

Abstract
Objective
Resuscitation of hemorrhagic hypotension after traumatic brain injury (TBI) is challenging. A hemoglobin (Hb)-based oxygen carrier (HBOC) may offer advantages. The novel therapeutic HBOC, polynitroxylated pegylated Hb (PNPH) may represent a neuroprotective HBOC for TBI resuscitation.
Hypotheses
1) PNPH is a unique non-neurotoxic HBOC in neuronal culture and is neuroprotective in in vitro neuronal injury models. 2) Resuscitation with PNPH would require less volume to restore mean arterial blood pressure (MAP) than lactated Ringer’s (LR) or Hextend (HEX) and confer neuroprotection in a mouse model of TBI plus hemorrhagic hypotension.
Design
Prospective randomized controlled experimental study.
Setting
University center.
Measurements and Main Results
In rat primary cortical neuron cultures, control bovine Hb was neurotoxic (LDH release; MTT assay) at concentrations from 12.5 to 0.625µM, while polyethylene-glycol (Peg)-conjugated Hb showed intermediate toxicity. PNPH was not neurotoxic (p<0.05 vs bovine Hb and Peg-Hb; all concentrations). PNPH conferred neuroprotection in in vitro neuronal injury (glutamate/glycine exposure and neuronal stretch), as assessed via LDH, and MTT all p<0.05 vs control. C57BL6 mice received controlled cortical impact followed by hemorrhagic hypotension (2mL/100g, MAP ~35–40 mmHg) for 90 min. Mice were resuscitated (MAP >50 mmHg for 30 min) with LR, HEX, or PNPH, then shed blood was re-infused. MAPs, resuscitation volumes, blood gasses, glucose and lactate were recorded. Brain sections at 7d were examined via H&E and Fluoro-Jade C (FJC, identifying dying neurons) staining in CA1 and CA3 hippocampus. Resuscitation with PNPH or HEX required less volume than LR (both p<0.05). PNPH but not HEX improved MAP vs. LR (p<0.05). Mice resuscitated with PNPH had fewer FJC+ neurons in CA1 vs. HEX and LR, and CA3 vs. HEX (p<0.05).
Conclusion
PNPH is a novel neuroprotective HBOC in vitro and in vivo that may offer unique advantages for TBI resuscitation.
Keywords: head injury, hemoglobin based oxygen carrier, nitroxide, oxidative stress, nitric oxide, polyethylene glycol, hemorrhagic shock, blast injury, polytrauma, blood substitute, polynitroxylation, superoxide
INTRODUCTION
Hypotension from hemorrhage complicating traumatic brain injury (TBI) contributes to secondary injury and poor outcome, including both morbidity and mortality. The deleterious effects of hypotension have been demonstrated in both civilian (1) and military settings such as blast injury (2). Effective resuscitation of combined TBI and hemorrhage is essential to improving outcomes in the increasing numbers of individuals exposed to these combined injuries, particularly, in military theaters abroad (3).
Traditional pre-hospital strategies for TBI resuscitation involve the administration of isotonic fluids, such as Lactated Ringer’s (LR) solution, or colloids, such as Hextend (HEX) (the civilian and military standards of care, respectively) after injury to restore blood pressure, and potentially, cerebral perfusion pressure. However, large volumes of resuscitation fluid may exacerbate cerebral edema (4), and fail to increase oxygen delivery. In addition, these solutions fail to specifically target deleterious cascades associated with ischemia reperfusion injury. In the pre-hospital setting, hemoglobin (Hb) based oxygen carriers (HBOCs) may offer specific advantages over crystalloids or colloids as a resuscitation fluid in the setting of hemorrhagic hypotension after TBI. HBOC resuscitation may allow for volume expansion using small amounts of fluid with the potential advantage of restoring oxygen delivery to the injured brain—which exhibits markedly increased metabolic demands and excitotoxicity early after TBI (5). However, human clinical trials have shown increased morbidity and mortality with the use of HBOCs (6). Current generation HBOCs and cell-free Hb scavenge nitric oxide (NO), resulting in vasoconstriction and decreased oxygen delivery to myocardium and other vascular beds (7). This could also impair posttraumatic cerebral blood flow (CBF), since NO levels and endothelial nitric oxide synthase (eNOS; NOS1) activity are compromised early after TBI in injured brain regions (8). Additional putative deleterious side effects of HBOCs have also been suggested. Winslow (9) proposed that increased oxygen delivery caused by free Hb juxtaposed to vascular endothelium causes local hyperoxia with coupled vasoconstriction which could further impair CBF in injured brain. Similarly, HBOCs and cell-free Hb can also directly contribute to oxidative stress in both the microcirculation and parenchyma. Specifically, extravasation of cell free Hb into tissue could result in direct cytotoxiciy and this could be of considerable importance in TBI, since vascular disruption occurs and classic studies have shown that cell free Hb is highly neurotoxic in cell culture (10, 11).
Chemical modification of the Hb structure has been studied to minimize the harmful side effects of HBOCs. Nitroxyl- groups have well-known antioxidant effects including potent superoxide dismutase (SOD) mimetic activity, along with other favorable effects on oxidative stress, such as limiting autoxidation (12). Polynitroxylation of Hb also creates redox coupling between the convalently labeled nitroxides and the heme iron to provide additional catalase mimetic activity (13). Pegylation (i.e., adding polyethylene glycol [Peg] moieties to the Hb molecule) provides several potential favorable effects including limiting the Hb from direct interaction with the endothelium—attenuating oxygen mediated vasoconstriction (14) and enhancing nitrite reductase activity—with increased NO production and blunted vasoactivity (15). Pegylation also adds a “super-colloid” effect to an HBOC, due to the potent water scavenging effect of Peg moieties (Personal communication, A. Abuchowski, PhD), and prolongs HBOC half life. Polynitroxylated-pegylated Hb (PNPH) is a novel bovine HBOC developed by SynZyme Technologies that uses Prolong Pharmaceutical’s Peg-Hb as the starting material, and thus has both of these chemical modifications. Specifically, it has ~14 nitroxyl radicals (nitroxides) and 8–10 Peg moieties (5KDa molecular weight each) per Hb tetramer molecule. It is prepared as a 4% Hb solution.
We recently published (16) details of a mouse model in which a mild controlled cortical impact (CCI) injury to brain is followed by 90 min of volume-controlled hemorrhagic hypotension resulting in an exacerbation of neuronal death in the vulnerable CA1 hippocampus vs that seen in either CCI or hemorrhage alone. Blood flow is compromised in the hippocampus beneath the contusion during the hemorrhage. The model is perfectly suited for exploratory evaluation of novel HBOCs in the setting of TBI resuscitation in that it is allows for the assessment of acute hemodynamics, and long term outcome including neuropathology. Mortality rates in the model with standard resuscitation solutions range between 25 and 30%. Unlike large animal models, novel therapies can be explored both with extremely small volumes of novel resuscitation fluids and with relatively limited expense.
We tested two hypotheses: 1) PNPH is a unique non-neurotoxic HBOC in neuronal culture and is neuroprotective in in vitro neuronal injury models, and 2) Resuscitation with PNPH would require less volume to restore mean arterial blood pressure (MAP) than LR or HEX and confer neuroprotection in a mouse model of TBI plus hemorrhagic hypotension. In the present communication, we present results that show that PNPH is a unique neuroprotective Hb both in vitro and in vivo and that support its further development and evaluation as a neuroprotective HBOC for small volume pre-hospital TBI resuscitation.
METHODS
Primary Cortical Neuronal Cultures
Primary cortical neuron-enriched cultures were prepared from the cerebral cortex of fetal Sprague-Dawley rats (embryonic day 16). Dissociated cell suspensions were plated in 96-well plates (5 × 104 cells/well) or in silicon dishes coated with poly-D-lysine (3 × 106). Cells were cultured using Neurobasal medium, supplemented with B27 (Invitrogen) and penicillin/streptomycin. The medium was changed every 2–3 days. Cells were maintained in incubators at 37 °C in a humidified atmosphere of 5% CO2 in air. Experiments were performed at 7–10 days in vitro. All studies in primary neuronal culture were replicated in at least three separate experiments using primary neurons cultured from separate litters and include assay of between 9 and 22 wells from 3 to 5 different 96-well plates.
