

Abstract
Obesity, especially visceral obesity, is associated with insulin resistance and metabolic syndrome. We previously identified the cell surface proteoglycan glypican-4 as differentially expressed in subcutaneous versus visceral white fat depots. Here we show that glypican-4 is released from cells and adipose tissue explants of mice, and that circulating glypican-4 levels correlate with BMI and insulin sensitivity in humans. Furthermore, glypican-4 interacts with the insulin receptor, enhances insulin receptor signaling, and enhances adipocyte differentiation. Conversely, depletion of glypican-4 results in reduced activation of the insulin receptor and prevents adipocyte differentiation in vitro by inhibiting insulin-mediated C/EBPβ phosphorylation. These functions of glypican-4 are independent of its glycosylphosphatidylinositol membrane anchorage, as a nonmembrane–bound mutant of glypican-4 phenocopies the effects of native glypican-4 overexpression. In summary, glypican-4 is a novel circulating insulin sensitizing adipose-derived factor that, unlike other insulin sensitizers, acts directly on the insulin receptor to enhance signaling.
Obesity is the main cause of insulin resistance in humans and, in many individuals, the first step in the development of type 2 diabetes and metabolic syndrome. The adverse metabolic effects of increasing fat mass depend heavily on its anatomical distribution, with visceral white adipose tissue (WAT) driving the development of insulin resistance and associated metabolic diseases (1). In contrast, increased subcutaneous WAT is not associated with insulin resistance, and in some circumstances has even been shown to have protective effects (1,2).
Expansion of adipose tissue is achieved by increased lipid storage in existing adipocytes and de novo differentiation of preadipocytes. Various autocrine, paracrine, and endocrine factors control adipocyte differentiation (3). Among them, insulin is important in regulation of differentiation and lipid accumulation in vitro and in vivo (4). WAT is also an important endocrine organ, secreting various cytokines and hormones (adipokines) regulating whole body metabolism and insulin sensitivity (5–7).
We previously identified a set of developmentally regulated genes that are differentially expressed in subcutaneous and visceral adipose tissue of mice and men (8). Among these, the patterning gene glypican-4 (Gpc4) is not only differentially expressed in these depots, but its expression in human WAT is also highly correlated with BMI and adipose distribution as measured by waist-to-hip ratio (WHR). Gpc4 belongs to a six-member family of glycosylphosphatidylinositol (GPI)-anchored heparan sulfate proteoglycans. Lacking transmembrane and intracellular domains, glypicans function as coreceptors for a variety of growth factors including Wnt, bone morphogenetic proteins (BMPs), fibroblast growth factors, and Hedgehog (9–11). Little is known about the signaling functions of Gpc4. Mammalian Gpc4 has been reported to bind to fibroblast growth factor 2 via its heparan sulfate chains in neuronal cells and to function as a low-affinity receptor for endostatin (12,13). The role of Gpc4 in adipocytes and its relationship to metabolic regulation remain unknown.
In this study, we demonstrate that Gpc4 is important for adipocyte differentiation by interacting with and regulating insulin receptor activation and its downstream signaling. This interaction is preserved in a soluble nonmembrane–anchored mutant of Gpc4. Furthermore, we provide evidence that Gpc4 is released from adipose tissue, and that serum Gpc4 is a marker for BMI and insulin sensitivity in mice and human. Thus, Gpc4 can serve as a novel adipokine being released from adipose tissue with the ability to enhance insulin sensitivity.
RESEARCH DESIGN AND METHODS
Human subjects.
Paired samples of visceral and subcutaneous adipose tissue were obtained from 160 subjects as previously described (8). All subjects gave written informed consent before taking part in the study.
Mice.
All protocols were approved by the Institutional Animal Care and Use Committee of the Joslin Diabetes Center and in accordance with National Institutes of Health (NIH) guidelines. Mice (Jackson Laboratory) were maintained on a 12-h light/dark cycle and fed a chow diet (9F5020; PharmaServ) or high-fat diet (HFD) (OpenSource Diet D12492, Research Diet).
Constructs.
Gpc4 cDNA clones were obtained from Open Biosystems. An HA-tag was inserted after the signal peptide for native Gpc4, and the cDNA was cloned into the pCDH-puro lentiviral vector (Systems Biosciences). Gpc4Δ529SAG531::HHHHHH (ΔGpc4) was created by site-directed mutagenesis (Stratagene) using the primers fwd:CGAGAAAGCTGACCACCATCACCATCACCATGGTGCCCATGCAG rev:CTGCATGGGCACCATGGTGATGGTGATGGTG-GTCAGCTTTCTCG. A 6xHis-tag was inserted at the N-terminus after the signal peptide and cloned into the pCDH-puro vector. All constructs were sequence verified. Short hairpin RNA (shRNA) lentiviral vectors (pLKO.1) were obtained from Open Biosystems. shGpc4 shRNA was targeted against the sequence GCCACTGGTTTAAGCAATGTT. A scrambled shRNA (shScr) targeting the sequence AGGTTAAGTCGCCCTCG served as control.
Oligonucleotide pull-down assays.
Pull downs were performed as previously described (14).
Cell culture.
3T3-L1 cells were cultured in Dulbecco’s modified Eagle’s medium 4.5 g/L glucose, 10% FBS and 2.5 μg/mL puromycin. Differentiation was induced with 170 nmol/L insulin, 500 µmol/L isobutylmethylxanthine, 400 ng/mL dexamethasone with or without 1 µmol/L troglitazone. Oil Red O staining was performed as previously described (14). Lentiviruses were produced in 293FT cells using the packaging plasmids psPAX2 and pMD2.G.
Quantitative real time PCR.
cDNA synthesis and quantitative real-time PCR (qPCR) were performed as previously described (1). Relative expression levels were calculated by the ΔΔCt method using TBP as reference. The primers used are described in references 8 and 14.
Western blots.
Cells were lysed in 150 mmol/L NaCl, 50 mmol/L Tris-HCl (pH 7.4), 1 mmol/L EDTA, and 1% Triton X-100 with protease and phosphatase inhibitors (Sigma). The following antibodies were used: HRP-Actin (Santa Cruz Biotechnology), pTyrosine (4G10), pIRS-1Y896 (Biosource), pIRSY612 (Invitrogen), insulin receptor substrate 1 (IRS-1) (BD Biosciences), pC/EBPβThr188, C/EBPβ, C/EBPδ, pAktS473, Akt, pERK, Erk, and IRβ (all Cell Signaling Technology). The Gpc4 antibody was raised against the peptide: EVRRLYVSKGFNKNDAPLYE (aa 32–52) in rabbits and affinity purified against the peptide.
