from the vicinity of the polymer chain by ≈1 nm, electron transfer to the acceptor will be so rare that the radiative recombination of the electron and the hole on the polymer chain (luminescence) will dominate. In the MV2+–biotin example, the quencher is presumably pulled away from the polymer chain by complexing with the high molecular weight protein, avidin. Whether this hypothesis is indeed correct remains to be formally tested.
The potential for adapting the biosensor described by Chen et al. for use with antibody:antigen pairs or DNA:DNA (or DNA:RNA) interactions is particularly interesting (Fig. 2). Presently used approaches for detection of such biologically relevant molecules include the enzyme-linked immunosorbent assay (ELISA), an antibody-based heterogeneous assay technique (13, 14). Although this now standard technology is both sensitive and specific, it requires two antibodies specific for different antigenic regions (epitopes) of the molecule to be detected and is a multistep process with several washes needed to separate bound from unbound antigen. Moreover, an ELISA requires a significant amount of time (hours to days) to complete. The PCR methodology, capable of detecting even single copies of specific RNA or DNA sequences, has further revolutionized molecular diagnostics (15). Newer modifications using a fluorescent oligonucleotide primer have even been developed such that it is now possible to quantify the results in real time (16). Nonetheless, the procedure requires multiple steps and at least several hours to obtain results.
The polymer-based biosensor proposed by Chen et al. has the potential to be a significant improvement over these standard technologies. If it were possible to link an antibody or a specific oligonucleotide to the quencher (Fig. 2), then it might be possible to rapidly and specifically detect protein, carbohydrate, nucleic acid, or other antigens under a variety of conditions. One important advantage to the approach is that the polymer-based biosensor represents a homogeneous assay. The quencher/ligand maintains the polymer in an “off” state, and detection (fluorescence) occurs only when the molecule of interest binds to and removes the quencher/ligand. The assay therefore does not require differentiating bound from unbound antigen; only the bound antigen removes the quencher from the polymer and induces a signal (fluorescence), eliminating the need for washing steps (i.e., in ELISAs). The induced fluorescent signal seems to be readily quantifiable using a standard fluorimeter (1). Finally, and perhaps most importantly, the reaction occurs essentially instantly. This feature would not only save time (and therefore money), but could potentially be extremely useful for “real time” detection of life threatening toxic substances in air or water.
Although these ideas are extremely provocative, there are many questions to be addressed regarding the practical use of such polymer-based biosensors. How does the size and charge of the ligand affect the ability of the quencher/ligand to quench polymer fluorescence? If the ligand tethered to the quencher is significantly larger than biotin (i.e., antibodies or antibody subunits have molecular weights of 26,000–150,000 Da), it might interfere with the complexing of the quencher to the polymer chain. In addition, the amino acid composition of antibodies can vary significantly, consequently affecting their overall charge. Quenching may be ineffective if the charge or size of the ligand prevents effective interactions with the polymer. Similarly, it will be important to determine the size and charge restrictions of antigens that can be detected in this manner. If the antigen to be detected is too small, it might be ineffective in pulling the quencher/ligand away from the luminescent polymer. This last point is particularly relevant to the studies by Chen et al. in that the detected unquenching of fluorescence was likely due, in part, to steric effects; the large size of the avidin may have prevented avidin-bound quencher/biotin from interacting with the polymer.
The reported studies have further demonstrated detection of only a single interaction between biotin and avidin under ideal experimental conditions (1). As noted, the avidin–biotin interaction is high affinity (binding constant, Kd, of 10−15) whereas typical antigen–antibody interactions are of much lower affinity [Kd of 10−5–10−10 (17)]. It therefore remains to be determined whether a quencher/antibody can be effectively removed from the polymer to induce a detectable signal. Similar issues are raised for the potential detection of DNA:DNA interactions by this method. Moreover, to be truly useful as a biosensor, the assay needs to be tested under less ideal conditions: i.e., detection of relevant molecules from unpurified water or air samples, or from even blood or serum. Finally, although the work of Chen et al. demonstrate that their assay can be quantitative, it remains to be determined whether it is sensitive enough to detect relevant molecules at concentrations useful for medical diagnostics or toxicology studies.
Despite these numerous unanswered questions, the fundamental discovery that polymer luminescence can be manipulated quantitatively is an important one. The investigators have opened a new direction of research that, if generalizable, has the potential for producing a wide range of biosensor applications.
Acknowledgments
We thank Neil Greenspan (Department of Pathology, Case Western Reserve University School of Medicine) for his helpful discussions and insightful comments.
Footnotes
See companion article on page 12287.
References
- 1.Chen L, McBranch D W, Wang H-L, Helgeson R, Wudl F, Whitten D G. Proc Nat Acad Sci USA. 1999;96:12287–12292. doi: 10.1073/pnas.96.22.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bredas J L, Silbey R, editors. Conjugated Polymers. Dordrecht, The Netherlands: Kluwer; 1991. [Google Scholar]
- 3.Salaneck W R, Lundström I, Rånby B, editors. Conjugated Polymers and Related Materials: The Interconnection of Chemical and Electronic Structure. Oxford: Oxford Univ. Press; 1993. [Google Scholar]
- 4.Heeger A, Kivelson S, Schrieffer J, Su W. Rev Mod Phys. 1988;60:781–850. doi: 10.1103/PhysRevLett.60.72. [DOI] [PubMed] [Google Scholar]
- 5.Heeger A. Solid State Commun. 1998;107:673–679. [Google Scholar]
- 6.Hide F, Diaz-Garcia M, Schwartz B, Heeger A. Acc Chem Res. 1997;30:430–436. [Google Scholar]
- 7.Drury C, Mutsaers C, Hart C, Matters M, de Leeus D. Appl Phys Lett. 1998;73:108–110. [Google Scholar]
- 8.Yu G, Gao J, Hummelen J, Wudl F, Heeger A. Science. 1995;270:1789–1791. [Google Scholar]
- 9.Heeger A, Diaz-Garcia M. Curr Opin Solid State Phys Mat Sci. 1998;3:16–22. [Google Scholar]
- 10.Sariciftci N, Smilowitz L, Heeger A, Wudl F. Science. 1992;258:1474–1476. doi: 10.1126/science.258.5087.1474. [DOI] [PubMed] [Google Scholar]
- 11.Kraabel B, McBranch D, Sariciftci N, Moses D, Heeger A. Phys Rev B Solid State. 1994;50:18543–18552. doi: 10.1103/physrevb.50.18543. [DOI] [PubMed] [Google Scholar]
- 12.Swager T. Acc Chem Res. 1998;31:201–207. [Google Scholar]
- 13.Margulies D. In: Current Protocols in Immunology. Coligan J, Kruisbeek A, Margulies D, Shevach E, Strober W, editors. Vol. 1. New York: Wiley; 1994. pp. 2.1.2–2.1.20. [Google Scholar]
- 14.Matesic D, Lehmann P, Heeger P. Transplantation. 1998;65:906–914. doi: 10.1097/00007890-199804150-00008. [DOI] [PubMed] [Google Scholar]
- 15.Wang A, Doyle M, Mark D. Proc Nat Acad Sci USA. 1989;86:9717–9721. doi: 10.1073/pnas.86.24.9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higuchi R, Dollinger G, Walsh P, Griffith R. Bio/Technology. 1992;10:413–417. doi: 10.1038/nbt0492-413. [DOI] [PubMed] [Google Scholar]
- 17.Karush F. In: Immunoglobulins. Litman G, Good R, editors. New York: Plenum; 1978. pp. 85–116. [Google Scholar]