

Abstract
An alternative strategy for inhibition of the cyclin dependent kinases in anti-tumor drug discovery is afforded through the substrate recruitment site on the cyclin positive regulatory subunit. Critical CDK substrates such as the Rb and E2F families must undergo cyclin groove binding before phosphorylation and hence inhibitors of this interaction also block substrate specific kinase activity. This approach offers the potential of generating highly selective and cell cycle specific CDK inhibitors and to reduce the inhibition of transcription mediated through CDK7 and 9, commonly observed with ATP competitive compounds. While highly potent peptide and small molecule inhibitors of CDK2/cyclin A, E substrate recruitment have been reported, little information has been generated on the determinants of inhibitor binding to the cyclin groove of the CDK4/cyclin D1 complex. CDK4/cyclin D is a validated anti-cancer drug target and continues to be widely pursued in the development of new therapeutics based on cell cycle blockade. We have therefore investigated the structural basis for peptide binding to its cyclin groove and have examined the features contributing to potency and selectivity of inhibitors. Peptidic inhibitors of CDK4/cyclin D of pRb phosphorylation have been synthesized, and their complexes with CDK4/cyclin D1 crystal structures have been generated. Based on available structural information, comparisons of the cyclin grooves of cyclin A2 and D1 are presented and provide insights into the determinants for peptide binding and the basis for differential binding and inhibition. In addition, a complex structure has been generated in order to model the interactions of the CDKI, p27KIP1, with cyclin D1. This information has been used shed light onto the endogenous inhibition of CDK4 and also to identify unique aspects of cyclin D1 and which can be exploited in the design of cyclin groove based CDK inhibitors. Peptidic and non-peptidic compounds have been synthesized in order to explore structure-activity relationship for binding to the cyclin D1 groove which to date has not been carried out in a systematic fashion. Collectively, the data presented provides new insights into how compounds can be developed that function as chemical biology probes to determine the cellular and anti-tumor effects of CDK inhibition. Furthermore, such compounds will serve as templates for structure-guided efforts to develop potential therapeutics based on selective inhibition of CDK4/cyclin D activity.
INTRODUCTION
CDKs, the cyclin regulatory subunits and their natural inhibitors, the CDK tumor suppressor proteins (CDKIs), are central to cell cycle regulation and their functions are commonly altered in tumor cells. Deregulation of CDK2 and CDK4 through inactivation of CDKIs such as p16INK4a, p21WAF1, p27KIP1, and p57KIP2 can override the G1 checkpoint [1, 2] and lead to transformation. CDKs interact with critical cell cycle substrates through the cyclin binding motif (CBM) which form a complex with the cyclin groove of the G1 and S phase cyclins, a surface binding site involving a protein-protein interaction. It has been shown that CDK isoform and substrate selective inhibition can be achieved through the use of peptides that block recruitment of both pRb and E2F and potently inhibit CDK2/CA kinase activity[3]. The cyclin binding motif represents a consensus of sequences found in many cell cycle and tumor suppressor proteins [3, 4]. Cyclin Groove Inhibitor (CGI) peptides in transducible form have been shown to induce cell cycle arrest and selective apoptosis in tumor cells in vitro [5–7]. These permeabilized peptides also act as anti-tumor agents as when administered directly to a SVT2 mouse tumor model, significant tumor growth inhibition was obtained through apoptosis[8].
Inhibition of CDKs though the cyclin provides an approach overcome CDK redundancy (refs), to obtain selectivity against other protein kinases and inhibit only the G1 and S phase CDKs (only these contain a functional cyclin binding groove). In particular, CDKs that regulate the RNA polymerase-II transcription cycle should be unaffected by CGI compounds. Although it has been shown that cancer cells depend on the RNAPII cycle to express anti-apoptotic genes and that inhibition of transcriptional CDKs leads to potent anti-tumor agents [9], it is at the same time likely that this will lead to effects in normal cells and may be responsible for toxicities observed with current CDK inhibitors being clinically evaluated. The ATP competitive CDK inhibitors developed to date are generally non specific against the single variants in the CDK family and in general potently inhibit the transcriptional CDKs [10]. The most selective CDK inhibitor described to date is a CDK4, 6 selective compound, PD0332991 although it has apparently not been tested against the transcriptional CDKs. Regardless, this compound is a potent antiproliferative agent against retinoblastoma (Rb)-positive tumor cells and induces a G1 arrest, with concomitant reduction of phospho-Ser780/Ser795 on pRb [11]. Oral administration to mice bearing the Colo-205 human colon carcinoma xenografts resulted in marked tumor regression suggesting that it has significant therapeutic potential and that targeting CDK4/cyclin D may be a viable strategy[12]. In addition to cyclins A and E, the D-type cyclins also contain a functional cyclin groove and CDK4/cyclin D dependent kinase activities can therefore be blocked by cyclin groove inhibitors.
CDK4/cyclin D1 is an extensively researched cancer target and numerous recent studies warrant continued investigation. Mice lacking Cyclin D are highly resistant to mammary carcinomas induced by erbB-2 oncogene[13]. Further research into the role of Cyclin D in tumor formation made use of a mutant form which binds to CDK4/6 but cannot promote catalytic activity[14]. This kinase-defective Cyclin D/CDK complex results in more evidence of resistance to erbB-2 induced tumorigenesis in mice. Combination of these two studies strongly indicates that Cyclin D1/CDK4 kinase activity is required for erbB-2-driven tumorigenesis and therefore confirms that Cyclin D1/CDK4 is a promising oncology target. While there are several reports of potent and selective inhibitors of the CDK2/cyclin A, E substrate recruitment, with both peptidic and peptidomimetic compounds being identified[15–17], very little has been reported with respect to either inhibitors or on the requirements for binding to the cyclin groove of CDK4,6/cyclin D1[4]. Here, structural and functional analysis of peptide inhibitor and p27 binding to the cyclin D1 groove has been investigated and used to design peptides that provide the basis for structure-activity relationships and have improved binding. Due to the critical roles of cyclin groove dependent CDK substrates in cell cycle regulation, such compounds will have considerable potential as chemical biology probes and in anti-tumor drug development.