Immunoprecipitations.
Protein lysates were incubated with mouse insulin receptor antibody (Cell Signaling Technology) overnight. Coimmunoprecipitation was performed using magnetic protein-A microbeads and µ Columns (Miltenyi Biotec). For the quantification of insulin receptor phosphorylation, insulin receptor was precipitated using protein A/G-agarose (Santa Cruz Biotechnology).
ELISA.
Serum Gpc4 was assessed by ELISA (USCNK Life Science), using 50 μL murine or human serum according to the manufacturer’s recommendation.
ΔGpc4 purification.
ΔGpc4 was purified from conditioned Opti-MEM (Invitrogen) of ΔGpc4 overexpressing 3T3-L1 cells. Medium from shScr cells was used as control. After 48 h, 400 mL medium was pooled and concentrated to 50 mL, dialyzed against PBS/10% glycerol and incubated with 500 μL Ni-NTA agarose (Qiagen) overnight. ΔGpc4 was eluted in 300 mmol/L NaCl, 50 mmol/L NaH3PO4, 10 mmol/L imidazole, and 0.05% Tween (pH 8.0) containing 250 mmol/L imidazole. Eluates were dialyzed overnight to PBS/10% glycerol and concentrated with Centricon filters to 150 μL.
Serum proteoglycan purification.
Anion exchange chromatography was performed as described (15), dialyzed against PBS/10% glycerol, concentrated using Centricon filters (Millipore) to 50 μL, and analyzed by SDS-PAGE.
Mass spectrometry.
Serum proteoglycan preparations from five 4-month-old male C57BL/6 mice were reduced and denatured in buffer containing 2.5% β-mercaptoethanol and resolved on 4–12% gradient acrylamide gels (Invitrogen). Gels were stained with Safestain (Invitrogen), and the gel fragment between 30 and 75 kDa was submitted for mass spectrometric analysis to the Joslin Proteomics Core.
Insulin binding assay.
125I Insulin (MP Biomedicals) binding to adherent cells was measured as previously described (16).
Statistical analysis.
Statistical analysis was performed using GraphPad Prism and presented as mean ± SEM. Significance was tested with unpaired t test, one-way or two-way ANOVA. A P value < 0.05 was considered significant. Multivariate regression analysis was performed using StatView.
RESULTS
Gpc4 expression in fat of humans correlates with body fat content and insulin sensitivity.
We previously showed that Gpc4 is differentially expressed between visceral and subcutaneous fat in rodents and humans, and that expression in adipose tissue of humans is strongly correlated with BMI and WHR (8). Further analysis revealed that Gpc4 expression in subcutaneous fat was markedly decreased in both males and females when comparing lean (BMI <25) to overweight (BMI 25–30) and obese (BMI >30) subjects (Fig. 1A). In contrast, expression of Gpc4 in visceral fat was increased in overweight and obese males and females. When grouped by BMI, Gpc4 expression in visceral adipose tissue was highest in overweight subjects with high visceral fat, defined by a CT or MRI ratio between subcutaneous and visceral fat areas >0.4. Interestingly, in both females and males, this relationship was bell-shaped, with the highest levels of Gpc4 expression in overweight individuals with a visceral fat distribution and lower levels in individuals with frank visceral obesity, who expressed Gpc4 at almost the same levels as lean individuals.
FIG. 1.

Gpc4 is differentially regulated in subcutaneous and visceral WAT upon weight gain. A: Gpc4 expression in subcutaneous (SCW) and visceral (Visc.) fat of 77 female and 83 male nondiabetic subjects, ranging from lean to obese, grouped by BMI. Visc. BMI 25–30 and visc. BMI >30 indicates subjects with a CT or MRI ratio between subcutaneous and visceral fat areas >0.4 in the given BMI range. B: Western blot for Gpc4 from 6-week-old C57BL/6 male mice. Actin is used as loading control. C: qPCR for Gpc4 from the indicated fat depots of C57BL/6 mice fed an HFD for 8 weeks, db/db and control mice. Control mice are C57BL/6 chow diet–fed mice and db/+ mice combined (HFD, n = 4; db/db, n = 6; controls, n = 4–6). BAT, brown adipose tissue; PGF, perigonadal fat; SCF, subcutaneous flank fat. *P < 0.05; **P < 0.01; ***P < 0.001.
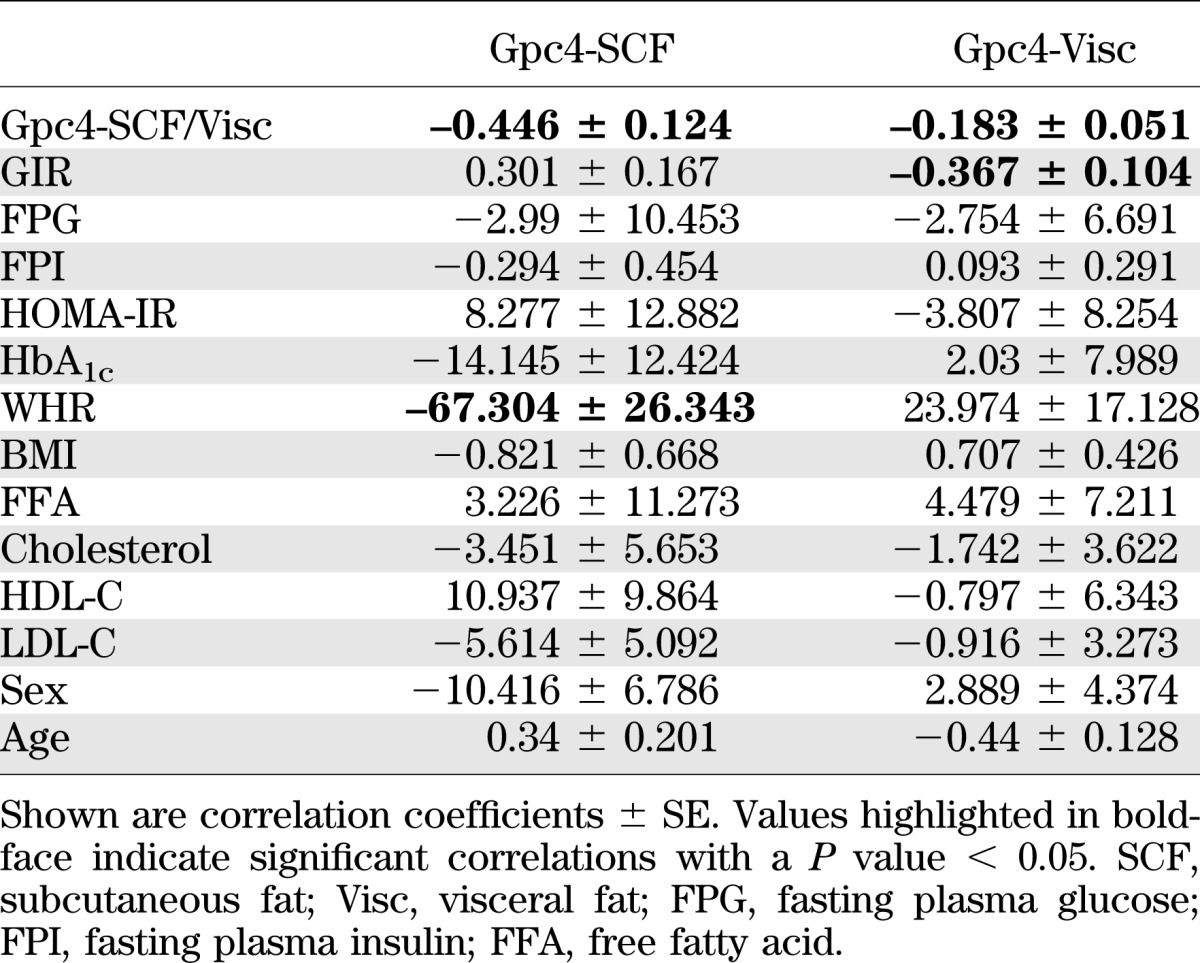