Preparation of Hb solutions
Stroma-free bovine Hb was prepared by cross-flow ultrafiltration (UF) using a modified published procedure (17): Fresh bovine red blood cells were purchased from Innovative Research Novi, MI. To match with PNPH, each Hb used in the experiments was the carboxy (CO) Hb formulation. Peg-Hb was prepared as follows. The starting material, deoxy Peg-Hb was obtained from Prolong Pharmaceutical Monmouth Junction, NJ, converted to carbon monoxide ligand form as a first step, and then the Peg-Hb(CO) was diafiltrated with 8-volumes saline using Sartoflow Alpha filtration system equipped with 50 kDa molecular-weight cut-off membrane cassettes. PNPH was prepared as follows. The polynitroxylation was performed by allowing Peg-Hb(CO) to react with the nitroxide compound 4-(2-bromoacetamido)-2,2,6,6-tetramethylpiperidine-1-oxyl (BrAcTPO) which was custom synthesized according to SynZyne’s SOP by Chunghwa Chemical Synthesis & Biotech Company. The reaction was carried out at pH 9.0 and 37°C for 4 hours under 3-psi CO pressure. A molar ratio of 16 nitroxides per Hb was used to start the reaction. The reaction mixture was then ultrafiltrated with saline. Under optimum conditions, about 14 nitroxide moieties can be covalently linked to each Peg-Hb tetramer. All samples, Hb, Peg-Hb, and PNPH, were adjusted to 4% Hb concentration which are equivalent to 0.62 mM of tetramer or 2.5 mM of heme as determined spectrophotometrically. MetHb was determined to be less than 5% spectrophototmetrically. Endotoxin was determined to be less than 1 EU/ml by Limulus Amebocyte Lysate (LAL) method. Labeled nitroxide concentration was measured by electron paramagnetic resonance spectroscopy and the labeling number of 14 nitroxides per Hb tetramer in PNPH was calculated from the ratio of nitroxide concentration to Hb concentration.
Assessment of Hb-induced Neurotoxicity
The toxicity of PNPH was compared to its aforementioned parent Hb molecules, specifically, bovine Hb and Peg-Hb in the primary neuronal culture system. For these studies, the Hb dilutions used included 1/50, 1/100, 1/500 and 1/1000. These dilutions yielded the following approximate Hb concentrations (expressed as the concentration of tetramer) for the three preparations, namely 12.50 µM, 6.25 µM, 1.25 µM, and 0.625 µM, respectively.
In Vitro Neuronal Injury Models
Glutamate/Glycine (Glu/Gly)-Induced Neuronal Death: Neurons were exposed to 10 µM L-Glu/10 µM Gly with or without bovine Hb, bovine Peg-Hb, or PNPH in the culture medium for 24 h. In Vitro TBI was produced by neuronal stretch (18). A computer controlled apparatus to stretch neurons at a defined strain magnitude and rate was used. Briefly, primary cortical neurons were grown on silicone membranes (0.002–0.005 inch thick, Specialty Manufacturing) secured to stainless steel rings that were polished and passivated prior to use. At 8 days in vitro (DIV), cultures were pre-treated with varying concentrations of Hb for 30 min. The membranes were then placed over a hollowed platform in a custom-made, sealed stainless steel chamber. The membranes were then stretched with a pre-set strain rate (10 s−1) and membrane deformation (50%) using an air pressure pulse. The pressure waveform is measured and collected on a data acquisition system to verify the degree of insult. Severe stretch was chosen in order to simulate a strain field similar to that seen in animal models of TBI (18). Neuronal cultures were then returned to the incubator. Cultures were pre-treated with varying concentrations of Hb for 30 min. Between 6 and 8 independent stretch insults were carried out for each condition tested.
Cytotoxicity Detection (LDH release)
Cortical neuronal cell injury was quantified by the measurement of lactate dehydrogenase (LDH) at 24 h after the insult. An aliquot of bathing media was combined with NADH and pyruvate solutions. LDH activity is proportional to the rate of pyruvate loss, which was assayed by absorbance change using a microplate reader (Spectra MAX 340). Blank LDH levels were subtracted from insult LDH values, and results normalized to 100% neuronal death caused by 0.5% Triton-100 exposure.
Cell Viability (MTT assay)
Cell viability was evaluated using the MTT (3-[4, 5-dimethylthiazol-2-yl-]-2, 5-diphenyltetrazolium bromide) colorimetric assay. MTT (Sigma) is a water soluble tetrazolium salt that yields a yellowish solution when prepared in medium lacking phenol red. Dissolved MTT is converted to an insoluble purple formazan by cleavage of the tetrazolium ring by the active mitochondrial dehydrogenases of living cells. The MTT solution (5 mg MTT/mL medium) was added to each well 24 h after the insult at the final concentration of 250 µM, and incubated for 3 h. The media was removed, and cells dissolved in DMSO. Formation of formazan was assessed by measuring the amount of reaction product (absorbance change) using a microplate reader (Spectra MAX 340).
Groups and Experimental Protocol for In Vivo Studies
The Institutional Animal Care and Use Committee of the University of Pittsburgh School of Medicine approved all experiments. C57BL6 male mice (Jackson Laboratories, Bar Harbor, ME), 12–15 weeks of age, and weighing 27.7±0.5 g were housed in controlled environmental conditions and allowed ad libitum food and water until the start of the study. The details of this experimental model of CCI and hemorrhage have been previously published (16).
Anesthesia was induced with isofluorane 4% in 2:1 N2O/O2 via nose cone. Femoral venous and arterial catheters were inserted via inguinal cut-down. Animals were placed in a stereotactic frame, a 5mm craniotomy was performed, and the bone flap removed. A brain temperature probe (Physitemp, Clifton, NJ) was inserted into the right parietal cortex via a burr hole. A rectal probe was inserted to monitor body temperature. Brain temperature was controlled with a heat lamp at 37.0±0.5°C throughout the study. After the craniotomy, anesthesia was changed to 1% isofluorane in room air for a 10 min stabilization period.
The time course of CCI and resuscitation is shown in Figure 1. CCI was performed with a pneumatic impactor (Bimba, Monee, IL) using a flat 3-mm tip at a velocity of 5 m/s to a depth of 1 mm. This level of injury produces a mild-moderate level of injury with no appreciable loss of CA1 hippocampal neurons and no mortality (16). After injury, the bone flap is replaced and the craniotomy is immediately re-sealed with dental cement and beginning within 5 min, blood was withdrawn from the femoral arterial catheter over 15 min to a total volume of 2.0ml/100g, a volume shown to produce a MAP between ~35–40 mmHg (Hemorrhage phase). Shed blood was stored for later infusion. This phase continued for 90 total min (75 min after the completion of blood withdrawal).
Figure 1.

Diagram depicting the time course of the experimental protocol used in this study (CCI, controlled cortical impact; TBI, traumatic brain injury; CCI, controlled cortical impact; MAP, mean arterial blood pressure, ABG, arterial blood gas.
Mice were then resuscitated with 0.1 mL aliquots of LR, HEX (Hospita, Lake Forest, IL), or PNPH to a goal MAP ≥ 50 mmHg for 30 min duration (Pre-hospital phase). Mice continued to receive 1% isofluorane with room air for anesthesia. At the conclusion of this phase, mice were switched to isofluorane 1% in oxygen and shed blood was rapidly re-infused (Definitive Care phase). After the shed blood was re-infused, a goal MAP ≥ 60 mmHg was obtained by administration of additional resuscitation fluid given in 0.1-mL aliquots, if necessary. Total volume of resuscitation fluid administered in each phase was recorded. After the completion of the Definitive Care phase, catheters were removed, incisions were closed, anesthesia was discontinued, and mice were placed in supplemental oxygen until they were recovered from anesthesia at which time they were then returned to their cage.
Monitoring
Arterial blood pressure was continuously monitored via femoral venous cannula. Heart rate was monitored continuously and was recorded every 5 min. Arterial blood gasses were obtained at baseline, 30 min after hemorrhage, Post-resuscitation and at the end of the Definitive Care phase. Evaluation of hematocrit, arterial lactate, and blood glucose was also performed at these time points.