RESULTS AND DISCUSSION
Structural comparison of cyclin A2 and D1 binding grooves
While numerous experimental structures exist for CDK2/cyclin A2 and other cyclin structures have been solved[15–18], for many years CDK4 in complex with the D type cyclins proved refractory to crystallization. The structures for CDK4 in complex with cyclin D1 were recently solved in complex with ligands binding to the ATP cleft[19, 20]. These data provided the opportunity to gain new insights into the cyclin groove of the D cyclins and also to determine the basis for their interactions with cyclin groove inhibitory (CGI) peptides. A limited body of data has been generated for CDK4 inhibition[4] where a series of peptides explored biologically as CDK2/cyclin A, E inhibitors were also characterized in terms of their inhibition of cyclin D1 mediated substrate recruitment. These results determined that highly potent peptidic CDK2 inhibitors were in general, significantly less potent against CDK4[4]. In order to determine the structural and functional differences of these compounds, their interactions with the cyclin D1 recruitment site were modeled and compared with known cyclin A complex structures. In terms of cyclin A binding, optimized peptides (i.e. the octomer, HAKRRLIF, p21 sequence) contain three major determinants which are required for high affinity binding. These include a primary hydrophobic pocket which interacts predominantly with leucine and phenylalanine residues of the peptide (Figure 1 left), an acidic region which forms ionic contacts with basic peptide residues and a secondary hydrophobic pocket occupied by either alanine or valine of the cyclin binding motif (CBM).While the majority of CGI peptide contacting residues are identical or semi-conserved in both cyclin isotypes, two notable exceptions were observed. In cyclin D1, Val60 (interacts with Phe8) and Thr62 (close to Arg4) are substituted for Leu214 and Asp216 in cyclin A2 respectively. As these residues in the cyclin A context, make contacts with major determinants of cyclin A binding, it is expected that even semi-conservative replacements would lead to significant effects on cyclin groove inhibition.
Figure 1.

Left Panel. Overlay of crystal structures of cyclin D1 (orange carbon atoms; 2W96) and cyclin A2 (1OKV) illustrating similarities and differences of CBM contacting residues. The Leu and Phe residues of the CBM interacting with the primary hydrophobic pocket are shown. E220 and D216 comprise the acidic region and the secondary hydrophobic pocket is to the left of W217. Right Panel. Ribbon representation of the overlay highlighting the differences in the cyclin box helices. Cyclin D1 is shown in yellow and the CGI peptide in blue. . The region displaying the largest structural differences after superimposing the backbone atoms is labeled (residues 116–136 of cyclin D1).
Upon overlay of the corresponding alpha carbons of the two cyclins, other semi-conserved and non-conservative differences were observed in the structural comparison. These residues are not as significant for binding of the octapeptide however their proximity to the cyclin binding groove suggests that they have potential for exploiting in the design of selective CDK inhibitors targeting cyclin D1. Of the non peptide contacting residues, the largest structural variation is in the exchange of Y286 of cyclin A for I132 of cyclin D1. Overlay and comparison of the Calpha trace of the two structures (Figure 1 right panel) indicates that this variation, coupled with the relative movement of a helix-loop segment (residues 119–136 of 2W96) leads to a significant conformation variation proximal to the cyclin groove. This region as a consequence is considerably more open in cyclin D1 and provides an extension to the primary hydrophobic pocket (Figure 2 bottom panel; Figure 3). This additional pocket could therefore accommodate larger ligand groups than would be feasible for cyclin A inhibitors.
Figure 2.

Comparison of the solvent accessible surface of the cyclin grooves of A2 (top panel) and D1 (bottom). The following regions displaying structural differences in cyclin binding determinants are labeled: 1) the top panel illustrates the more acidic region of cyclin A compared to cyclin D1 which results in differential interactions with Arg4. 2) the primary hydrophobic extension of cyclin D1 (top right), 3) the shallower cyclin D1 primary hydrophobic pocket (the Leu214 – Val60 interchange; bottom right) and 4) the narrower cyclin D1 secondary hydrophobic pocket (left). Each of these structural features explains the differential requirements for cyclin groove inhibitor binding.
Figure 3.

Modeled complex of p27 residues 25–49 (green carbons) with Cyclin D1 (2W96) overlayed with SAKRNLFGM. The P35 and V36 interacting site on cyclin D1 is the region shown to provide a more extensive hydrophobic pocket than in the cyclin A2 context and which was exploited by methionine substitution (Met is the C-terminal residue in the p27 peptide colored with orange carbon atoms).
Another consequence of the differing conformation and composition of the 116–136 region affects the secondary hydrophobic pocket with which the CGI peptide Ala2 interacts. I281 of cyclin A2 is a Tyrosine residue (Y127) in D1. The kinked helix containing this residue is shifted towards the groove, bringing this residue closer to the peptide and decreasing the volume of the lipophilic pocket on the peptide N-terminal side of the W63 (Figure 2, bottom panel).
Structural basis for cyclin D1 inhibition
Prior to detailed analysis of modeled peptide-cyclin D1 complexes, the structural and energetic basis for potencies of cyclin A inhibitors was examined. With regards to generating models of peptide binding to the cyclin groove, two factors were taken into consideration. Firstly, it is known that very little rearrangement of the groove occurs upon substrate and inhibitor binding. Comparison of the numerous cyclin A complexes available with the non-liganded cyclin A reveals that only minor backbone and side chain movements are observed and generally only involve exposed hydrophilic residues. This removes a significant part of the complexity of modeling peptide-cyclin complexes and allowed the use of relatively simple minimization protocols allowing for movement of ligand and receptor side chains where necessary. Secondly, it is likely that pre-organization of the peptide conformation in solution contributes to the thermodynamics of binding. Inter-residue interactions between Leu6 and Phe7(8) are observed in the bound conformation and therefore would contribute significantly to binding if occurring in the free peptide. Cyclic peptides previously described, and which display less than expected improvements in affinity, suggest that this is the case [16].
Since a complete set of cyclin A crystal structures for peptides with cyclin D1 affinity is not available, a cyclin A complex for HAKRRLIF was first constructed. This was completed by building on existing pentapeptide (1OKV) and octapeptide structures to supplement those available for PVKRRLDL (E2F) and SAKRRLFG (p107) CBM sequences. . This and subsequent modeled complexes were generated using energy minimization protocols and validated by running molecular dynamics simulations. The non-bonded interactions of these modeled and crystallographic complexes were estimated by calculation of per residue and total interaction energy values (DS 2.5, Accelrys) to determine individual contributions and to establish if these were reflective of the observed affinities (approximated by inhibition constants). These values (Table 1, supplementary material) delineated a relationship in terms of both previous SAR of individual residues and CGI potency. As determined through sensitivity to major potency loss by alanine substitution and other residue replacement, the energetic analysis shows the critical Arg4 of the octapeptide makes an extensive contribution to binding, whereas that of the less sensitive Arg5 is lower [3, 4]. Truncation of the His-Ala-Lys N-terminal sequence has been shown to result in a decreased affinity for cyclin A with the potency decreasing approximately 100 fold [3, 4, 15, 21]. The contribution of these three residues to binding is confirmed through the energetic analysis where His1 and Lys3 especially provide favorable interactions with the acidic region of the binding pocket. The total binding energies of both HAKRRLIF and RRLIF calculated (−298 vs −191) correlate well with the inhibition constants of these two compounds. Further analysis of the cyclin residue energetics determined that acidic residues, including Asp216, Glu220, Glu224 and Asp283 allow favorable electrostatic contacts with the basic peptide N-terminal sequence. In addition, the energetics of the contribution of Ala2 for binding (to the secondary hydrophobic pocket) correlates well with observed potency increase of the Ser-Ala mutation in the p21 C-terminal context [4].
Table 1.