Multiple clinical parameters differed between these groups (Supplementary Table 1). Therefore, we performed multivariate analysis of Gpc4 expression in visceral and subcutaneous fat versus 14 different clinical parameters, which revealed a significant negative correlation of subcutaneous Gpc4 expression with WHR, and a negative correlation of Gpc4 expression in visceral fat with glucose infusion rate (GIR) during euglycemic hyperinsulinemic clamps (Table 1). These correlations were independent from the association of Gpc4 expression with body fat content and distribution, suggesting a link between Gpc4 expression and insulin sensitivity.
TABLE 1.
Multivariate regression analysis of Gpc4 WAT expression with clinical parameters
Gpc4 expression in fat of rodents at the mRNA and protein level.
We have previously shown that in mice Gpc4 mRNA expression is twofold higher in perigonadal than in subcutaneous fat (8). To better understand Gpc4 physiology in the rodent, we raised a peptide antibody against murine Gpc4 and used this to assess Gpc4 protein levels in tissues and serum of mice. As expected, Western blots of extracts from 3T3-L1 preadipocytes that are run under nonreducing conditions for native Gpc4 revealed a broad smear from ∼100 to >170 kDa, representing the 63-kDa core protein with the attached heparan sulfate chains of varying lengths (Supplementary Fig. 1). As previously described, the core protein of Gpc4 undergoes furin-mediated cleavage, creating two disulfide-linked subunits of Gpc4 (11). Thus, when these same extracts were run under reducing conditions, we detected the proteolytically cleaved N-terminal α-subunit of Gpc4 as a sharp band at 37 kDa, allowing more precise quantitation (Supplementary Fig. 1).
Using this assay, we found that the difference in expression of Gpc4 between the murine fat depots was even more marked at the protein than at the mRNA level, and that perigonadal fat had ∼fivefold higher Gpc4 levels than subcutaneous and brown adipose tissue (Fig. 1B). As in humans, Gpc4 expression in perigonadal fat of mice showed a bell-shaped relationship with level of obesity with upregulation of Gpc4 expression in mice with mild obesity due to an HFD, and lower levels in the very obese db/db mice. In subcutaneous fat, Gpc4 expression was also increased in mice fed an HFD and increased even further in db/db mice in this depot. This regulation by obesity state was specific to WAT with no change in Gpc4 in brown adipocyte tissue (BAT) in either the HFD or db/db mice (Fig. 1C).
Role of Gpc4 in adipocyte differentiation and insulin signaling.
To better understand the functional link between Gpc4 and adipogenesis, we created 3T3-L1 preadipocytes with stable knockdown of Gpc4 using lentivirally expressed shRNA (shGpc4). This resulted in a >95% depletion of Gpc4 mRNA (Fig. 2A) and a reduction of Gpc4 protein below the limits of detection when compared with control cells infected with scrambled shRNA (shScr) (Fig. 2B). The control 3T3-L1 cells differentiated efficiently into adipocytes within eight days after induction as visualized by Oil Red O (Fig. 2C). In contrast, Gpc4 knockdown cells failed to accumulate lipids. Furthermore, although stimulation by thiazolidinediones enhanced the differentiation of control cells, this had no significant effect on shGpc4 cells (Fig. 2C).
FIG. 2.

Gpc4 is essential for adipocyte differentiation. A: qPCR for Gpc4 from shGpc4 and control 3T3-L1 cells (n = 9). B: Western blot for Gpc4 and actin as loading control, from control and shGpc4 3T3-L1 preadipocytes. C: Oil Red O staining of shScr and shGpc4 cells at day 8 of differentiation with or without troglitazone (TZD). D: qPCR for key transcription factors of adipocyte differentiation during 8 days of differentiation (n = 9). E: Western blots from nuclear extracts of shScr and shGpc4 cells 24 h after induction of differentiation. F: Quantification of phospho-C/EBPβ on Thr188 normalized to total C/EBPβ (n = 3), 24 h after induction. G: Western blots from oligonucleotide pull downs with a wild-type C/EBP binding motif (wt) or a mutant that is not bound by C/EBPβ as control (mut) 24 h after induction of differentiation. **P < 0.01; ***P < 0.001; ****P < 0.0001. (A high-quality color representation of this figure is available in the online issue.)