Histology
At 7 days post-injury, animals were anesthetized with 4% isofluorane and killed by transcardial perfusion fixation with ice-cold saline followed by 10% buffered formalin. Brain tissue was fixed with 10% buffered formalin and embedded in paraffin. Multiple 5µm sections, 200µm apart from bregma −1.86 to −2.26, were prepared from each brain. Sections were stained with Fluoro-Jade C (FJC; Chemicon, Temecula, CA) and hematoxylin and eosin (H&E; Thermo Scientific, Pittsburgh, PA), and hippocampal neuronal degeneration was quantified in the dorsal CA1 and CA3 hippocampus underlying the cortical contusion in brain sections stained with FJC by an evaluator blinded to treatment group using ImageJ software (NIH). The number of surviving neurons in CA1 and CA3 were also quantified by a blinded observer in the H&E-stained sections.
Statistical Analysis
Data are expressed as mean ± SEM or median [range] where appropriate. Analysis of variance was used to compare continuous physiologic variables between groups. Appropriate post-hoc analysis was performed including Student-Neuman-Keuls or Tukey test when a significant ANOVA result was discovered. Kruskal-Wallis non-parametric analysis was used to determine the significance of non-parametric data.
RESULTS
In Vitro Studies
Hb toxicity studies
Exposure of neurons to bovine Hb produced anticipated neurotoxicity based on prior reports (10), and neurotoxicity was seen at all concentrations and dose dependent as assessed by either LDH or MTT assays (Figures 2 and 3). Based on LDH release, a significant dose response (a ~20–40% cell loss relative to Triton lysis) was observed across concentrations for bovine Hb, with the 1:50 dilution showing significant neuronal death vs all other concentrations (p<0.05, Figure 2). In the MTT assay, native bovine Hb also showed a dose response for toxicity with the greatest loss of neuronal viability at the highest two concentrations (again p<0.05, Figure 3). Peg-Hb was also neurotoxic, but less (p<0.05) than native bovine Hb at the two highest concentrations in the LDH assay, and across all concentrations in MTT assay (Figures 2 and 3). Remarkably, PNPH was devoid of any neurotoxicity as assessed with either LDH or MTT assays across all concentrations. In both the LDH and MTT assays, all PNPH concentrations were different (p<0.05) from both bovine Hb and Peg-Hb at all concentrations studied (Figures 2 and 3).
Figure 2.

Effect of various hemoglobins (Hbs) on lactate dehydrogenase (LDH) release from primary rat cortical neurons in culture. Hbs were added to the culture medium for 24 h at concentrations (based on the tetramer) ranging between 0.625 µM and 12.50 µM. Percent cytotoxicity (LDH release) relative to Triton exposure (corrected for background LDH) is graphed. Native bovine Hb (Hb) showed dose-dependent neurotoxicity. Polyethylene glycol (Peg) conjugated bovine Hb (Peg-Hb) showed less toxicity than native Hb while polynitroxylated pegylated Hb (PNPH) was devoid of neurotoxicity across all concentrations tested. Data are mean ± SEM from at least three separate experiments and include assay of between 9 and 30 wells from between 3 and 5 different 96 well plates. ap<0.05 vs all Hb and Peg-Hb values at all dilutions; bp<0.05 vs Hb values at the 12.50 and 6.25 µM concentrations; cp<0.05 vs Hb at 12.50 µM concentration; dp<0.05 vs Hb at 6.25µM concentration.
Figure 3.

Effect of various hemoglobins (Hbs) on cell viability assessed with the MTT assay in primary rat cortical neurons in culture. Hbs were added to the culture medium for 24 h at concentrations ranging between 0.625 µM and 12.50 µM. MTT as a percent of baseline is shown. Native bovine Hb (Hb) showed dose-dependent neurotoxicity with reduced neuronal viability. Polyethylene glycol (Peg) conjugated bovine Hb (Peg-Hb) showed less toxicity than native Hb while polynitroxylated pegylated Hb (PNPH) was devoid of neurotoxicity across all concentrations tested. Data are mean ± SEM from at least three separate experiments and include assay of between 9 and 30 wells from between 3 and 5 different 96 well plates. ap<0.05 vs all Hb and Peg-Hb values at all concentrations; bp<0.05 vs Hb values at all concentrations; cp<0.05 vs Hb at 12.50 µM and 6.25 µM concentrations.
In Vitro Neuronal Injury Models
PNPH also attenuated Glu/Gly-induced neuronal death at all concentrations as determined by LDH and MTT assays (p<0.05, Figures 4 and 5). The reduction in LDH release by PNPH after Glu/Gly exposure was similar to baseline LDH release in normal neurons in our preparation (Figure 4). Native bovine Hb, at the highest two concentrations further reduced neuronal viability after Glu/Gly as assessed in the MTT assay (Figure 5). Native bovine Hb reduced LDH release induced by Glu/Gly, but only at 0.625 µM (Figure 4). Bovine Peg-Hb did not exacerbate neuronal death induced by Glu/Gly, and in fact showed intermediate attenuation of Glu/Gly toxicity assessed by MTT and LDH assays (Figures 4 and 5). The effect of the three Hb preparations was also assessed in the neuronal stretch model at the 1.25µM concentration with cell death assessed again using both the LDH and MTT assays at 24 h (Figures 6A–B). In the stretch model, between 30 and 40% neuronal death was observed (by both assays). Neuronal death from stretch was significantly exacerbated by exposure to native bovine Hb (p<0.05 vs stretch alone by both LDH and MTT assays). Peg-Hb modestly reduced neuronal death vs stretch alone in the LDH but not MTT assay, while a marked reduction in neuronal death was produced by PNPH (to ≤12% in both LDH and MTT assays, p<0.05 vs all stretch injury groups including stretch plus Peg-Hb).
Figure 4.

Effect of various hemoglobins (Hbs) on lactate dehydrogenase (LDH) release in a primary rat cortical neuron culture model of glutamate/glycine (Glu/Gly)-induced excitotoxicity. The test Hb was added to the culture medium and 30 min later Glu/Gly (10 µM) exposure was begun and continued for 24 h. Hb concentrations ranged between 0.625 µM and 12.50 µM. Percent cytotoxicity (LDH release) relative to Triton exposure (corrected for background LDH) is graphed. PNPH showed surprising neuroprotection at all Hb concentrations. Peg-Hb showed intermediate protection, while native bovine Hb (Hb) was not neuroprotective. Data are mean ± SEM from at least three separate experiments and include assay of between 9 and 30 wells from between 3 and 5 different 96 well plates. ap<0.05 vs Glu/Gly and both respective Hb and Peg-Hb value; bp<0.05 vs Glu/Gly and respective Hb value; cp<0.05 vs Glu/Gly value.
Figure 5.

Effect of various hemoglobins (Hbs) on cell viability (MTT assay) in a primary rat cortical neuron culture model of glutamate/glycine (Glu/Gly)-induced excitotoxicity. The test Hb was added to the culture medium and 30 min later Glu/Gly (10 µM) exposure was begun and continued for 24 h. Hb concentrations ranged between 0.625 µM and 12.50 µM. MTT as a percent of baseline is graphed. PNPH showed surprising neuroprotection at all Hb concentrations. Peg-Hb showed intermediate protection, while native bovine Hb (Hb) exacerbated Glu/Gly toxicity reducing neuronal viability at the highest concentrations. Data are mean ± SEM from at least three separate experiments and include assay of between 9 and 30 wells from between 3 and 5 different 96 well plates. ap<0.05 vs Glu/Gly and both respective Hb and Peg-Hb value; bp<0.05 vs Glu/Gly and respective Hb value; cp<0.05 vs Glu/Gly value.
Figure 6.


A–B. Effect of bovine Hb, Peg-Hb and PNPH (1.25 µM) on rat cortical neurons in culture exposed to in vitro trauma by neuronal stretch. The test Hb was added to the medium and 30 min later neuronal stretch was induced. A) % cytotoxicity (LDH release) relative to Triton exposure (corrected for background LDH). Stretch produced ~34% neuronal death at 24 h assessed by LDH. PNPH showed surprising neuroprotection. Peg-Hb showed intermediate protection, while native bovine Hb exacerbated neuronal death after stretch. Data are mean + SEM from 6–8 separate experiments and include assay of 18–22 wells for each Hb. ap<0.05 vs stretch; bp<0.05 vs native bovine Hb value; cp<0.05 vs Peg-Hb value. B) MTT as % baseline. Stretch again produced ~35% neuronal death at 24 h by MTT. PNPH again showed surprising neuroprotection. Peg-Hb showed no neuroprotection by MTT, while native bovine Hb again exacerbated neuronal death induced by stretch. Data are mean + SEM from 6–8 separate experiments and include assay of 18–22 wells for each Hb. ap<0.05 vs stretch; bp<0.05 vs bovine Hb value; cp<0.05 vs Peg-Hb value.