Structure-activity relationship for C-terminal amino acid subsitution in the cyclin binding motif of p21 and p107 derived peptides
 | ||||||
|---|---|---|---|---|---|---|
| SEQUENCE | IC50 CDK2/A2(µM) |
Potency ratio |
IC50 CDK4/D1(µM) |
Potency ratio |
IC50 CDK2/E(µM) |
|
| SAKRRLFG | 3.3 | 2.9 | ||||
| SAKRRLX1G | 9.1 | 2.8 | 7.5 | 2.6 | 4 | |
| SAKRRLX2G | 27 | 8.2 | 11.4 | 3.9 | ||
| SAKRRLX3G | 1 | 0.3 | 6 | 2.1 | ||
| p107 | SAKRRLX4G | 100 | 30.3 | 74 | 25.5 | |
| SAKRRLX5G | 18 | 5.5 | 28 | 9.7 | ||
| SAKRRLX6G | 83 | 25.2 | 36 | 12.4 | ||
| SAKRRLX7G | 80 | 24.2 | 51 | 17.6 | ||
| SAKRRLX8G | 750 | 227.3 | 143 | 49.3 | ||
| HAKRRLIF | 1.3 | 1.5 | 0.3 | |||
| HAKRRLIX1 | 6.1 | 4.7 | 11.4 | 7.6 | 1.3 | |
| HAKRRLIX2 | 3.6 | 2.8 | 6.5 | 4.3 | ||
| HAKRRLIX3 | 25 | 19.2 | 100 | 66.7 | ||
| p21 | HAKRRLIX4 | 25 | 19.2 | 100 | ||
| HAKRRLIX5 | 20 | 15.4 | 90 | 60.0 | ||
| HAKRRLIX6 | 58 | 44.6 | 6.3 | 4.2 | ||
| HAKRRLIX7 | 29 | 22.3 | 28 | 18.7 | ||
Further correlation of the interactions and contributions of the C-terminal sequence of the CBM interacting with the primary hydrophobic pocket (Figure 1, left) corroborates the structural and energetic differences. In varying peptide sequence contexts, the p21 Leu-Ile-Phe (LIF ‘motifette’) sequence has been demonstrated to be more potent than the p107 (and p27) LFG and E2F, LDL motifettes [3, 4, 15, 21]. While the Leucine contributions in each context are similar, the Phe side chain provides increased complementarity in the p21 sequence (−23.5 kcal/mol vs. −12.2) resulting in its 2–3 fold greater affinity compared to the LFG sequence. More favorable contacts are observed due to the geometrical arrangement of the aromatic side chain allowed by the spacer residue between the Leu and Phe in the p21 context[21]. Overall, the energetic analysis of peptide binding to cyclin A confirms that a relationship exists between calculated binding enthalpy and experimental affinity and additionally that individual residue energetics closely correlate with the SAR and contribution of CBM determinants. This relationship provides the basis to perform an analysis of peptide binding to cyclin D1 and to determine the structural basis for decreased affinity of cyclin D1 inhibitors and therefore to facilitate the design of more potent compounds.
The intermolecular complexes of cyclin D1 with the above peptides were formed by superposition of the apo-cyclin D1 structure (2W96) with the crystallographically derived cyclin A bound structure of the CBM containing peptides and followed by deletion of cyclin A. The energy-minimized structure was then calculated using the CHARMm molecular forcefield, and the similarities and differences of cyclin binding motif – cyclin interactions were examined. In order to further probe the molecular consequences of variations in binding residues, the intermolecular energies were calculated for the interactions of each of the peptides with cyclins A and D1. In line with the observed potencies of each compound and selectivity for cyclin A, a correlation was determined between affinity (kinase inhibition) and total calculated interaction energy (CIE) for 4 peptides ranging in IC50 from 0.021 to 99 µM (Supplementary Figure 1). For this relationship, an R2 of 0.91 indicated that the both the crystal and modeled structural complexes are accurate and that the established correlation is useful as a predictive tool for design and synthesis of more potent and selective compounds. Comparison of the predicted affinities of each peptide also demonstrates that the CIE correlates well with the selectivity of the compound for cyclin A (Table 1, supplementary material). This was additionally confirmed by a second method for estimation of binding affinity. Calculation of Ludi Scores provided results directly in line with the relative potencies on cyclin A vs. D1. As a further validation of the protocol used, the RMSD values of the minimized RRLIF complex were compared with the crystal structure coordinates. The structures were found to be highly similar with an RMSD value of 0.21 A). Further analysis of the individual energetic contributions of residues of both the peptide and cyclin in each context revealed further evidence for the structural basis of CGI selectivity. Not surprisingly, it was observed that the cyclin D binding site variations described above contributed extensively to the selectivity of each peptide for cyclin A2.
The optimized p21 derived peptide, HAKRRLIF is highly selective for A (0.021 µM) vs. D1 (6 µM). In addition to the total interaction energy describing the non-bonded interactions of the peptide – cyclin interaction, the individual contributions of residues from both molecules was determined. These results indicate that the highly basic N-terminal residues interact much more favorably with the cyclin A groove. As no crystal structure is available for this peptide, a cyclin A complex was modeled on the basis of the residue contacts of RRLIF (1OKV) and SAKRRLFG (1H28). Analysis of protein-peptide contacts and interaction energies reveals that a greater concentration of acidic residues in A2 compared to D1 contributes extensively to this selectivity. In particular Asp216 of cyclin A2 (which is aligned with Thr62 of cyclin D1) provides a favorable addition of 17 kcal/mol to the binding energy in its interactions with Arg4. This contribution is largely absent in the cyclin D1 complexes modeled where the hydroxyl group of Thr62 weakly interacts with Arg4. When the interaction of both Arg4 and Arg5 are considered, the calculated binding energy of these two residues for cyclin A is more than twice that observed for cyclin D1. Glu220 in Cyclin A2 interacts with Arg4 similarly to the corresponding residue (Glu66) in Cyclin D suggesting that the energetic differences are mainly due to the absence of the second acidic residue in D. Desolvation penalties for peptide cyclin complexes were examined in order to determine how the protein environment around the Arg4 binding site affects peptide interactions in each context (see supplementary table 2). Direct comparisons for each peptide indicate that the penalty is similar (generally slightly higher with cyclin D1) and therefore does not affect the above conclusions regarding side chain contributions obtained through analysis of interaction energies
As mentioned above, comparison of the CGI peptide binding residues in cyclin D1 revealed that a valine residue (Val60) occupied the position observed as a leucine in A2 (Leu214Val). As this residue is located in the lipophilic pocket interacting with the LIF motif of p21, the immediate conclusion is that this contributes significantly to peptide selectivity for A vs D. Initially, this appears to be counterintuitive since valine is a smaller residue and might be expected to provide a larger binding pocket. Close examination of Val60 indicates that the shorter and less flexible sidechain brings the interacting methyl groups closer to the phenylalanine of the peptide and therefore decreases the volume of the hydrophobic pocket (Figure 1 left, 2 bottom panel). This was confirmed upon overlay of cyclin A2 bound to HAKRRLIF with the cyclin D1 modeled complex, where a significant steric clash with the Phe8 side chain was observed (Figure 1). This suggests that the binding mode of Phe8 with cyclin A2 is not compatible for interaction with cyclin D. In order to determine the consequences of the overlap, the complex formed between cyclin D1 and HAKRRLIF was subjected to energy minimization to relieve this overlap. A significant displacement of the phenylalanine was observed and which did not come at the expense of Leu6, whose position was not affected. Further analysis of the interaction energy and comparison with the values calculated for octapeptide inhibition of both cyclins, indicated a reasonable correlation between predicted and calculated per-residue affinity of the C-terminal motifette. These data suggest that displacement of the aromatic side chain comes at the expense of its complementarity with the primary hydrophobic pocket and that the valine substitution is responsible for the significant decrease in affinity for cyclin D1.