Failure to accumulate lipids was due to a blockade in differentiation. qPCR revealed that Gpc4 knockdown cells induced early adipogenic markers C/EBPβ and C/EBPδ at levels comparable with control. By contrast, treatment of knockdown cells with an induction cocktail did not induce the key downstream transcription factors for adipogenesis C/EBPα and peroxisome proliferator–activated receptor γ (PPARγ), which were robustly increased in control cells (Fig. 2D) (17). Western blots from nuclear extracts 24 h after induction confirmed similar protein levels of C/EBPβ and C/EBPδ between control and knockdown cells (Fig. 2E); however, the important regulatory phosphorylation of C/EBPβ Thr188 was reduced by 54% in Gpc4 knockdown cells compared with controls (Fig. 2E and F). Pull downs from nuclear lysates from these cells with oligonucleotides containing a C/EBP binding site revealed similar binding of C/EBPβ from control and shGpc4 cells; however, the bound C/EBPβ from Gpc4 knockdown cells showed greatly reduced Thr188 phosphorylation, indicating diminished activation of this key transcription factor (Fig. 2G). In addition to its role as activator of C/EBPα and PPARγ transcription, C/EBPβ is essential for clonal expansion in 3T3-L1 preadipocytes (18), and consistent with the diminished phosphorylation/activation of C/EBPβ, we also observed reduced mitotic clonal expansion in knockdown cells (Supplementary Fig. 2A).
Phosphorylation of C/EBPβ on Thr188 is mediated by MAPK and PI3-kinase signaling (19). Assessment of the phosphorylation/activation of ERK and Akt during the first 49 h of differentiation revealed a tendency for lower AktS473 phosphorylation, but no alterations of ERK phosphorylation (Supplementary Fig. 2B). Phosphorylation of IRS-1 on Y612 and Y896, sites required for insulin-mediated Akt and ERK activation, showed reduced phosphorylation, suggesting an effect of Gpc4 deletion on insulin signaling (Supplementary Fig. 2C).
Insulin stimulation of 3T3-L1 preadipocytes revealed 33% reduction in insulin receptor and reduced IGF-1 receptor (IGF1R) phosphorylation of Gpc4 knockdown cells compared with control (Fig. 3A and B). The reduced IR/IGF1R activation resulted in a reduction of IRS-1 phosphorylation and a 40–45% reduction in ERK activation (P < 0.01) and phosphorylation of Akt on Ser473 (P < 0.001) in Gpc4 knockdown cells (Fig. 3C and D). This was not caused by reduced insulin binding, as shGpc4 preadipocytes showed higher binding of the 125I insulin tracer, but lower affinity as judged by a rightward shift of the competition curve by unlabeled insulin (Supplementary Fig. 3A). Furthermore, AktS473 phosphorylation declined more rapidly in the Gpc4 knockdown cells during the 60 min time course (Fig. 3D), resulting in a ∼50% reduction of AktS473 phosphorylation over the time course in Gpc4 knockdown cells as quantified by the area under the curve (Fig. 3E). This decreased AktS473 and ERK phosphorylation in Gpc4-depleted cells was observed in a wide range of insulin concentrations (Supplementary Fig. 3B). However, these changes were specific to insulin and not observed after stimulation with 10% FBS (Supplementary Fig. 3C).
FIG. 3.

Gpc4 regulates insulin receptor activation and downstream signaling. A: Western blots from insulin- and IGF1R β−subunit immunoprecipitations of confluent shScr and shGpc4 preadipocytes, blotted for insulin/IGF1R β and pTyrosine before and after 5 min of 10 nmol/L insulin stimulation. B: Quantification of tyrosine phosphorylated insulin receptor in 3T3-L1 preadipocytes, normalized to total insulin receptor levels (n = 6). C: Western blots of confluent shScr and shGpc4 preadipocytes from total cell lysates before and after 5-min stimulation with 10 nmol/L insulin. D: Quantification of ERK and AktS473 phosphorylation at 0, 5, 10, 20, 40, and 60 min after insulin stimulation. pERK and pAktS473 were normalized to total ERK and Akt levels (n = 8). E: Area under the curve of AktS473 phosphorylation shown in D. F: Coimmunoprecipitation of Gpc4 with insulin and IGF1R β-subunit in 3T3-L1 cells. For all stimulation experiments, confluent undifferentiated preadipocytes were serum-starved for 3 h and stimulated with 10 nmol/L insulin. **P < 0.01; ***P < 0.001. (A high-quality color representation of this figure is available in the online issue.)
Gpc4 interacts with the insulin receptor and enhances adipocyte differentiation independent of membrane anchorage.
Gpc4 does not possess transmembrane or intracellular domains but is anchored to the cell membrane via a GPI anchor. Thus, Gpc4 itself cannot signal, but mediates its intracellular functions via interaction with other transmembrane proteins. Because depletion of Gpc4 resulted in reduced insulin/IGF1R activation (Fig. 3A and B), we tested for a possible interaction of Gpc4 with these receptors by performing coimmunoprecipitation experiments. This revealed coimmunoprecipitation of Gpc4 with the insulin receptor under basal growth conditions, which was lost upon insulin stimulation, indicating that Gpc4 interacts with the unoccupied insulin receptor, but dissociates upon insulin binding and receptor activation. Interestingly, interaction with the IGF1R showed a reciprocal pattern, as Gpc4 associated with the IGF1R after, but not prior to, insulin stimulation (Fig. 3F).
WAT is an endocrine organ secreting various adipokines, regulating metabolic function and glucose homeostasis (5). Glypicans can be released from the cell surface by cleavage of the GPI anchor (20). To determine if Gpc4 is released from adipocytes and acts as a soluble modulator of insulin signaling, we created 3T3-L1 cell lines with stable overexpression of native Gpc4 and a soluble mutant form of Gpc4 lacking the GPI anchor attachment site (ΔGpc4). Western blots confirmed moderate overexpression of native Gpc4 and ΔGpc4 (Fig. 4A). Analysis of conditioned medium confirmed Gpc4 protein in the medium of ΔGpc4 cells, as well as smaller amounts of Gpc4 in the medium of control and cells overexpressing wild-type Gpc4, demonstrating that endogenous Gpc4 is released from the cell surface to the medium (Fig. 4B). Overexpression of Gpc4 or ΔGpc4 opposed the results of Gpc4 depletion during adipocyte differentiation with slightly increased PPARγ and C/EBPα expression and C/EBPβ phosphorylation compared with control cells (Supplementary Fig. 4A and B). This led to an increased adipocyte differentiation when compared with control cells (Fig. 4C). Interestingly, overexpression of ΔGpc4 also resulted in enhanced adipocyte differentiation, indicating that membrane anchorage is not required for the pro-adipogenic effect of Gpc4. Expression of perilipin and Glut4, both markers of mature adipocytes, were also significantly increased after differentiation of ΔGpc4 cells and trended toward increasing expression in Gpc4 overexpressing cells (Fig. 4D).
FIG. 4.