In Vivo Experimental TBI plus Hemorrhagic Hypotension in Mice
To obtain n=5 survivors in each of the three groups, a total of 20 mice were used. A total of n=6 animals were resuscitated with PNPH, n=5 mice were resuscitated with HEX, and n=9 mice were resuscitated with LR. The trend toward higher mortality in the LR group did not reach significance.
Physiology
Table 1 provides a summary of the resuscitation volume delivered during the Pre-hospital phase of the experiment and the mean and peak MAPs resulting in each phase of the experiment. Resuscitation with PNPH and HEX required less volume than LR (p < 0.0001 for PNPH vs. LR and p≤ 0.05 for HEX vs LR). When expressed in volume per kilogram dose, PNPH, HEX, and LR treated groups required 6.7 ± 1.8 mL/kg, 9.6 ± 2.6 mL/kg, and 36.3 ± 11.5 mL/kg, respectively.
Table 1.
Physiologic Data (Resuscitation Volume, Mean Arterial Pressures)
| PNPH | HEX | LR | |
|---|---|---|---|
| Resuscitation Volume (mL) | 0.18±0.03* | 0.26±0.04* | 0.98±0.16 |
| Initial MAP (mmHg) | 79.3±1.7 | 76.4±2.9 | 78.3±1.4 |
| Mean Hemorrhage Phase MAP (mmHg) | 35.5±1.8 | 39.2±2.2 | 37.8±1.6 |
| Peak Hemorrhage Phase MAP (mmHg) | 43.8±2.9 | 48.2±3.4 | 46.5±2.6 |
| Mean Pre-hospital Phase MAP (mmHg) | 64.4±1.5*† | 58.8±1.5* | 50.5±1.7 |
| Peak Pre-hospital Phase MAP (mmHg) | 73.5±2.0* | 65.8±3.2* | 58.0±2.2 |
| Mean Definitive Care Phase MAP (mmHg) | 73.5±2.6 | 67.9±1.9 | 69.4±1.7 |
| Peak Definitive Care Phase MAP (mmHg) | 83.6±3.4*† | 73.0±1.8 | 76.0±1.7 |
p<0.05 vs. LR,
p<0.05 vs. Hex
Before resuscitation, all three groups had similar initial MAPs (p=0.61). Each group had similar mean and peak MAPs during the Hemorrhage phase (p=0.42 mean MAP, p=0.61 peak MAP). The mean MAP for each group during the Hemorrhage phase was within a target range of 30–40 mmHg and no differences were detected between groups (p=0.61). During the Pre-hospital phase, all groups attained mean MAPs greater than 50 mm Hg. However, the PNPH and HEX groups attained mean MAPs significantly greater than the LR group (p<0.05 for both PNPH vs. LR and HEX vs. LR). Peak MAPs were also higher in the PNPH group vs LR (p< 0.05), but did not differ vs HEX. Mice resuscitated with PNPH reached goal MAPs by 5 min into the Pre-hospital phase (Figure 7), while mice in the HEX and LR groups took between 10 and 15 min despite ongoing fluid administration. After the initial difference between groups at 5 min, mice in the PNPH group trended towards higher MAP vs both HEX and LR until 25 min into the Pre-hospital phase when the mean MAPs were statistically higher. During the Definitive Care phase, which began after reinfusion of shed blood, the mean MAPs between groups were not different (p=0.19). However, the PNPH group attained higher peak MAPs during the 30 min Definitive Care phase than HEX (p<0.05 for PNPH vs. HEX).
Figure 7.

Time course of the response in mean arterial pressure to resuscitation fluid administered during the 30 min Pre-hospital resuscitation phase for each group. Time points 0 min and 30 min below correspond to experiment time 90 min to 120 min, respectively, in Figure 1. Data indicate mean ± SEM at each time point, n=5 per group. (* p<0.05, ANOVA)
Table 2 provides a summary of hematocrit, lactate, base deficit, and glucose levels throughout the study. In all groups, the hematocrit decreased significantly from baseline values to the Hemorrhage phase (p<0.05). Pre-hospital resuscitation with PNPH, HEX, and LR resulted in post-resuscitation hematocrit values that trended lower than, but were not different from values in the Hemorrhage phase. Infusion of shed blood in the Definitive Care phase appropriately yielded a rise in hematocrit from Post-Resuscitation values in all groups, but resulted in hematocrits still lower than baseline values (p<0.05). There were no between group differences in hematocrit noted in any of the phases. There were also no between group differences in lactate levels, though the lactate did decrease in each group in the Definitive Care phase vs earlier phases. There was a significant difference in base deficit in LR treated mice vs baseline during hemorrhage, but there were no differences between treatment groups during hemorrhage, and the effect was extremely small and unlikely to be physiologically significant. Glucose levels were also similar between groups at all points during the experiment. Though there was a trend towards lower glucoses in the Post-Resuscitation and Definitive Care phases, these levels did not reach statistical significance.
Table 2.
Physiologic Data (Hematocrit, Lactate, Base Deficit, Glucose) at defined time points in the experimental protocol
| Baseline | Hemorrhage | Post-resuscitation | Definitive Care | |
|---|---|---|---|---|
| Hematocrit (%) | ||||
| PNPH | 37.5±0.4 | 25.8±1.8* | 23.3±1.2* | 27.7±0.6* |
| HEX | 37.4±0.6 | 28.2±0.9* | 23.0±1.2* | 28.2±0.7* |
| LR | 37.9±0.5 | 27.6±1.3* | 22.5±1.0* | 29.6±0.8* |
| Lactate (mmol/L) | ||||
| PNPH | 2.6±0.1 | 2.4±0.2 | 2.6±0.2 | 1.8±0.1*‡ |
| HEX | 2.9±0.2 | 2.6±0.2 | 2.5±0.2 | 1.9±0.1* |
| LR | 2.8±0.2 | 2.7±0.2 | 3.0±0.5 | 1.7±0.1‡ |
| Base Deficit (mmol/L) | ||||
| PNPH | 4.9±0.8 | 7.0±0.8 | 6.2±0.7 | 6.2±0.9 |
| HEX | 4.1±0.4 | 5.7±0.4 | 5.0±0.3 | 4.7±0.4 |
| LR | 4.4±0.4 | 6.2±0.4* | 5.9±0.5 | 5.8±0.4 |
| Glucose (mg/dL) | ||||
| PNPH | 78.3±4.7 | 70.3±9.5 | 53.8±5.7 | 56.5±2.7 |
| HEX | 89.5±4.2 | 81.8±7.4 | 62.4±1.6 | 59.4±2.2 |
| LR | 90.6±10.9 | 92.6±9.1 | 72.3±7.1 | 55.9±2.7*† |
p<0.05 vs Baseline,
p<0.05 vs. Hemorrhage,
p<0.05 vs. Post-resuscitation
Table 3 summarizes pH, PaCO2, and PaO2 values during the study. There was no significant change in pH during the study phases or difference between groups. In the each group, there was a significant rise in PaCO2 between the Hemorrhage and Definitive Care phases and between the Post-Resuscitation time point and the Definitive Care phase. There was a significant decrease in the PaO2 across groups between the Baseline measurements and the Hemorrhage phase. There was also a significant increase in PaO2 in all groups between the Post-Resuscitation time point and the Definitive Care phase values as expected with the increase in FiO2 from 0.21 to 1.0.
Table 3.