Examination of the intermolecular contacts and interaction energies for SAKRRLFG (p107 cyclin binding motif) with cyclin D1 reveals a similar pattern of residue energetics for the basic region of the peptide as in the HAKRRLIF context. SAKRRLFG has a lower affinity for cyclin A, with the less optimal geometry of the LFG motifette resulting in a reduced contact surface area of the phenyl ring with the pocket. Calculation of the individual residue interaction energies suggests that the presence of Val60 has a markedly smaller impact on affinity of the p107 peptide for cyclin D1 than in the p21 context due to the different approach angle of the interacting side chain.
Comparison of the E2F CBM, PVKRRLDL reveals further insights into the structural basis for CGI selectivity for cyclin A and after comparison of the binding energetics again indicates less favorable contacts with the peptide in the cyclin D1 context. As has been previously described, the LDL containing inhibitors generally have a decreased binding relative to the LIF compounds and in this case is reflected in the 50 fold increased IC50 value. In contrast to the LFG sequence, the LDL sequence has a substantially lower predicted affinity for hydrophobic pocket of cyclin D1, consistent with the observed inhibition constants.
Further analysis of peptide SAR and Insights into the design of selective cyclin D1-CDK4 inhibitors
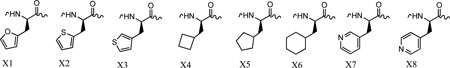
The insights into the structural basis for peptide recognition for cyclin A and for the decreased potency against cyclin D1, provided further opportunity to expand inhibitor structure activity relationships. As suggested from the above structural analysis, differences in the primary hydrophobic pocket were the major determinants in cyclin A selectivity of the studied peptides. These observations predicted that analogs with variant C-terminal groups may interact with the cyclin D pocket with differing affinity than to the cyclin A groove. Based on this observation, peptides were designed to exploit these structural differences and generate compounds with increased affinity for cyclin D1. Due the decreased volume of the primary pocket in cyclin D1, a series of non-proteinogenic cyclic replacements for Phe7 (p107) and Phe8 (p21) cyclin binding motif containing octapeptides were designed. A series of 5 and 6 membered ring systems were incorporated into the p21 (HAKRRLIX) and p107 (SAKRRLXG) contexts (Table 1). These included 2-furylalanine (X1), 2-thienyl alanine (X2), 3-thienylalanine (X3), cyclobutylalanine (X4), cyclopentylalanine (X5), cyclohexylalanine (X6) and 3 and 4 pyridyl alanine residues (X7 and X8) providing for the most part isosteric functionalities mimicking the interactions of the phenylalanine. The inhibition of CDK activity was determined through a standard filter capture assay involving a GST-labeled Rb protein and quantification of the incorporation of 32P into the substrate. Activities of peptides previously tested against CDK2A and CDK4D were determined using this assay format. Although similar constructs and substrate was used, significant differences in potency were observed. In particular the IC50 for HAKRRLIF was approximately 10 fold higher than previously determined[4, 15, 21] (1.3 vs. 0.14 µM) and the inhibition of CDK4/D1 was more pronounced than before (1.6 vs. 6 µM). These differences can be accounted for in slight differences in amount of cyclin in the protein prep and excess cyclin or CDK would result in data variation. As a consequence, it was decided that structure-activity relationships determined using the kinase assay were best interpreted by functional comparisons calculated relative to the native p21 or p107 sequence in each assay. Data is therefore presented as a ratio of each C-terminal and other analogs activity in addition to the IC50s presented for each compound (Table 1).
For the 2-furylalanine replacement (X1) in the p107 context, it was found that kinase activity induced by this compound decreased a similar amount in both CDK2A (2.8 fold) and CDK4D (2.6 fold) although slightly less so for the latter. In the p21 context more of a differential was observed (4.7 and 7.6 fold decrease respectively). The p107 X2 derivative (2-thienylalanine) data indicates that the potency decrease against cyclin D was considerably reduced (3.9 fold) relative to cyclin A (8.2 fold decrease). A similar differential was observed for the p21 X2 derivative (2.8 vs. 4.3 fold respectively). The X3 amino acid, 3-thienylalanine was found to be considerably more potent than X2 in the p107 peptide but lower potency in the p21 context.
Examination of the results for aliphatic cyclic amino acid replacements, including cyclobutyl (X4), cyclopentyl (X5) and cyclohexylalanine (X6), indicated that depending on the CBM context, different selectivity profiles were observed. X4 resulted in dramatic potency decreases in both contexts however significantly more so with cyclin A. Both the p21 and p107 versions incorporating X5, indicate that it is tolerated to a larger degree in binding to cyclin A. Conversely, the p21 derivative of X6 is tolerated to a significantly larger degree in binding to cyclin D1 with only a 4 fold drop-off observed compared to 45 fold with CDK2/cyclin A. If the IC50s of this compound are considered, it is significantly more potent towards CDK4/cyclin D1 than against CDK2/cyclin A (6.3 vs. 58 µM). A similar trend was shown for the p107 X6 sequence although was not as dramatic. An interesting set of results was obtained for the pyridylalanine derivatives where one carbon of the native Phe residue is replaced with nitrogen. A large decrease in activity was observed for these compounds in both p21 and p107 variants. Binding to cyclin D1 for these analogs was again tolerated to a larger degree, especially with the 3-pyridylalanine derivative (X7) in the p107 context. Unexpectedly, the activity of 4-pyridylalanine (X8) incorporated in SAKRRLXG decreases 200 fold relative to the native sequence in terms of cyclin. Further analysis of the p21 analog binding to cyclin D1 indicates that the X8 containing peptide loses all activity towards CDK4/cyclin D1. The binding of X7 to cyclin D1 decreases 17.6 fold relative to the phenylalanine in the LXG motif and 18.7 fold in the LIX context.