Overexpression of Gpc4 enhances adipocyte differentiation and insulin signaling. A: Western blot for Gpc4 of 3T3-L1 stably infected with control lentivirus, native Gpc4, shGpc4, or ΔGpc4. In the ΔGpc4 mutant, the GPI attachment motif 529SAG531 was replaced with a 6xHis-tag. Actin was used as loading control. B: Western blot for Gpc4 from serum-free Opti-MEM conditioned for 24 h by the indicated cell lines. C: Oil Red O staining and brightfield images from control, Gpc4, and ΔGpc4 expressing cells taken at day 8 of differentiation. D: qPCR for Glut4 and perilipin during an 8-day time course of differentiation of control, Gpc4, and ΔGpc4 overexpressing cells. **Indicates significantly higher expression in ΔGpc4 versus control cells (n = 5). E: Ni-NTA pull downs of His-tagged ΔGpc4 from total cell lysates during normal growth conditions or after 5 min of 10 nmol/L insulin stimulation. F: Quantification of ERK and AktS473 phosphorylation at 0, 5, 10, 20, 40, and 60 min after 10 nmol/L insulin stimulation of confluent 3T3-L1 preadipocytes. pERK and pAktS473 were normalized to total ERK and Akt levels (n = 3). G: Western blot for Gpc4 of purified ΔGpc4 and control eluate. H: Insulin stimulation in presence or absence of purified recombinant ΔGpc4. Cells were pretreated with ΔGpc4 or control eluate during the 1-h serum starvation before 10 nmol/L insulin stimulation. All samples were run on one SDS gel; time points were separated for better visualization. *P < 0.05; **P < 0.01. (A high-quality color representation of this figure is available in the online issue.)
To determine if soluble ΔGpc4 could interact with the insulin receptor, we pulled down His-tagged-ΔGpc4 using Ni-NTA-agarose from cell lysates with or without insulin stimulation (Fig. 4E). Similarly to endogenous membrane-anchored Gpc4, the insulin receptor coprecipitated with ΔGpc4 under basal conditions, but this interaction was lost upon insulin stimulation. Interestingly, we did not pull down ΔGpc4 after insulin stimulation, indicating that not only is Gpc4 binding to the insulin receptor abolished upon insulin stimulation, but also the sequestration of ΔGpc4 to the cell surface is lost.
Depletion of Gpc4 resulted in reduced insulin signaling. Overexpression of native Gpc4 or ΔGpc4 enhanced insulin-stimulated ERK (100 and 67%, respectively) and Akt-Ser473 (140 and 94%, respectively) peak phosphorylation (Fig. 4F) and ΔGpc4 increased 2-deoxy glucose uptake by cells (Supplementary Fig. 4C). Furthermore, when 3T3-L1 cells were pretreated with affinity-purified ΔGpc4 or control eluate during serum starvation (Fig. 4G), ΔGpc4 enhanced ERK, Akt, and IRS-1Y896 phosphorylation, after stimulation with insulin (Fig. 4H).
Gpc4 is released from adipose tissue and is a circulating marker for BMI and insulin resistance.
To determine whether Gpc4 can be released from adipocytes into the circulation, we separated adipocytes from the stromal vascular fraction (SVF) of subcutaneous, perigonadal, and brown fat, cultured them in vitro, and assayed the media for Gpc4 by Western blotting. The release of Gpc4 from intra-abdominal (perigonadal) adipocytes was greater than that of subcutaneous adipocytes, and there was no release from either SVF or brown adipocytes (Fig. 5A). Gpc4 mRNA expression was also significantly higher in isolated perigonadal adipocytes compared with the corresponding SVF (Supplementary Fig. 5). To determine whether Gpc4 is also released in vivo, we purified glycoproteins from mouse serum and assayed these samples by Western blotting for Gpc4. As shown in Fig. 5B, we detected Gpc4 in sera from both male and female C57BL/6 mice. Mass spectrometric analysis confirmed this with three tryptic peptides for Gpc4 (Supplementary Fig. 6A). ELISA assays for Gpc4 revealed circulating levels of around 2 ng/mL in lean C57BL/6 and ob/+ mice, which increased to ∼4 ng/mL in mice subjected to 8 weeks of HFD feeding, mirroring the gene expression data. Serum Gpc4 levels were ∼1 ng/mL in the markedly obese ob/ob mice (Fig. 5C). Random-fed blood glucose and insulin measurements revealed that HFD-fed mice were still able to maintain normal glycemia and normal insulinemia, with much higher serum Gpc4 levels than controls, whereas ob/ob mice had elevated blood glucose levels despite hyperinsulinemia, which was accompanied with reduced serum Gpc4 levels (Supplementary Fig. 6B).
FIG. 5.