Physiologic Data (pH, PaCO2, PaO2) at defined time points in the experimental protocol
| Baseline | Hemorrhage | Post-resuscitation | Definitive Care | |
|---|---|---|---|---|
| pH | ||||
| PNPH | 7.38±0.01 | 7.39±0.02 | 7.40±0.03 | 7.32±0.02 |
| HEX | 7.38±0.01 | 7.40±0.02 | 7.42±0.01 | 7.35±0.01 |
| LR | 7.39±0.01 | 7.40±0.01 | 7.40±0.02 | 7.35±0.01 |
| PaCO2 (torr) | ||||
| PNPH | 31.7±1.1 | 27.0±1.8 | 27.2±1.2 | 35.6±2.0†‡ |
| HEX | 32.9±1.8 | 28.0±2.0 | 27.5±1.2 | 35.2±1.8†‡ |
| LR | 31.9±1.0 | 27.0±1.2 | 28.3±1.3 | 33.3±1.5†‡ |
| PaO2 (torr) | ||||
| PNPH | 164.2±3.3 | 93.0±4.3* | 87.8±4.4*† | 444.9±3.7*†‡ |
| HEX | 161.3±4.8 | 92.7±5.5* | 87.4±5.6*† | 447.2±1.2*†‡ |
| LR | 157.6±3.5 | 97.3±3.1* | 86.8±3.9*† | 446.4±10.7*†‡ |
p<0.05 vs Baseline,
p<0.05 vs. Hemorrhage,
p<0.05 vs. Post-Resuscitation
Neuropathology
Hippocampal sections taken from mice resuscitated with LR and HEX in our combined injury model typically exhibited a focal area of neuronal degeneration in the CA1 region of the hippocampus at 7 d after the insult as reflected by FJC staining (Figure 8). As anticipated, FJC-positive neurons were markedly greater in CA1 than in CA3 in this model (16). However, the number of FJC positive neurons found in CA1 was significantly less in mice resuscitated with PNPH (median 3, range [0–6]) than those resuscitated with HEX (18, [7–48]) or LR (21, [4–34]) (p<0.05). Mice resuscitated with PNPH (0, [0–1]) also had fewer FJC positive neurons in CA3 compared to HEX (12; [0–15]) (PNPH vs. HEX, p<0.05)
Figure 8.

Photomicrographs of representative sections of CA1 stained with Fluoro-Jade C depict degenerating neurons in mice resuscitated with (A) PNPH, (B) HEX, or (C) LR. Scatter plot (D) of number of FJC positive neurons in both CA1 and CA3 for each group with median represented by solid bar. (p<0.05, post hoc Tukey test vs. *HEX, and †LR)
Hippocampal sections taken from mice resuscitated with LR and HEX typically exhibited thinning of the hippocampus as well as a focal area of neuronal death, as noted in H&E-stained sections by the defect in the contour in the CA1 region of the hippocampus (Figure 9). Sections taken from those animals resuscitated with PNPH showed little evidence of neuronal eosinophilia with pyknotic nuclei indicative of the typical injury to CA1 seen in this model. The linear density of neurons in CA1 (Figure 10) did not significantly differ between groups: PNPH median 18.95 cells/0.1 mm [13.08–20.82 cells/0.1mm] vs. HEX 14.23 cells/0.1mm [7.79–17.42 cells/0.1mm] vs. LR 13.58 [8.13–17.97 cells/0.1mm] (p=0.16). There was no significant effect of the resuscitation fluid on neuronal survival in CA3: PNPH 9.34 cells/0.1mm [7.57–9.80 cells/0.1mm] vs. HEX 9.04 cells/0.1mm [8.15–10.04 cells/0.1mm] vs. LR 8.89 cells/0.1mm [7.68–9.74 cells/0.1mm]. The degree of injury seen on H&E staining in HEX and LR groups is comparable to our prior report in this model (16).
Figure 9.
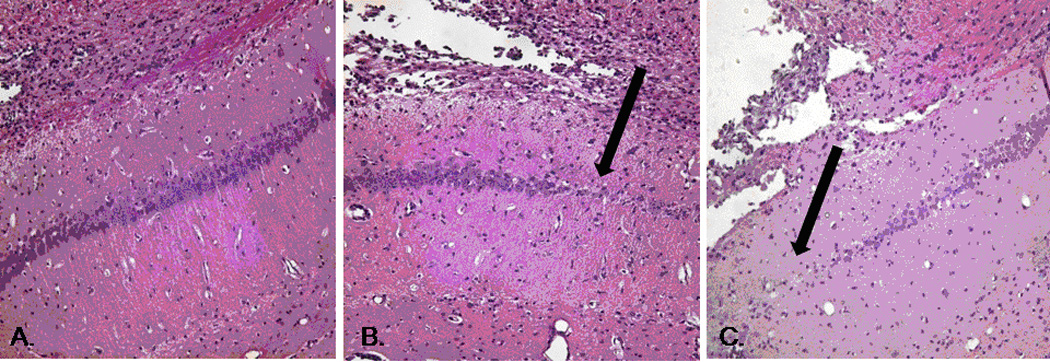
Photomicrographs (20× objective) of Hematoxylin and Eosin stained representative sections of CA1 hippocampus in mice resuscitated with (A) PNPH, (B) HEX, and (C) LR. Dark arrows mark focal deficits seen in multiple sections of mice treated with HEX and LR, but not those treated with PNPH.
Figure 10.

Scatter plot showing density of viable neurons stained with H&E in CA1 and CA3 for each group with median represented by solid bar. There were no significant differences between groups.
DISCUSSION
This study is the first to examine PNPH as a therapeutic HBOC. Based on our work in in vitro models, we report that unlike native Hb, PNPH exhibits a unique lack of neurotoxicity as assessed with multiple assays of cell viability in rat primary neuronal culture, and surprisingly, PNPH is neuroprotectve in in vitro models of both excitotoxicity and TBI (neuronal stretch). In vivo, using an established murine model of TBI plus hemorrhagic hypotension (16, 19), the use of a PNPH solution as a resuscitation fluid, like the colloid HEX, conferred an advantage over LR as being a small volume resuscitation solution, but PNPH also produced a potentially more favorable hemodynamic response than either LR or HEX. Importantly, mirroring the in vitro studies, PNPH produced unique neuroprotection in selectively vulnerable CA1 beneath the contusion in our model. Each of these effects deserves further discussion.
Specific properties of PNPH
The PNPH used in these studies is a covalently modified (polynitroxylated) version of a well-characterized bovine Peg-Hb (Prolong) (20). It has a P50 of 11 and it contains ~14 nitroxide moieties and 8–10 Peg moieties that are covalently linked. It is prepared as a 4% solution. Based on a formulation containing a 4% solution, it is being developed as a small volume, antioxidant, neuroprotective resuscitation fluid, rather than a current generation blood replacement therapy. With regard to in vivo resuscitation, LR, HEX, and PNPH have an osmolarity of 273, 307, and ~308, respectively.
Lack of neurotoxicity of PNPH in vitro
As anticipated, native bovine Hb showed dose-dependent neurotoxicity in our in vitro system (10, 11). Neurotoxicity was blunted by pegylation, although it was still evident at all concentrations for Peg-Hb. In contrast, PNPH was devoid of neurotoxicity across concentrations and assays in our culture system. Regan and Panter (10) reported dose dependent neurotoxicity (LDH release) in a mixed cortical culture containing both neurons and glia exposed to purified human Hb concentrations between 0.025 and 25 µM (based on the tetramer) for 24–48 h. Supporting a key potential role for oxidative injury, neurotoxicity was markedly attenuated by the antioxidant Trolox, the 21-aminosteroid U745000A, or the iron chelators deferoxamine or phenanthroline (10, 21). Oxidative damage to the erythrocyte cell membrane or membrane bound enzymes was suggested to mediate these findings, and iron release from Hb may be important (22). We noted similar results for bovine Hb at concentrations of 0.625 – 12.50 µM (based on the tetramer) at 24 h exposure. Thus, neurotoxicity of Hb may be abrogated by PNPH via its covalently linked nitroxides, which confer SOD mimetic effects and other antioxidant actions. However, based on our data, pegylation also partially blunts the neurotoxicity. Thus other effects such as accessibility or stability of the Hb moiety could also contribute to this favorable effect of PNPH. The unique lack of neurotoxicity of PNPH could be quite important in TBI resuscitation if an HBOC is contemplated as a therapy since blood-brain barrier (BBB) damage and hemorrhage are well-known complications of TBI and thus resuscitation with a conventional cell free Hb could cause direct neurotoxicity locally in the injured brain.