Structure-activity relationship for peptide binding to cyclin D1
For the CDK4/cyclin D1/pRb SAR of the Phe replacements in the SAKRRLXG context, the most potent analog is the 3-thienylalanine, X3 derivative with an IC50 of 6 µM whereas X2, the 2- thiophene containing peptide is slightly less potent (11.4 µM). The order of potency is reversed in the p21 CBM since HAKRRLIX2 peptide has approximately 2 fold greater inhibition than the furylalanine containing peptide (6.5 and 11.4 µM respectively). The 3-thienyl analog X3 undergoes a significant potency increase relative to X2 in the p107 context. Cyclobutylalanine incorporation into the p107 peptide retained a level of binding, as do HAKRRLIX5 and SAKRRLX5G although this is weak relative to the native sequences. The cyclohexylalanine replacement, X6 was of equivalent potency to the 2-thiophene containing peptide in the HAKRRLIX context, however of notably higher inhibition than the p107 derivative (6.3 µM vs 36 µM). The 3-pyridylalanine peptides (X7) were considerably more significant inhibitors when incorporated C-terminal to the Ile containing spacer residue and which has previously been shown to allow more favorable geometry for binding. The 4-substituted derivative (X8) is a weak binder with an IC50 of 143µM observed in the CDK4/cyclin D1 kinase assay for SAKRRLX8G. For the most part, the p21 sequences follow the previously observed trend as being more potent than the p27[4] and p107 peptides. Two C-terminal analogs however have higher affinity when incorporated with the p107 residues, these being the furylalanine (X1) and 4-pyridylalanine (X8) containing peptides.
Additional insights into cyclin groove interactions in cyclin D1 are provided by C-terminal and other derivatives incorporated into HAKRRLIF. The p-fluorophenylalanine (4FPhe) derivative has been previously shown to significantly increase the inhibitory potential of peptide cyclin A inhibitors with respect to the native residue[15, 22]. In contrast to these results, synthesis and testing of RRLI(4FPhe) resulted in decreased inhibition of CDK4/cyclin D1 kinase activity (compared to HAKRRLIF, a 160 fold decrease) vs. only a 20 fold decease in CDK2/cylin A activity). Incorporation of 3-ClPhe into the E2F sequence, PVKRRLFG was beneficial for CDK4/cyclin D1 inhibition giving greater than a 6 fold increase in potency relative to the native Phe residue.
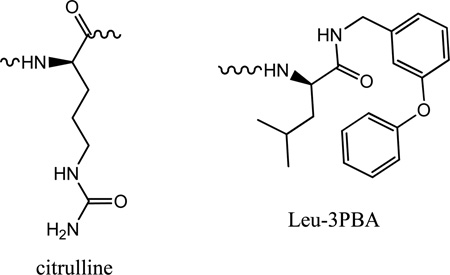
As discussed in above sections, there are differences in the Arg4 interacting residues in cyclin D1 vs. cyclin A2 and that these variations contribute to decreased binding of peptides to cyclin D1. Specifically, cyclin A has two acidic residues that interact with the positively charged side chain compared to only one in cyclin D1. This residue has previously been shown to be critical for cyclin A binding activity. It would therefore be predicted that replacement of the Arg with an isosteric residue would have less of an impact on cyclin D binding. Incorporation of citrullene (Table 2.) into p21 (HAKCitRLIF) confirmed that Arg4 is significant for interaction with cyclin D1. The ratio for the activities of the Cit and Arg containing peptides in both contexts revealed that its effect on cyclin D1 activity (14 fold potency decrease) was similar to that observed in cyclin A. This result was corroborated by comparison of the activities of citrullene incorporated into pentapeptide, RCitLIF. Compared to the octapeptide sequence, the 5mer potency decreased roughly 120 fold for cyclin A (1.3 vs. 164µM) and cyclin D1 (1.5 vs. 179 µM).
Table 2.
Structure-activity relationship for various cyclin D1 binding peptides
 | ||||
|---|---|---|---|---|
| SEQUENCE | IC50 CDK2/A2(µM) |
Potency ratio |
IC50 CDK4/D1(µM) |
Potency ratio |
| SAKRRLFG | 3.3 | 2.9 | ||
| HAKRRLIF | 1.3 | 1.5 | ||
| RRLIpfF | 26 | 20.0 | 250 | 166.7 |
| HAKCitRLIF | 18 | 13.8 | 21 | 14.0 |
| HAKTRLIF | 50 | 38.5 | 25 | 16.7 |
| CitRLIF | 164 | 126.2 | 179 | 119.3 |
| SCCP10 | 25 | 19.2 | 8 | 5.3 |
| SCCP 5624 | >100 | 60 | 20.7 | |
| SAKRNLFGM | 146 | |||
| SAKRNLFG | 75 | |||
| SAKRALFGM | 68 | |||
| PAKRRLFG | 8 | 6.7 | ||
| PVKRRLFG | 3 | 28 | ||
| PVKRRL3CFG | 1 | 3.2 | ||
Insights into SAR for interaction of cyclin D1 inhibitors of the secondary hydrophobic was revealed through synthesis of peptides containing the E2F and p107 CBMs. A preference for smaller side chains was indicated by the increased inhibition of PAKRRLFG compared to that of PVKRRLFG (3 vs. 20 µM). This result is in agreement with the structural analysis which shows a decreased volume of this subsite in cyclin D1 compared to A. (Figure 2 bottom panel; Figure 3)
From previous studies into the replacement of peptide determinants with fragment alternatives [17], compounds were identified where the p21 LIF motif was replaced with a Leu-bis-aryl ether system (Table 2), while maintaining a similar potency level for cyclin A2 inhibition. A compound was synthesized incorporating 3-phenoxybenzylamide replacing the Phe and also N-terminally capped with 1-(3,5-dichlorophenyl)-5-methyl-1H-1,2,4-triazole-3-carboxylic acid and subsequently tested for inhibition of CDK4/D1 (Table 2). SCCP10 was found to have a respectable inhibition of cyclin D1 and in addition, its relative potency compared to the p21 octapeptide was enhanced compared to cyclin A inhibition. SCCP10 is 5 fold less potent than HAKRRLIF towards cyclin D1, however undergoes a 20 fold drop off when cyclin A2 activity is considered. In order to further examine the SAR of the 3-phenoxybenzylamine (3-PBA) system, the peptide – small molecule hybrid, Arg-Arg-β-homoleucyl-3-phenoxybenzylamide (SCCP 5624) was synthesized. This compound adds a methylene group between the α-carbon and α-carboxyl of the leucine residue and probes the spacing requirement of the 3-PBA system. The lower potency of SCCP 5624 (although with greater affinity for cyclin D1) suggests that the native leucine residue is optimal in this context. The results obtained for the two 3-PBA containing peptides suggest that the smaller hydrophobic pocket of cyclin D1 can provide more optimal interactions in the right context.