Gpc4 is released from adipocytes and correlates with markers of body fat and insulin resistance. A: Western blot for Gpc4 from conditioned serum-free Opti-MEMI of cultured isolated subcutaneous, perigonadal, and brown adipocytes and the corresponding SVF. Ponceau-S staining shows equal loading of proteins. Cells were isolated by collagenase digest and medium was conditioned for 12 h. B: Western blot of serum Gpc4. Glycoproteins from serum of 4-month-old C57BL/6 male and female mice were purified using anion exchange chromatography. Western blots from concentrated eluates were probed for Gpc4. C: Gpc4 ELISA from serum of C57BL/6 mice fed an HFD for 8 weeks, ob/ob and control mice. Control mice are C57BL/6 chow diet–fed mice and ob/+ mice combined (n = 6 per genotype). D: Gpc4 ELISA from serum of nondiabetic females (n = 77) and males (n = 83) grouped according to BMI and body fat distribution. Visceral overweight and obesity is defined by a CT or MRI ratio >0.4 between subcutaneous and visceral fat areas. E: Comparison of BMI, WHR, and GIR during a euglycemic hyperinsulinemic clamp and HOMA-IR of the lowest and highest quartile of serum Gpc4 levels of females and males (n = 19 and 20 per quartile, respectively). F: Comparison of GIR from nonobese (BMI <30) and obese (BMI >30) subjects divided into groups with low serum Gpc4 levels (≤5 ng/mL) and high serum Gpc4 levels (≥9 ng/mL). G: Serum Gpc4 levels in 30 obese age-, sex-, and BMI-matched insulin-sensitive and insulin-resistant subjects. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
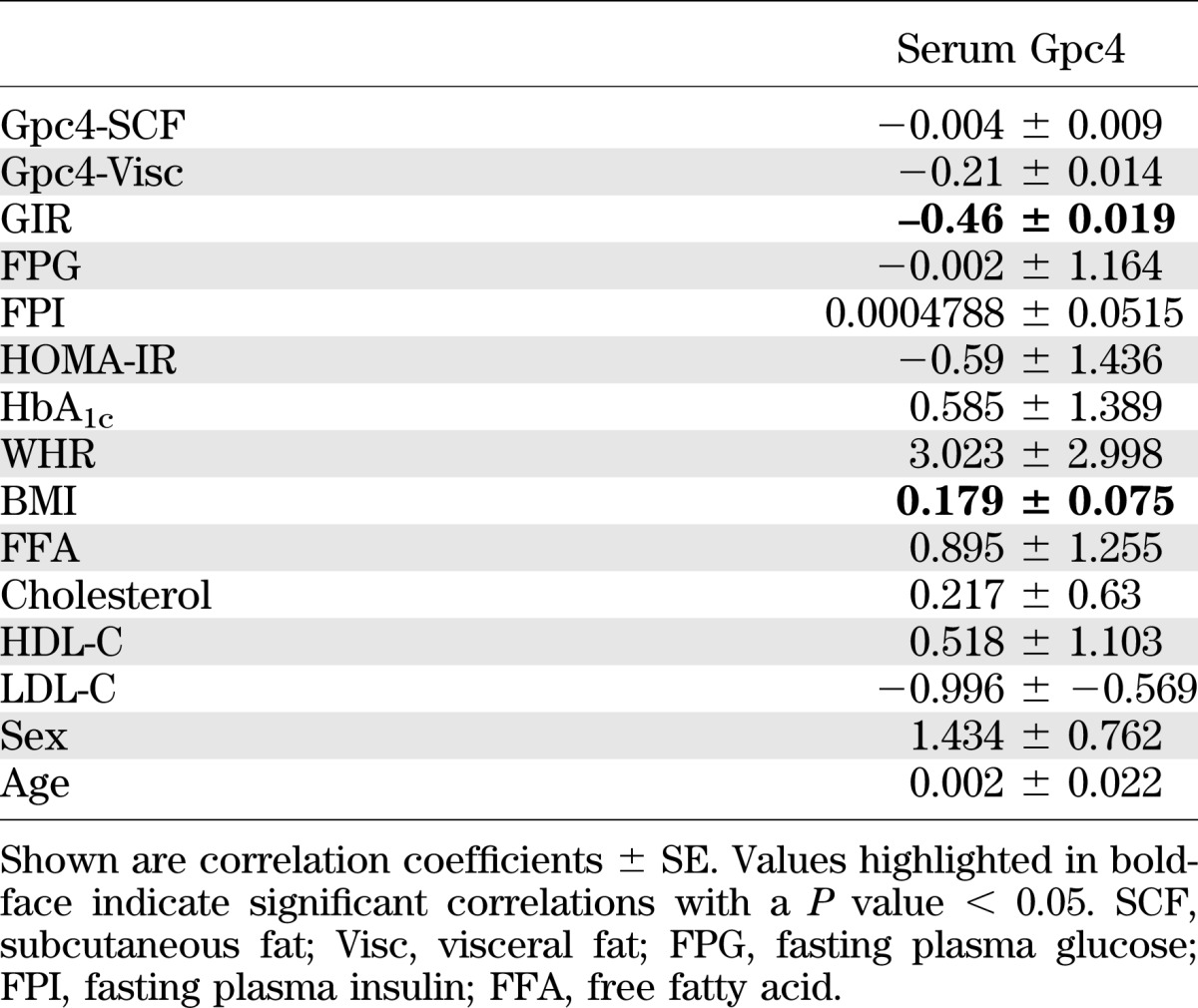
To determine whether Gpc4 was circulating in humans, we used a human Gpc4 ELISA assay to assess serum Gpc4 levels in the same cohort that had been used for expression analysis of Gpc4 mRNA in adipose. In males, serum Gpc4 levels paralleled the gene expression data from visceral fat (Fig. 5D), with the highest serum Gpc4 levels in individuals who were overweight with a visceral distribution and lower levels in both lean and viscerally obese subjects. By contrast, females showed a continuous increase in serum Gpc4 levels from lean to overweight and obese. When both male and female subjects were divided into the lowest and highest quartile of serum Gpc4 levels, those individuals with highest serum Gpc4 had significantly higher percentage body fat, higher BMI, larger WHR, and higher levels of free fatty acids and leptin, all markers of body fat content. Additionally, high serum Gpc4 was associated with increased markers of insulin resistance, including high homeostasis model assessment of insulin resistance (HOMA-IR), high fasting plasma insulin, and insulin resistance as assessed by decreased GIR (Fig. 5E and Supplementary Fig. 6C). We did not observe any association with fasting plasma glucose, cholesterol, HDL-C, LDL-C, or serum adiponectin, although in this group of nondiabetics, those with high serum Gpc4 did have significantly higher HbA1c values, although still within the normal range (Supplementary Fig. 6C). Multivariate analysis of 15 parameters including Gpc4 expression in subcutaneous and visceral fat confirmed a positive correlation of BMI and a negative correlation of GIR with serum Gpc4 levels (Table 2 and Supplementary Fig. 7A). When subjects were divided into subgroups of nonobese and obese subjects with either low serum Gpc4 (≤5 ng/mL) or high serum Gpc4 (≥9 ng/mL), nonobese subjects with high serum Gpc4 levels showed the same degree of insulin resistance, measured by fasting plasma insulin, GIR, and HOMA-IR, as obese subjects with either low or high serum Gpc4 levels (Fig. 5F and Supplementary Fig. 7B). In an independent set of 30 age-, sex-, and BMI-matched obese insulin-sensitive and insulin-resistant patients (21), we observed ∼2 times higher sGpc4 levels in insulin-resistant compared with insulin-sensitive patients (Fig. 5G).
TABLE 2.
Multivariate regression analysis of serum Gpc4 with clinical parameters and Gpc4 expression in WAT
DISCUSSION
Glypican-4 belongs to the family of GPI-anchored heparan sulfate proteoglycans, which includes six members in mammals (10). We previously found that Gpc4 is differentially expressed between fat depots and is highly regulated in obesity (8). We now show that Gpc4 regulates insulin signaling via interaction with the insulin receptor. As a result, reducing levels of Gpc4 diminishes insulin signaling. In preadipocytes, this results in blunted activation of C/EBPβ and a block in adipocyte differentiation. We also demonstrate that Gpc4 is released from adipose tissue and that circulating Gpc4 in rodents and humans positively correlates with body fat content and insulin resistance.