In vitro neuroprotection by PNPH
Based on prior work (11), we did not anticipate dramatic augmentation of glutamate-mediated neuronal death by the Hb preparations that we examined. Minimal increases in LDH release by human Hb were seen in mixed murine neuron and glial cultures exposed to the excitotoxic agent N-methyl-D-aspartate (NMDA) at a 9 µM concentration. Our data with Glu/Gly exposure at 10 µM also suggest only modest augmentation of excitotoxic neuronal death by bovine Hb at the highest concentrations tested. We were, however, surprised that PNPH was neuroprotective against Glu/Gly toxicity at all concentrations and that even Peg-Hb showed some protection. Although the mechanisms underlying the neuroprotective effect of PNPH remain to be defined, oxidative damage is a key facet of excitotoxicity. Relevant to protection by PNPH, excitoxic injury was recently shown to involve a critical role for neuronal membrane bound NADPH oxidase-mediated oxidative damage (23). Since PNPH would be expected to be confined to the extracellular space, this is an attractive target. Redox effects directly on the NMDA receptor could also reduce excitotoxicity (24). Finally, since Peg-Hb conferred intermediate neuroprotection, other effects could be operating. PNPH also showed protection in the in vitro TBI model system as shown again using both LDH and MTT assays. Given our results in the Glu/Gly model, this finding might be anticipated since excitotoxicity plays a major role in mediating neuronal death in the stretch model (18).
Small volume resuscitation effects of PNPH on hemodynamics
PNPH required significantly less volume than LR to achieve target MAP in mice subjected to TBI plus hemorrhage. PNPH required ~1/5th of the volume as LR, while HEX required ~1/3rd the volume as LR. Small volume resuscitation with HEX or HBOCs may be quite effective in the setting of acute hemorrhage after TBI (25–27). However, the ability to consistently maintain the pre-hospital target MAP of 50 mmHg was better with PNPH than with either LR or HEX—even when using this conservative MAP goal. The ability of an HBOC to more rapidly achieve a target MAP of 50 mmHg, however, could be viewed as either beneficial or detrimental. Small volume resuscitation after TBI confers protection against the development of brain edema and intracranial hypertension (4). Better recovery of MAP could be beneficial by more promptly restoring CBF if cerebral perfusion pressure (CPP) is below the autoregulatory limit, or if blood pressure autoregulation of CBF is impaired, which is common after TBI (28). However, in the setting of polytrauma with uncontrolled bleeding, the target MAP may need to be conservative to limit bleeding (29). We chose a MAP of 50 mmHg in the pre-hospital phase to simulate a clinical scenario where ongoing bleeding outside of the brain might limit the ability to attain a “normal” MAP while providing the lowest acceptable MAP for cerebral perfusion. Also, scavenging of NO by an HBOC could lead to inappropriate vasoconstriction that could compromise both systemic recovery from hemorrhage and CBF (7). Our findings, however, argue against this possibility since PNPH did not worsen Post Resuscitation lactate or pH and also produced neuroprotection, unlike either LR or HEX. Potentially favorable properties related to pegylation could play a role in mediating some of the observed hemodynamic effects of PNPH. Pegylation of proteins confers “supercolloid” effects. Theoretically, each strand of PEG binds 132 molecules of water (personal communication, A. Abuchowski), thus each PNPH molecule can potentially bind >1000 water molecules. This would be anticipated to provide significant oncotic advantages to PNPH and Peg-Hb vs either HEX, which is generally felt to have oncotic effects similar to 5% albumin (30, 31), or the crystalloid LR. This could attenuate brain edema while expanding vascular volume. Thus, our data suggest that for a reasonable, albeit conservative, target pre-hospital MAP, resuscitation with PNPH confers important advantages over LR or HEX.
Neuroprotective effects of PNPH in vivo
We observed neuroprotection by PNPH resuscitation at 7 d after injury that was not seen with either LR or HEX. We previously examined resuscitation with LR, HEX, or 3% saline in this model and none of these solutions conferred neuroprotection in CA1 (19). Our current report confirms this finding for both LR and HEX. Several other studies have shown beneficial effects of HBOC-201 based resuscitation on CPP, ICP, brain edema, brain tissue oxygen levels, and neuropathology in pig models of TBI plus hemorrhage (25–27). Unique in our studies in mice is the assessment of neuropathology at a 7 d after injury. Studies in large animal models have focused on very short-term outcomes (generally 6–8 h), and it has long been known that ischemia-induced neuronal death in CA1 can be delayed, often beyond 2 d (32). We observed neuroprotection by PNPH in the hippocampus which is beneath the contusion and may represent a penumbra in the injured hemisphere. It is a region where the BBB is disrupted (33) and where there would be concerns over HBOC extravasation into injured brain. In contrast, prior reports with other HBOCs in large animal models have seen protection limited to sites remote from the impact (27).
The neuroprotection could be partially explained by the improved MAP seen in mice resuscitated with PNPH vs HEX or LR. However, since both PNPH and HEX represented small volume resuscitation solutions in our model but only PNPH conferred neuroprotection, limitation of volume administration alone cannot explain the observed benefit. Nevertheless, the ability of PNPH to promptly and consistently achieve the target pre-hospital MAP of 50 mmHg may prevent a cascade of secondary injury.
PNPH may facilitate oxygen delivery to the hippocampus beneath the contusion. However, the amount of oxygen carrying capacity from ~6 mL/kg of a 4% PNPH solution during resuscitation is modest when compared to the amount of Hb lost via hemorrhage in our model (~30% blood volume). The P50 of 11 (although lower than the normal P50 of ~28 for Hb in erythrocytes) is relatively high for an HBOC (6). The ability of HBOCs to approach the vascular endothelium can yield greater than anticipated oxygen delivery. A 500 mL infusion of the non-nitroxylated parent PEG-Hb (Prolong) formulation used in our studies substantially improved oxygen delivery in a pig model of hemorrhagic shock (20). It is thus possible that some of the neuroprotection afforded by PNPH is related to enhanced cerebral oxygen delivery. We are currently examining hippocampal brain tissue PO2 during PNPH resuscitation.
PNPH also contains ~14 nitroxide moieties that provide antioxidant and SOD mimetic effects. Free nitroxides exhibit neuroprotective effects in experimental TBI. Zhang et al (12) reported a reduction in brain edema and BBB permeability in a rat model of TBI by treatment with Tempol. Deng-Bryant et al (34) reported that Tempol attenuated neurodegeneration after CCI in mice as assessed with silver staining. Leker et al (35) showed benefit from Tempol in experimental stroke. Further supporting a role of the nitroxides of PNPH, a polynitroxylated albumin solution reduced vascular injury in models of ischemia reperfusion (36, 37), conferred neuroprotection in stroke in rats (38), and improved survival in experimental hemorrhage (39). PNPH, by reducing superoxide may limit NO loss to peroxynitrite formation. This would confer a dual benefit—limiting NO consumption and attenuating peroxynitrite production. One might not expect that antioxidant moieties decorating a large molecule like Hb could contribute to neuroprotection in parenchyma. However, our in vitro data argue strongly against that premise. Further studies are needed to define the effects of PNPH on oxidative stress in our models.
Finally, the PNPH used in these studies was a carboxy (CO) Hb PNPH formulation. Carbon monoxide (CO) protects the Hb iron during the synthetic addition of the nitroxide moieties. CO inhalation confers cytoprotection in several model systems, possibly via effects on P38 kinase or oxidative stress (40). Koehler et al (41), in preliminary studies, reported that CO Hb levels increase to ~5% after a 20 mL/kg topload with PNPH and then decrease back to baseline over ~2h in rats. This suggests that PNPH may deliver CO to injured brain early in resuscitation. This would also argue that PNPH is unlikely to confer direct neuroprotection via enhanced oxygen delivery, at least during the pre-hospital phase. It has been shown; however, that unlike inhalation of CO, transfusion of blood with high levels of CO to dogs does not produce toxicity (42). The possible role of CO in mediating the neuroprotective effects of PNPH in TBI merits additional study.
Unique aspects of resuscitation in the setting of TBI plus hemorrhage
Free Hb binding to NO at the endothelium produces vasoconstriction and systemic hypertension (7). Deleterious effects of current generation HBOCs in humans likely result from coronary artery constriction via this mechanism. Studies have investigated the delivery of inhaled NO or other vasodilators during HBOC use to prevent vasoconstriction (43). However after TBI, some vasopressor action may be tolerated or even advantageous. The use of pressors after TBI may minimize resuscitation fluid volume (44). Use of large fluid volumes during resuscitation may exacerbate brain edema. Though the use of pressors in the setting of hemorrhagic shock is controversial, a resuscitation fluid with some intrinsic vasopressor action may limit fluid volumes and brain edema.