Modeling the interactions of p27 with cyclin D1 structures
CDK4/cyclin D1 have been shown to associate with p27 and that this interaction promotes the formation of the complex[23]. It is also known that different states of the ternary complex exist, where p27 can bind to generate inhibited and non-inhibited CDK4 species[24, 25]. A critical aspect of this process is the phosphorylation of p27 on Y88, sited on the 310 helix which inserts into the ATP binding site of CDK4 in the inhibited complex. Phosphorylation presumably leads to dissociation of the helix from the ATP cleft through disruption of hinge H-bonding interactions and through repulsion of the phosphate with nearby acidic residues. In this non-inhibited form, p27 however, must still maintain affinity for the complex in order to sequester the inhibitor from CDK2/cyclin E complexes and allow cell cycle progression [23]. A major contribution to this binding is through cyclin D1/p27 interactions and assisted by the CBM and other residues. So as to construct a model structure of p27/cyclin D1 interactions, cyclin D1 isolated from the 2W96 crystal structure was overlayed with the CDK2/cyclin A/p27 ternary complex (1JSU)[26]. After deletion of the CDK2, cyclin A and non cyclin D1 interacting p27 residues, the newly formed complex was subjected to energy minimization. After convergence of the structure to a suitable minimum, and examination of the resulting interactions, a plausible structural basis for the interactions of p27 with cyclin D1 interactions was described. Subsequent to generation of this structure, the interaction energies of individual p27 residues with cyclin D1 were generated and compared with those for cyclin A (Table 2, supplementary info). Significant differences in the intermolecular interactions are apparent for several residues, several of which are noted in the octapeptide complexes described in the above sections. These include, Ala28, Asn31, Phe33, Val36, Leu41 and Leu45. Comparison of the molecular surface for the p27 interacting residues of cyclin A vs. those of cyclin D1 indicated that profound differences exist specifically in the region where the C-terminus of the inhibitory protein exits from the primary hydrophobic pocket. As can be seen (Figure 3), the Pro35 and Val36 contacting site of cyclin D1 has a larger accessible volume and therefore has suboptimal interactions with p27. This was confirmed in the per residue interaction energy calculation which yielded values of −1.9 and −3.6 kcal/mol for D1 and A respectively. The more extensive cleft of cyclin D1, led to the hypothesis that incorporation of a suitable residue C-terminal to the glycine would lead to increased binding.. Computational design of a number of different residues suggested that methionine would be a good candidate for more optimal interactions and therefore synthesis and testing of the p27 sequences (Table 2), was completed and confirmed this conclusion. The lack of increase of the Asparagine containing sequence can be explained by the formation of an intramolecular H-bond observed in the crystal structure and which precludes optimal interactions of the methionine. Substitution of this residue with an alanine resulted in a 2 fold potency enhancement as predicted. As illustrated (Figure 3), the linear side chain of the P35M analog extends with a high degree of complementarity into the extension of the primary hydrophobic pocket. These results suggest that this extended binding site in cyclin D1 could be exploited in the design of small molecule cyclin groove inhibitors.
Conclusions from structural analysis of CGI amino acid substitutions
In this study, comparison of the cyclin binding grooves of cyclin D1 structures obtained recently through crystallographic studies provides considerable insight into the structural requirements for cyclin A2 vs. D1 selectivity and for differential binding of CGI peptide analogues. While the binding of peptide inhibitors of cyclin A and E substrate recruitment has been extensively characterized, little information has been made available describing the determinants of cyclin D inhibition. Structural analysis revealed that two key amino acid substitutions in the cyclin D1 groove have a major impact on peptide inhibitor binding. Exchange of one of the two acidic residues interacting with Arg4 (Asp216 and Glu220), with Thr62 significantly decreases the calculated enthalpic contribution to binding and is suggestive of a large decrease in affinity. In order to determine if the predicted decrease in the electrostatic interaction energy is significant in contributing to cyclin A selectivity, the arginine isostere citrullene was incorporated into the p21 8mer, HAKCitRLIF (Table 2),. It was predicted that due to the less acidic environment of the Arg contacting residues in cyclin D, that the potency decrease would be less marked in this context. In reality however, a similar drop off was demonstrated in both scenarios and thus indicating otherwise. Closer examination of the peptide-cyclin D1 structure suggests that the urea carbonyl of citrullene is within H-bonding distance of the OH group of Thr62. This interaction would therefore compensate for the decreased capacity to ion pair and result in a similar potency decrease. Replacement of Arg4 with threonine was beneficial with respect to cyclin D1 binding and therefore was in line with the above result.
The second major difference between the two cyclins is in the exchange of Leu214 in cyclin A for Val60 in cyclin D. The smaller valine sidechain projects down toward the base of this hydrophobic pocket with the net effect that the γ methyls are brought into closer proximity to the peptide inhibitor side chains which insert into this pocket. This substitution therefore decreases the volume of the primary hydrophobic pocket in the latter and thereby results in lower affinity of CGI peptides containing phenylalanine. Cyclin bound complexes were generated for a series of peptides previously determined to have varying affinities for cyclin A and cyclin D1 and possessing different C-terminal sequences. The calculated binding energies for these complexes correlated well not only for IC50s determined for cyclin A and D1 individually but also for the selectivity of the peptides observed (data not shown). These results therefore determined that in addition to the X-ray structures used, the model structures for the peptide-cyclin complexes gave valid results and that this information is useful in the potential design and optimization of improved cyclin D1 inhibitors. From these observations, the hypothesis was proposed that due to the decreased volume of the primary hydrophobic pocket relative to cyclin A, that the incorporation of non-natural amino acids with differing cyclic sidechains (relative to phenylalanine) might be tolerated to a greater degree. To this end, the results presented (Table 1) confirm that this is indeed the case however these are dependent on the ring size and peptide context. As has been previously structurally characterized, the presence of a spacer residue between the critical Leu and Phe functions to allow a geometrical arrangement of the two side chains that interacts with a greater degree of complementarity and therefore increases binding affinity relative to peptides with no spacer[21]. The results suggest that the non-spacer containing peptide, SAKRRLXG, has a binding mode which is more conducive and tolerant of smaller cyclic sidechains. In order to probe this further, a 3D structure for each of the synthesized analogs in complex with both cyclins was generated and further to this, their non-bonded interaction energy calculated. The structural basis for the greater affinity of the furylalanine (X1) vs. the 2-thienylalanine (X2) in the p107 context is apparent from the modeled structure. The closer proximity of the heteroatom to Val60 in the peptide without the spacer residue results in displacement of the larger sulfur containing Phe replacement (2-thiophene ring) and lower relative affinity. In the p21 peptide, the conformational preference allowed by the spacer residue, results in the heteroatom pointing to the back wall of the primary hydrophobic pocket, away from Val60. As the heteroatom projects into more expansive region, the larger sulfur atom provides greater complementarity with K96 and Q100 resulting in increased affinity in the thienylalanine derivative. Changing the context of the heterocyclic sulfur atom as in X3, resulted in potency increase of SAKRRLX3G for cyclin A but an small decrease in cyclin D1 affinity. The larger hydrophobic pocket in cyclin A can accommodate the bulky sulfur atom more readily than can the cyclin D1 site decreased in volume by Val60. Examination of the intermolecular contacts for the cyclohexylalanine derivative X6, a bulkier Phe replacement as a result of the unsaturated ring, again provides insight into the differing potencies for peptides containing this residue with cyclin D1. Modeling of the complex of SAKRRLX6G with cyclin D1 (12 fold decrease in IC50), suggests that in order to maintain productive binding, the CHA sidechain is brought in close proximity to Val60 resulting in unfavorable contacts. For the HAKRRLIX6 inhibitor (4 fold loss in potency), the sidechain can adopt a more favorable position, contacting several residues of the primary binding site in line with its higher relative potency. The dramatic decreases in inhibition of the pyridylalanine derivatives X7 and X8 relative to the native phenylalanine cannot readily explained in terms of different interactions with the cyclin groove. A probable scenario is that the pyridyl ring is solvated to a greater degree relative to the phenyl and therefore a desolvation penalty would disfavor binding. A number of substitutions in the cyclin groove recognition motif have been incorporated in the N-terminal and arginine binding site and provide additional information on the tolerance of sequence changes upon binding to the secondary hydrophobic and acidic regions of cyclin D1 (Table 2).