Expansion of visceral adipose tissue, i.e., central obesity, is associated with insulin resistance, whereas expansion of subcutaneous adipose tissue, i.e., peripheral obesity, is not (5,7). Defining the mechanisms underlying body fat distribution and this differential link to insulin resistance is important for understanding the development of comorbidities associated with obesity, including type 2 diabetes, stroke, hypertension, and cardiovascular disease (22). We find that expression of Gpc4 is not only differential between subcutaneous and visceral fat, but that Gpc4 expression in visceral adipose positively correlates with both BMI and, independently, with insulin resistance as measured by euglycemic hyperinsulinemic clamps. Of greater significance, Gpc4 is present in serum of mice and humans, and serum Gpc4 levels are positively correlated with body fat content and insulin resistance. In nondiabetics, serum Gpc4 increases progressively with BMI, especially in viscerally obese women and viscerally overweight men. Multivariate analysis revealed an independent negative correlation of serum Gpc4 with GIR, i.e., higher serum Gpc4 levels are associated with greater insulin resistance. Indeed, nonobese subjects (BMI <30) with high serum Gpc4 (≥9 ng/mL) levels have the same degree of insulin resistance by euglycemic clamp, fasting insulin, and HOMA-IR as obese subjects, independent of serum Gpc4 levels. Furthermore, sGpc4 levels are doubled in insulin-resistant obese subjects compared with age-, sex-, and BMI-matched insulin-sensitive subjects. Thus, serum Gpc4 is not only a marker for BMI, but it is also an independent marker of insulin resistance.
This link between Gpc4 and changes in insulin sensitivity appear to involve two novel mechanisms. First, glypicans are released from the cell surface by an enzymatically regulated process mediated by GPI-lipases. Glycosylphosphatidylinositol-specific phospholipase D (GPLD1) has been suggested to cleave Gpc4 (20,23) and its activity is regulated by insulin (24,25). Similar to Gpc4, GPLD1 levels in serum are increased upon feeding a high-sucrose diet (26), but are decreased in ob/ob mice (27). This could explain the lack of direct correlation between expression of Gpc4 in fat and serum Gpc4 levels. We find no change in GPLD1 expression in adipose tissue of ob/ob mice, but increased expression of another GPI lipase, Notum, increased GPLD1 levels (Supplementary Fig. 8). In addition, Gpc4 is widely expressed with highest expression in kidney, pituitary, and WAT, indicating that other tissues could contribute to serum Gpc4. However, the strong association of serum Gpc4 levels with BMI in humans and the fact that Gpc4 can be released from cultured primary adipocytes make adipose tissue one likely source of serum Gpc4.
To date, no circulating factor has been shown to directly enhance the activation of the insulin receptor itself. Both the transmembrane glycoprotein plasma cell membrane glycoprotein-1 or ectonucleotide pyrophosphatase/phosphodieterase and circulating alpha 2-HS glycoprotein are known to interact with the extracellular domains of the insulin receptor and to negatively affect insulin binding and activation of the insulin receptor (28,29). By contrast, we find that both membrane- and nonmembrane–bound Gpc4 can interact with the insulin receptor and enhance insulin signaling. This interaction occurs with the unoccupied insulin receptor, and stimulation by insulin disrupts the interaction of Gpc4 with the insulin receptor. Thus, overexpression of native Gpc4 or ΔGpc4 or addition of recombinant ΔGpc4 enhances insulin signaling in 3T3-L1 cells, whereas the depletion of Gpc4 results in reduced insulin receptor phosphorylation and downstream signaling.
Insulin is an important regulator of adipocyte differentiation and function (4). In line with that, adipocyte differentiation is increased in Gpc4 or ΔGpc4 overexpressing cells and blocked in Gpc4 knockdown cells. The latter is due to an inability to induce C/EBPα and PPARγ, the key transcription factors required for differentiation, secondary to reduced phosphorylation of C/EBPβ at the ERK/GSK3β consensus site Thr188. Phosphorylation of Thr188 is essential for DNA binding and transactivation of C/EBPα and PPARγ (19,30). Block of adipocyte differentiation at this stage of differentiation is also seen in IRS-1/IRS-2 double knockout cells (31), further indicating a link between insulin signaling and the adipocyte differentiation defect. Overexpression of the Akt and ERK inhibitor TRB3 also prevents activation of C/EBPβ and thereby inhibits adipocyte differentiation (14). However, it is possible that Gpc4 could affect additional signaling pathways, or that other factors within the insulin signaling pathway contribute to the differentiation defect, as insulin signaling induces a variety of transcription factors that might regulate adipocyte differentiation (32).
Taken together, our data show that Gpc4 is an insulin-sensitizing “adipokine” that directly interacts with the insulin receptor to regulate its activation and downstream signaling. The importance of Gpc4 in modulating insulin signaling is underlined by the inability of Gpc4 knockdown cells to differentiate into adipocytes because of a lack of insulin signaling. In addition to its biological activity, serum levels of Gpc4 are correlated with insulin resistance. The role of Gpc4 as an insulin sensitizer and its higher serum levels in insulin-resistant individuals may seem counterintuitive at first. However, insulin itself shows a similar distribution, with lower levels in insulin-sensitive versus insulin-resistant individuals. Given that GPLD1 is the most likely candidate to cleave Gpc4 and is itself an insulin-regulated gene, it is possible that increasing levels of insulin early in obesity lead to increased Gpc4 cleavage, which results in increased circulating Gpc4 levels. With disease progression, as in the ob/ob mouse, increased insulin resistance in GPLD1-producing cells would result in a reduction of GPLD1 activity and a drop in circulating Gpc4 levels, further decreasing insulin sensitivity and accelerating disease progression. Thus, our data suggest that increased circulating Gpc4 levels could be a novel regulatory mechanism by which fat acts to counteract insulin resistance, and maintaining high serum Gpc4 levels in severely insulin-resistant or diabetic subjects could lower insulin demands. Although further studies are required to dissect the various function of soluble versus membrane-bound Gpc4, glypican-4 forms a novel adipokine and a novel mechanism by which adipose tissue can modulate insulin signaling.
ACKNOWLEDGMENTS
The work was supported by National Institutes of Health grants DK31036 and DK82654, the Joslin DERC core laboratories (DK36836), the Mary K. Iacocca Professorship and the Human Frontier Science Program, and the Deutsche Forschungsgemeinschaft, KFO 152 (project BL 833/1-1).
No potential conflicts of interest relevant to this article were reported.
S.U. wrote the manuscript and researched data. O.B. and M.B. researched data and reviewed and edited the manuscript. C.R.K. helped design experiments and edited the manuscript. C.R.K. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-1395/-/DC1.