Limitations of the study
We recognize that cell culture systems do not include a number of endogenous antioxidant defenses that are present in brain tissue (10). In our in vivo studies, although we showed benefit of PNPH on both hemodynamics and neuroprotection, there are several limitations. We focused on acute hemodynamics and 7 d neuropathology. We are currently assessing other outcomes including brain tissue oxygen, intracranial pressure, BBB, CBF, and behavior. It is also not clear what the optimal target MAP is in TBI plus hemorrhage (45). The chosen target MAP of 50 mmHg is reasonable, but other targets could yield different results. We did not re-infuse shed blood until completion of the pre-hospital phase to maximize clinical relevance and we recognize that the effect of immediate resuscitation with blood was not explored. Effects of volatile anesthetics such as isoflurane could influence the target MAP after TBI and neuroprotection (46), however it is the most commonly used anesthetic in experimental TBI (47). We did not monitor total Hb levels in this exploratory study with PNPH. Currently, using a similar protocol we noted that 20 mL/kg of 4% PNPH does not appreciably increase total Hb levels, suggesting that it is unlikely that use of only ~6–7 mL/kg of PNPH in this study would significantly increase arterial Hb level. This may also be related to plasma volume expansion produced by the Peg moieties. We noted greater neuroprotection assessed by FJC staining than with H&E, which may be related to greater sensitivity of FJC in assessing damage. Blood glucose levels were generally in the low normal range and a hyperglycemic response was not observed. However, glucose levels did not differ between groups. We used room air during the pre-hospital phase. Use of supplemental oxygen could alter the efficacy of an HBOC. Given that this work was supported by the United States Army, we chose to simulate the combat casualty care scenario where oxygen is not used in the field due to tactical factors.
CONCLUSIONS
PNPH is a unique non-neurotoxic therapeutic HBOC with novel neuroprotective effects in both in vitro and in vivo TBI models. Limited fluid resuscitation of TBI plus hemorrhagic hypotension in mice using PNPH offers advantages over LR or HEX including better normalization of MAP, and neuroprotection. The mechanisms underlying the neuroprotection produced by PNPH deserve further exploration.
ACKNOWLEDGEMENTS
Supported by grant PR054755 W81XWH-06-1-0247 from the United States Army (PMK), T-32 HD 040686 (JE), and NS 30318 (CED). We thank Dr. David Meaney at the University of Pennsylvania for assistance in establishing the in vitro neuronal stretch model in our center. We thank Marci Provins for assisting with manuscript preparation.
Dr. Kochanek received funding from the U.S. Army and from NIH. Dr. Du received funding from NIH. Dr. Hsia received U.S. patents and received funding from NIH and the U.S. Department of Defense. Dr. Hsia is also employed by Synzyme Technologies and holds stock ownership in Synzyme Technologies.
Footnotes
Disclosure: Drs. Ma and Hsia are officers at SynZyme Technologies, LLC. None of the other authors have any conflicts to disclose related to this work.
REFERENCES
- 1.Chesnut RM, Marshall SB, Piek J, et al. Early and late systemic hypotension as a frequent and fundamental source of cerebral ischemia following severe brain injury in the Traumatic Coma Data Bank. Acta Neurochir Suppl (Wien) 1993;59:121–125. doi: 10.1007/978-3-7091-9302-0_21. [DOI] [PubMed] [Google Scholar]
- 2.Nelson TJ, Wall DB, Stedje-Larsen ET, et al. Predictors of mortality in close proximity blast injuries during Operation Iraqi Freedom. J Am Coll Surg. 2006;202:418–422. doi: 10.1016/j.jamcollsurg.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 3.Ling G, Bandak F, Armonda R, et al. Explosive blast neurotrauma. J Neurotrauma. 2009;26:815–825. doi: 10.1089/neu.2007.0484. [DOI] [PubMed] [Google Scholar]
- 4.Ramming S, Shackford SR, Zhuang J, et al. The relationship of fluid balance and sodium administration to cerebral edema formation and intracranial pressure in a porcine model of brain injury. J Trauma. 1994;37:705–713. doi: 10.1097/00005373-199411000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Hovda DA, Lee SM, Smith ML, et al. The neurochemical and metabolic cascade following brain injury: moving from animal models to man. J Neurotrauma. 1995;12:903–906. doi: 10.1089/neu.1995.12.903. [DOI] [PubMed] [Google Scholar]
- 6.Natanson C, Kern SJ, Lurie P, et al. Cell-free hemoglobin-based blood substitutes and risk of myocardial infarction and death: a meta-analysis. JAMA. 2008;299:2304–2312. doi: 10.1001/jama.299.19.jrv80007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alayash AI. Hemoglobin-based blood substitutes: oxygen carriers, pressor agents, or oxidants? Nat Biotechnol. 1999;17:545–549. doi: 10.1038/9849. [DOI] [PubMed] [Google Scholar]
- 8.Hlatky R, Lui H, Cherian L, et al. The role of endothelial nitric oxide synthase in the cerebral hemodynamics after controlled cortical impact injury in mice. J Neurotrauma. 2003;20:995–1006. doi: 10.1089/089771503770195849. [DOI] [PubMed] [Google Scholar]
- 9.Winslow RM. Targeted O2 delivery by low-p50 hemoglobin: a new basis for hemoglobin-based oxygen carriers. Artif Cells Blood Substit Immobil Biotechnol. 2005;33:1–12. doi: 10.1081/bio-200046634. [DOI] [PubMed] [Google Scholar]
- 10.Regan RF, Panter SS. Neurotoxicity of hemoglobin in cortical cell culture. Neurosci Lett. 1993;153:219–222. doi: 10.1016/0304-3940(93)90326-g. [DOI] [PubMed] [Google Scholar]
- 11.Regan RF, Panter SS. Hemoglobin potentiates excitotoxic injury in cortical cell culture. J Neurotrauma. 1996;13:223–231. doi: 10.1089/neu.1996.13.223. [DOI] [PubMed] [Google Scholar]
- 12.Zhang R, Shohami E, Beit-Yannai E, et al. Mechanism of brain protection by nitroxide radicals in experimental model of closed-head injury. Free Radic Biol Med. 1998;24:332–340. doi: 10.1016/s0891-5849(97)00267-0. [DOI] [PubMed] [Google Scholar]
- 13.Krishna MC, Samuni A, Taira J, Goldstein S, Mitchell JB, Russo A. Stimulation by Nitroxides of Catalase-like Activity of Hemeproteins. The Journal of Biological chemistry. 1996;271:26018–26025. doi: 10.1074/jbc.271.42.26018. [DOI] [PubMed] [Google Scholar]
- 14.Winslow RM. Cell-fee oxygen carriers: scientific foundations, clinical development, and new directions. Biochim Biophys Acta. 2008;1784:1382–1386. doi: 10.1016/j.bbapap.2008.04.032. [DOI] [PubMed] [Google Scholar]
- 15.Lui FE, Dong P, Kluger R. Polyethylene glycol conjugation enhances the nitrite reductase activity of native and cross-linked hemoglobin. Biochemistry. 2008;47:10773–10780. doi: 10.1021/bi801116k. [DOI] [PubMed] [Google Scholar]
- 16.Dennis AM, Haselkorn ML, Vagni VA, et al. Hemorrhagic shock after experimental traumatic brain injury in mice: effect on neuronal death. J Neurotrauma. 2009;26:889–899. doi: 10.1089/neu.2008.0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chapman KW, Snell SM, Jesse RG, et al. Pilot scale production of pyrogen-free human hemoglobin for research. Biomater Artif Cells Immobilization Biotechnol. 1992;20:415–421. doi: 10.3109/10731199209119661. [DOI] [PubMed] [Google Scholar]