The results obtained in this study provide new insights into the cyclin regulatory subunits that bind to CDK family members. The major structural variations in the cyclin groove between cyclin D1 and A2 have been described and the key interaction determinants for the peptide inhibitor and the receptor have been defined. A cyclin D1 complex of the endogenous CDK2 and CDK4 inhibitor, p27KIP has been generated and used to identify a unique groove that could be exploited to generate specific inhibitory compounds. Non-natural amino acid and fragment based mimetics of the key hydrophobic pocket determinants have been designed and give initial insights into compounds that have potential as highly CDK4/cyclin D1 selective compounds. This progress will provide the basis for further development of cyclin groove inhibitors as cell cycle specific anti-tumor therapeutics.
METHODS
Solid Phase Peptide Synthesis
Peptides were assembled by using standard solid phase synthesis methods [4] on a Argonaut Quest 210 semi-automated solid phase synthesizer. 10 equivalents of the C-terminal amino acid were coupled to Rink resin at the first place using DIEA (0.082ml) and HBTU (189.6 mg) in 5ml DMF for 1 h. Fmoc of the C-terminus amino acid was removed using 20% piperidine in 5ml DMF for 10mins before assembly of 10 equivalents of the next amino acid using DIEA (0.082ml) and HBTU (189.6 mg) in 5ml DMF. Wash cycles (5* 10ml DMF + 5* 10ml DCM) were applied to each step in between coupling and deprotection of Fmoc. Upon completion of assembly, side chain protecting groups were removed and peptides were finally cleaved from Rink resin using 90:5:5 mixtures of TFA/H2O/TIS. Crude peptides were purified using reverse phase flash chromatography and semi-preparative reversed-phase HPLC methods. Pure peptides were lyophilized and characterized using mass spectrometry and analytical HPLC (See Table3, supplementary info). All peptides used in this study contained free amino termini and were either C-terminal carboxylates or carboxamides.
Computational chemistry
Modeled complexes of peptidic cyclin groove inhibitors bound to either Cyclin A or Cyclin D were generated as follows: SAKRRLXG series were modeled from the crystal structure the p107 peptide bound to cyclin A (PDB: 1H28). The HAKRRLIX series were obtained by hybridizing the peptide conformation of RRLIF (PDB: 1OKV) and SAKRRLFG (PDB: 1H28). The Cyclin A structure in this complex was taken from 1OKV. Cyclin D1/SAKRRLXG and Cyclin D/HAKRRLIX were generated in a similar manner using cyclin D1 crystal structures (PDB: 2W96) where the peptidic inhibitor bound to Cyclin A was superimposed with cyclin D1 and followed by deletion of Cyclin A from further minimization of the complex. After applying the CHARMm forcefield in Discovery Studio 2.5 (Accelrys, San Diego), the Smart Minimizer algorithm comprised of steepest descent and conjugate gradient and an implicit solvent model of Generalized Born with a simple Switching (GBSW) were applied to the complex. In general, all peptide residues were flexible, for cyclin A, all protein residues were restrained and for cyclin D1, the backbone atoms were fixed and approximately 300 steps of minimization were required for convergence to a energy minimum. Molecular dynamics simulations (with backbone atoms fixed) carried out in order to validate the minimization protocols were completed using the following parameters: minimization; 500 steps steepest descent following by 500 steps conjugate gradient; heating stage (2000 steps, 0.001 ps timestep), equilibration stage (1000 steps, 0.001 ps timestep) and production stage (1000 steps, 0.001 ps timestep). A target temperature of 300 K was used for each stage. The calculate interaction energy protocol of DS 2.5 was used to generate non-bonded energy values between peptidic inhibitor and its associated cyclin. This included calculation of van der Waals and electrostatic energies to provide an estimation of the affinity of inhibitors. The calculate binding energy protocol of DS 2.5 enabled the generation of desolvation penalty data for each peptide- cyclin complex. Interaction energy calculations were performed on low and high energy frames from the molecular dynamics simulations and were found to be closely in line with the values obtained solely using minimization protocols.
In Vitro Kinase Assay
CDK2/Cyclin A2 and CDK4/Cyclin D1 kinase assays were performed using full-length recombinant CDK2/cyclin A2 and CDK4/Cyclin D1 co-expressed by baculovirus in Sf9 insect cells using an N-terminal GST tag on both proteins. The kinase assay buffer I consisted of 25mM MOPS, pH7.2, 12.5mM beta-glycerol-phosphate, 25mM MgCl2, 5mM EGTA, 2mM EDTA and 0.25mM of DTT was added prior to use. The [32P]-ATP Assay cocktail was prepared in a designated radioactive working area by adding 150ul of 10mM ATP stock solution, 100ul[32P]-ATP (1mCi/100ul), 5.75ml of kinase assay buffer I. 10mM ATP Stock Solution was prepared by dissolving 55mg of ATP in 10ml of kinase assay buffer I. Store 200ul aliquots at −20°C. The substrate used is Rb (773–928) protein with 0.2 mg/ml concentration. The blank control was set up by adding 10ul of diluted active CDK/Cyclin with 10ul of distilled H2O. Otherwise, adding 10ul of diluted active CDK/Cyclin with 10ul of 0.2mg/ml stock solution of Rb (773–928). The reaction was initiated by the addition of 5 µl [32P]-ATP assay cocktail bringing the final volume up to 25 µl and incubating the mixture in a water bath at 30° C for 15 minutes. After the incubation period, the reaction was terminated by spotting 20 µl of the reaction mixture onto individual pre-cut strips of phophocellulose P81 paper. The pre-cut P81 strip was air-dried and sequentially washed in a 1% phosphoric acid solution with constant gentle stirring. Radioactivity on the P81 paper was counted in the presence of scintillation fluid on a scintillation counter. The corrected cpm was determined by subtracting the blank control value for each sample and calculating the kinase specific activity as follows: Calculation of [P32]-ATP specific activity (SA) (cpm/pmol) Specific activity (SA) = cpm for 5 ul [32P]-ATP/pmoles of ATP (in 5 ul of a 250 uM ATP stock solution). Kinase Specific Activity (SA) (pmol/min/ug or nmol/min/mg)Corrected cpm from reaction / [(SA of 32P-ATP in cpm/pmol)*(Reaction time in min)*(Enzyme amount in ug or mg)]*[(Reaction Volume) / (Spot Volume)] (SignalChem, Richmond, Canada).