REFERENCES
- 1.Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell 2007;131:242–256 [DOI] [PubMed] [Google Scholar]
- 2.Tran TT, Yamamoto Y, Gesta S, Kahn CR. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab 2008;7:410–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butterwith SC. Regulators of adipocyte precursor cells. Poult Sci 1997;76:118–123 [DOI] [PubMed] [Google Scholar]
- 4.Blüher M, Michael MD, Peroni OD, et al. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev Cell 2002;3:25–38 [DOI] [PubMed] [Google Scholar]
- 5.Deng Y, Scherer PE. Adipokines as novel biomarkers and regulators of the metabolic syndrome. Ann N Y Acad Sci 2010;1212:E1–E19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahima RS, Flier JS. Adipose tissue as an endocrine organ. Trends Endocrinol Metab 2000;11:327–332 [DOI] [PubMed] [Google Scholar]
- 7.Kralisch S, Bluher M, Paschke R, Stumvoll M, Fasshauer M. Adipokines and adipocyte targets in the future management of obesity and the metabolic syndrome. Mini Rev Med Chem 2007;7:39–45 [DOI] [PubMed] [Google Scholar]
- 8.Gesta S, Bluher M, Yamamoto Y, et al. Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc Natl Acad Sci USA 2006;103:6676–6681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Cat B, David G. Developmental roles of the glypicans. Semin Cell Dev Biol 2001;12:117–125 [DOI] [PubMed] [Google Scholar]
- 10.Fico A, Maina F, Dono R. Fine-tuning of cell signaling by glypicans. Cell Mol Life Sci 2011;68:923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filmus J, Capurro M, Rast J. Glypicans. Genome Biol 2008;9:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karumanchi SA, Jha V, Ramchandran R, et al. Cell surface glypicans are low-affinity endostatin receptors. Mol Cell 2001;7:811–822 [DOI] [PubMed] [Google Scholar]
- 13.Hagihara K, Watanabe K, Chun J, Yamaguchi Y. Glypican-4 is an FGF2-binding heparan sulfate proteoglycan expressed in neural precursor cells. Dev Dyn 2000;219:353–367 [DOI] [PubMed] [Google Scholar]
- 14.Bezy O, Vernochet C, Gesta S, Farmer SR, Kahn CR. TRB3 blocks adipocyte differentiation through the inhibition of C/EBPbeta transcriptional activity. Mol Cell Biol 2007;27:6818–6831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woods A, Couchman JR. Proteoglycan isolation and analysis. Curr Protoc Cell 2001;10.7.1–10.7.19 [DOI] [PubMed]
- 16.Grunfeld C, Van Obberghen E, Karlsson FA, Kahn CR. Antibody-induced desensitization of the insulin receptor. Studies of the mechanism of desensitization in 3T3-L1 fatty fibroblasts. J Clin Invest 1980;66:1124–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farmer SR. Transcriptional control of adipocyte formation. Cell Metab 2006;4:263–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang YY, Li X, Qian SW, et al. Transcriptional activation of histone H4 by C/EBPβ during the mitotic clonal expansion of 3T3-L1 adipocyte differentiation. Mol Biol Cell 2011;22:2165–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park BH, Qiang L, Farmer SR. Phosphorylation of C/EBPbeta at a consensus extracellular signal-regulated kinase/glycogen synthase kinase 3 site is required for the induction of adiponectin gene expression during the differentiation of mouse fibroblasts into adipocytes. Mol Cell Biol 2004;24:8671–8680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Traister A, Shi W, Filmus J. Mammalian Notum induces the release of glypicans and other GPI-anchored proteins from the cell surface. Biochem J 2007;410:503–511 [DOI] [PubMed] [Google Scholar]
- 21.Klöting N, Fasshauer M, Dietrich A, et al. Insulin-sensitive obesity. Am J Physiol Endocrinol Metab 2010;299:E506–E515 [DOI] [PubMed] [Google Scholar]
- 22.Grundy SM. Obesity, metabolic syndrome, and cardiovascular disease. J Clin Endocrinol Metab 2004;89:2595–2600 [DOI] [PubMed] [Google Scholar]
- 23.Brunner G, Metz CN, Nguyen H, et al. An endogenous glycosylphosphatidylinositol-specific phospholipase D releases basic fibroblast growth factor-heparan sulfate proteoglycan complexes from human bone marrow cultures. Blood 1994;83:2115–2125 [PubMed] [Google Scholar]
- 24.Raikwar NS, Bowen-Deeg RF, Du XS, Low MG, Deeg MA. Glycosylphosphatidylinositol-specific phospholipase D improves glucose tolerance. Metabolism 2010;59:1413–1420 [DOI] [PubMed] [Google Scholar]
- 25.Saltiel AR, Cuatrecasas P. In search of a second messenger for insulin. Am J Physiol 1988;255:C1–C11 [DOI] [PubMed] [Google Scholar]
- 26.Kurtz TA, Fineberg NS, Considine RV, Deeg MA. Insulin resistance is associated with increased serum levels of glycosylphosphatidylinositol-specific phospholipase D. Metabolism 2004;53:138–139 [DOI] [PubMed] [Google Scholar]
- 27.Bowen RF, Raikwar NS, Olson LK, Deeg MA. Glucose and insulin regulate glycosylphosphatidylinositol-specific phospholipase D expression in islet beta cells. Metabolism 2001;50:1489–1492 [DOI] [PubMed] [Google Scholar]
- 28.Youngren JF. Regulation of insulin receptor function. Cell Mol Life Sci 2007;64:873–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Srinivas PR, Wagner AS, Reddy LV, et al. Serum alpha 2-HS-glycoprotein is an inhibitor of the human insulin receptor at the tyrosine kinase level. Mol Endocrinol 1993;7:1445–1455 [DOI] [PubMed] [Google Scholar]
- 30.Tang QQ, Gronborg M, Huang HJ, et al. Sequential phosphorylation of CCAAT enhancer-binding protein beta by MAPK and glycogen synthase kinase 3beta is required for adipogenesis. Proc Natl Acad Sci USA 2005;102:9766–9771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miki H, Yamauchi T, Suzuki R, et al. Essential role of insulin receptor substrate 1 (IRS-1) and IRS-2 in adipocyte differentiation. Mol Cell Biol 2001;21:2521–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boucher J, Tseng Y H, Kahn CR. Insulin and insulin-like growth factor-1 receptors act as ligand-specific amplitude modulators of a common pathway regulating gene transcription. J Biol Chem 2010;285:17235–17245 [DOI] [PMC free article] [PubMed] [Google Scholar]