- 18.Spaethling JM, Klein DM, Singh P, Meaney DF. Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J Neurotrauma. 2008;25:1207–1216. doi: 10.1089/neu.2008.0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Exo J, Shellington D, Bayir H, et al. Resuscitation of traumatic brain injury and hemorrhagic shock with polynitroxylated albumin, hextend, hypertonic saline and lactated ringers: Effects on acute hemodynamics, survival, and neuronal death in mice. J Neurotrauma. 2009 Aug 19; doi: 10.1089/neu.2009.0980. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abuchowski A. Clinical applications of pegylated bovine hemoglobin. Proceedings of the XII International Symposium on Blood Substitutes; August 25–28, 2009; Parma, Italy. p. P49. [Google Scholar]
- 21.Regan RF, Rogers B. Delayed treatment of hemoglobin neurotoxicity. J Neurotrauma. 2003;20:111–120. doi: 10.1089/08977150360517236. [DOI] [PubMed] [Google Scholar]
- 22.Rogers B, Yakopson V, Teng ZP, et al. Heme oxygenase-2 knockout neurons are less vulnerable to hemoglobin toxicity. Free Radic Biol Med. 2003;35:872–881. doi: 10.1016/s0891-5849(03)00431-3. [DOI] [PubMed] [Google Scholar]
- 23.Brennan AM, Suh SW, Won SJ, et al. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aizenman E, Lipton SA, Loring RH. Selective modulation of NMDA responses by reduction and oxidation. Neuron. 1989;2:1257–1263. doi: 10.1016/0896-6273(89)90310-3. [DOI] [PubMed] [Google Scholar]
- 25.Patel MB, Feinstein AJ, Saenz AD, et al. Prehospital HBOC-201 after traumatic brain injury and hemorrhagic shock in swine. J Trauma. 2006;61:46–56. doi: 10.1097/01.ta.0000219730.71206.3a. [DOI] [PubMed] [Google Scholar]
- 26.Stern S, Rice J, Philbin N, et al. Resuscitation with the hemoglobin-based oxygen carrier, HBOC-201, in a swine model of severe uncontrolled hemorrhage and traumatic brain injury. Shock. 2009;31:64–79. doi: 10.1097/SHK.0b013e3181778dc3. [DOI] [PubMed] [Google Scholar]
- 27.Rosenthal G, Morabito D, Cohen M, et al. Use of hemoglobin-based oxygen-carrying solution-201 to improve resuscitation parameters and prevent secondary brain injury in a swine model of traumatic brain injury and hemorrhage: laboratory investigation. J Neurosurg. 2008;108:575–587. doi: 10.3171/JNS/2008/108/3/0575. [DOI] [PubMed] [Google Scholar]
- 28.Golding EM, Robertson CS, Bryan RM., Jr The consequences of traumatic brain injury on cerebral blood flow and autoregulation: a review. Clin Exp Hypertens. 1999;21:299–332. doi: 10.3109/10641969909068668. [DOI] [PubMed] [Google Scholar]
- 29.Bickell WH, Wall MJ, Jr, Pepe PE, et al. Immediate versus delayed fluid resuscitation for hypotensive patients with penetrating torso injuries. N Eng J Med. 1994;331:1105–1109. doi: 10.1056/NEJM199410273311701. [DOI] [PubMed] [Google Scholar]
- 30.Moggio RA, Rha CC, Somberg ED, et al. Hemodynamic comparison of albumin and hydroxyethyl starch in postoperative cardiac surgery patients. Crit Care Med. 1983;11:943–945. doi: 10.1097/00003246-198312000-00009. [DOI] [PubMed] [Google Scholar]
- 31.Gold MS, Russo J, Tissot M, et al. Comparison of hetastarch to albumin for perioperative bleeding in patients undergoing abdominal aortic aneurysm surgery. A prospective, randomized study. Ann Surg. 1990;211:482–485. doi: 10.1097/00000658-199004000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deshpande JK, Siesjö BK, Wieloch T. Calcium accumulation and neuronal damage in the rat hippocampus following cerebral ischemia. J Cereb Blood Flow Metab. 1987;7:89–95. doi: 10.1038/jcbfm.1987.13. [DOI] [PubMed] [Google Scholar]
- 33.Whalen MJ, Carlos TM, Wisniewski SR, et al. Effect of neutropenia and granulocyte colony stimulating factor-induced neutrophilia on blood-brain barrier permeability and brain edema after traumatic brain injury in rats. Crit Care Med. 2000;28:3710–3717. doi: 10.1097/00003246-200011000-00029. [DOI] [PubMed] [Google Scholar]
- 34.Deng-Bryant Y, Singh IN, Carrico KM, et al. Neuroprotective effects of tempol, a catylatic scavenger of peroxynitrite-derived free radicals in a mouse traumatic brain injury model. J Cereb Blood Flow Metab. 2008;28:1114–1126. doi: 10.1038/jcbfm.2008.10. [DOI] [PubMed] [Google Scholar]
- 35.Leker RR, Teichner A, Lavie G, et al. The nitroxide antioxidant tempol is cerebroprotective against focal cerebral ischemia in spontaneously hypertensive rats. Exp Neurol. 2002;176:355–363. doi: 10.1006/exnr.2002.7910. [DOI] [PubMed] [Google Scholar]
- 36.Zhang S, Li H, Ma L, et al. Polynitroxyl-albumin (PNA) plus tempol attenuate lung capillary leak elicited by prolonged intestinal ischemia and reperfusion. Free Radic Biol Med. 2000;29:42–50. doi: 10.1016/s0891-5849(00)00295-1. [DOI] [PubMed] [Google Scholar]
- 37.Saetzler RK, Arfors KR, Tuma RF, et al. Polynitroxylated hemoglobin-based oxygen carrier: inhibition of free radical-induced microcirculatory dysfunction. Free Radic Biol Med. 1999;27:1–6. doi: 10.1016/s0891-5849(99)00037-4. [DOI] [PubMed] [Google Scholar]
- 38.Beaulieu C, Busch E, Röther J, et al. Polynitroxyl albumin reduces infarct size in transient focal cerebral ischemia in the rat: potential mechanisms studied by magnetic resonance imaging. J Cereb Blood Flow Metab. 1998;18:1022–1031. doi: 10.1097/00004647-199809000-00012. [DOI] [PubMed] [Google Scholar]
- 39.Kentner R, Safar P, Behringer W, et al. Early antioxidant therapy with Tempol during hemorrhagic shock increases survival in rats. J Trauma. 2002;53:968–977. doi: 10.1097/00005373-200211000-00025. [DOI] [PubMed] [Google Scholar]
- 40.Chung HT, Choi BM, Kwon YG, et al. Interactive relations between nitric oxide (NO) and carbon monoxide (CO): Heme Oxygenase-1/CO pathway is a key modulator in NO-mediated antiapoptosis and anti-inflammation. Methods Enzymol. 2008;441:329–338. doi: 10.1016/S0076-6879(08)01218-4. [DOI] [PubMed] [Google Scholar]
- 41.Koehler RC, Cao S, Zhang J, et al. Effect of cell-free hemoglobin on the cerebral circulation. Proceedings of the XII International Symposium on Blood Substitutes; August 25–28, 2009; Parma, Italy. p. P68. [Google Scholar]
- 42.Kao LW, Nanagas KA. Toxicity associated with carbon monoxide. Clin Lab Med. 2006;26:99–125. doi: 10.1016/j.cll.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 43.Yu B, Raher MJ, Volpato GP, et al. Inhaled nitric oxide enables artificial blood transfusion without hypertension. Circulation. 2008;117:1982–1990. doi: 10.1161/CIRCULATIONAHA.107.729137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dudkiewicz M, Proctor KG. Tissue oxygenation during management of cerebral perfusion pressure with phenylephrine or vasopressin. Crit Care Med. 2008;36:2641–2650. doi: 10.1097/CCM.0b013e3181847af3. [DOI] [PubMed] [Google Scholar]
- 45.DeWitt DS, Prough DS. Blast-induced brain injury and posttraumatic hypotension and hypoxemia. J Neurotrauma. 2009;26:877–887. doi: 10.1089/neu.2007.0439. [DOI] [PubMed] [Google Scholar]
- 46.Statler KD, Alexander H, Vagni V, et al. Comparison of seven anesthetic agents on outcome after experimental traumatic brain injury in adult, male rats. J Neurotrauma. 2006;23:97–108. doi: 10.1089/neu.2006.23.97. [DOI] [PubMed] [Google Scholar]
- 47.Statler KD, Jenkins LW, Dixon CE, et al. The simple model versus the super model: translating experimental traumatic brain injury research to the bedside. J Neurotrauma. 2001;18:1195–1206. doi: 10.1089/089771501317095232. [DOI] [PubMed] [Google Scholar]