Supplementary Material
REFERENCES
- 1.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 2.Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nature Reviews Cancer. 2001;1:222–231. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- 3.McInnes C, Andrews MJI, Zheleva DI, Lane DP, Fischer PM. Peptidomimetic design of CDK inhibitors targeting the recruitment site of the cyclin subunit. Current Medicinal Chemistry - Anti-Cancer Agents. 2003;3:57–69. doi: 10.2174/1568011033353506. [DOI] [PubMed] [Google Scholar]
- 4.Zheleva DI, McInnes C, Gavine A-L, Zhelev NZ, Fischer PM, Lane DP. Highly potent p21WAF1 derived peptide inhibitors of CDK-mediated pRb phosphorylation: delineation and structural insight into their interactions with cyclin A. Journal of Peptide Research. 2002;60:257–270. doi: 10.1034/j.1399-3011.2002.21014.x. [DOI] [PubMed] [Google Scholar]
- 5.Chen YN, Sharma SK, Ramsey TM, Jiang L, Martin MS, Baker K, Adams PD, Bair KW, Kaelin WG., Jr Selective killing of transformed cells by cyclin/cyclin-dependent kinase 2 antagonists. Proc Natl Acad Sci U S A. 1999;96:4325–4329. doi: 10.1073/pnas.96.8.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schulman BA, Lindstrom DL, Harlow E. Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proceedings Of The National Academy Of Sciences Of The United States Of America. 1998;95:10453–10458. doi: 10.1073/pnas.95.18.10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ball KL, Lain S, Fåhraeus R, Smythe C, Lane DP. Cell-cycle arrest and inhibition of Cdk4 activity by small peptides based on the carboxy-terminal domain of p21WAF1. Current Biology. 1996;7:71–80. doi: 10.1016/s0960-9822(06)00029-7. [DOI] [PubMed] [Google Scholar]
- 8.Mendoza N, Fong S, Marsters J, Koeppen H, Schwall R, Wickramasinghe D. Selective Cyclin-dependent Kinase 2/Cyclin A Antagonists that Differ from ATP Site Inhibitors Block Tumor Growth. Cancer Research. 2003;63:1020–1024. [PubMed] [Google Scholar]
- 9.Oelgeschlager T. Regulation of RNA polymerase II activity by CTD phosphorylation and cell cycle control. Journal Of Cellular Physiology. 2002;190:160–169. doi: 10.1002/jcp.10058. [DOI] [PubMed] [Google Scholar]
- 10.Fischer PM, Gianella-Borradori A. Recent progress in the discovery and development of cyclin-dependent kinase inhibitors. Expert Opin Investig Drugs. 2005;14:457–477. doi: 10.1517/13543784.14.4.457. [DOI] [PubMed] [Google Scholar]
- 11.Baughn LB, Di Liberto M, Wu K, Toogood PL, Louie T, Gottschalk R, Niesvizky R, Cho H, Ely S, Moore MA, Chen-Kiang S. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res. 2006;66:7661–7667. doi: 10.1158/0008-5472.CAN-06-1098. [DOI] [PubMed] [Google Scholar]
- 12.Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Molecular Cancer Therapeutics. 2004;3:1427–1438. [PubMed] [Google Scholar]
- 13.Yu Q, Sicinska E, Geng Y, Ahnstrom M, Zagozdzon A, Kong Y, Gardner H, Kiyokawa H, Harris LN, Stal O, Sicinski P. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9:23–32. doi: 10.1016/j.ccr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 14.Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006;9:13–22. doi: 10.1016/j.ccr.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 15.Kontopidis G, Andrews MJI, McInnes C, Cowan A, Powers H, Innes L, Plater A, Griffiths G, Paterson D, Zheleva DI, Lane DP, Green S, Walkinshaw MD, Fischer PM. Insights into cyclin groove recognition: complex crystal structures and inhibitor design through ligand exchange. Structure. 2003;11:1537–1546. doi: 10.1016/j.str.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Andrews MJI, McInnes C, Kontopidis G, Innes L, Cowan A, Plater A, Fischer PM. Design, synthesis, biological activity and structural analysis of cyclic peptide inhibitors targeting the substrate recruitment site of cyclin-dependent kinase complexes. Organic & Biomolecular Chemistry. 2004;2:2735–2341. doi: 10.1039/B409157D. [DOI] [PubMed] [Google Scholar]
- 17.Andrews MJ, Kontopidis G, McInnes C, Plater A, Innes L, Cowan A, Jewsbury P, Fischer PM. REPLACE: a strategy for iterative design of cyclin-binding groove inhibitors. Chembiochem. 2006;7:1909–1915. doi: 10.1002/cbic.200600189. [DOI] [PubMed] [Google Scholar]
- 18.Brown NR, Noble ME, Endicott JA, Johnson LN. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nature Cell Biology. 1999;1:438–443. doi: 10.1038/15674. [DOI] [PubMed] [Google Scholar]
- 19.Day PJ, Cleasby A, Tickle IJ, O'Reilly M, Coyle JE, Holding FP, McMenamin RL, Yon J, Chopra R, Lengauer C, Jhoti H. Crystal structure of human CDK4 in complex with a D-type cyclin. Proc Natl Acad Sci U S A. 2009;106:4166–4170. doi: 10.1073/pnas.0809645106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takaki T, Echalier A, Brown NR, Hunt T, Endicott JA, Noble ME. The structure of CDK4/cyclin D3 has implications for models of CDK activation. Proc Natl Acad Sci U S A. 2009;106:4171–4176. doi: 10.1073/pnas.0809674106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kontopidis G, Andrews MJ, McInnes C, Plater A, Innes L, Renachowski S, Cowan A, Fischer PM. Truncation and optimisation of peptide inhibitors of cyclin-dependent kinase 2-cyclin a through structure-guided design. ChemMedChem. 2009;4:1120–1128. doi: 10.1002/cmdc.200900093. [DOI] [PubMed] [Google Scholar]
- 22.Castanedo G, Clark K, Wang S, Tsui V, Wong M, Nicholas J, Wickramasinghe D, Marsters JC, Jr, Sutherlin D. CDK2/cyclinA inhibitors: targeting the cyclinA recruitment site with small molecules derived from peptide leads. Bioorg Med Chem Lett. 2006;16:1716–1720. doi: 10.1016/j.bmcl.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Blain SW. Switching cyclin D-Cdk4 kinase activity on and off. Cell Cycle. 2008;7:892–898. doi: 10.4161/cc.7.7.5637. [DOI] [PubMed] [Google Scholar]
- 24.James MK, Ray A, Leznova D, Blain SW. Differential modification of p27Kip1 controls its cyclin D-cdk4 inhibitory activity. Mol Cell Biol. 2008;28:498–510. doi: 10.1128/MCB.02171-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ray A, James MK, Larochelle S, Fisher RP, Blain SW. p27Kip1 inhibits cyclin D-cyclin-dependent kinase 4 by two independent modes. Mol Cell Biol. 2009;29:986–999. doi: 10.1128/MCB.00898-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.