

Abstract
Obesity-associated increases in adipose tissue (AT) CD11c+ cells suggest that dendritic cells (DC), which are involved in the tissue recruitment and activation of macrophages, may play a role in determining AT and liver immunophenotype in obesity. This study addressed this hypothesis. With the use of flow cytometry, electron microscopy, and loss-and-gain of function approaches, the contribution of DC to the pattern of immune cell alterations and recruitment in obesity was assessed. In AT and liver there was a substantial, high-fat diet (HFD)–induced increase in DC. In AT, these increases were associated with crown-like structures, whereas in liver the increase in DC constituted an early and reversible response to diet. Notably, mice lacking DC had reduced AT and liver macrophages, whereas DC replacement in DC-null mice increased liver and AT macrophage populations. Furthermore, delivery of bone marrow–derived DC to lean wild-type mice increased AT and liver macrophage infiltration. Finally, mice lacking DC were resistant to the weight gain and metabolic abnormalities of an HFD. Together, these data demonstrate that DC are elevated in obesity, promote macrophage infiltration of AT and liver, contribute to the determination of tissue immunophenotype, and play a role in systemic metabolic responses to an HFD.
Dendritic cells (DC) are a heterogeneous population of professional antigen-presenting cells (1) that play important roles in both innate and adaptive immunity. They have well-described functions in the activation of conventional and regulatory T cells and are also involved in the recruitment and activation of macrophages at sites of immune responses (2–6). A series of studies has established that macrophage activation and infiltration of adipose tissue (AT) and liver contributes to the proinflammatory status of obesity (7–13) and that these events are involved in the pathogenesis of the obesity-associated metabolic abnormalities of insulin resistance and dyslipidemia (13–15). A significant subset of proinflammatory AT macrophages express the integrin α-chain CD11c, defining these cells as “triple-positive” (triple+), since they also express the CD11b and F4/80 markers (16,17). However, CD11c is commonly recognized as a marker of DC, which can also express CD11b and F4/80 (18,19), raising the possibility that at least a proportion of the obesity-induced increase in CD11c+ cells is because of elevations in AT DC, a hypothesis that has been proposed (17) but not tested. Furthermore, given the role of DC in the activation and recruitment of immune cells, it is possible that DC may play an important role in determining immune cell infiltration in obesity. The current study addressed these hypotheses. Our data demonstrate that a substantial proportion of CD11c+ cells in liver and AT are DC, that obesity is associated with an elevation in DC in AT and the liver, that genetic and pharmacologic manipulation of DC influences macrophage infiltration of adipose and liver tissue, and that genetic deletion of DC protects against diet-induced obesity (DIO).
RESEARCH DESIGN AND METHODS
Animals.
C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed and bred in the University of Pittsburgh facility. Male C57BL/6-Flt3L null (Flt3l−/−) mice, derived from C57BL/6J, were purchased from Taconic Farms (Germantown, NY). Animals were housed under standard conditions with ad libitum access to water and food. Experiments were conducted in compliance with National Institute of Health guidelines, and all procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Study Design
DC contribution to the pattern of immune cell infiltration in obesity.
Male C57BL/6J mice were weaned at 3 weeks of age and fed either a standard chow diet (SCD; 65% carbohydrate, 11% fat, 24% protein per calories; No. 01351, Harlan Teklad, Madison, WI) or a high-fat diet (HFD; 40% carbohydrate, 41% fat, 19% protein; No. 96001, Harlan Teklad) for 3 or 26 weeks. Another group of mice was fed the HFD for 3 weeks, followed by 3 weeks on the SCD, while a control group was fed the SCD for a total of 6 weeks (“recovery studies”). Animal weights and food intake were recorded weekly. At the end of the dietary exposure period tissue immune cells were isolated and assessed as detailed below.
Effects of gain-and-loss of DC on AT and liver immune cell infiltration.
Seven-week-old Flt3l−/− and wild-type mice were placed on the HFD for 16 weeks. Another group of HFD-fed Flt3l−/− mice was treated with either human recombinant Flt3 ligand (CHO-cell derived, 10 μg/100 μL PBS, intraperitoneal [IP] injection every other day; Amgen, Seattle, WA) or PBS for 2 weeks. Six-week-old male mice were IP injected with either 0.5–1 × 106 CD11c+ bone marrow–derived DC (BMDC)/200 μL PBS or PBS only and animals maintained on the SCD for 1 week. The effects of these manipulations on tissue immune cell populations were then assessed.
Metabolic effects of an HFD.
Male Flt3l−/− mice and wild-type controls were exposed to HFD or SCD (see above) for 16 weeks. During this period body weight and caloric intake were monitored once a week. Glucose tolerance tests (GTT) and insulin tolerance tests were performed at weeks 13 and 14, respectively, as described previously (20). Metabolic rate and activity were assessed before obesity using the comprehensive laboratory animal monitoring system (CLAMS) (20). Body composition was assessed by densitometry at 15 weeks (20), and liver triglycerides were assessed as described (21).
Flow cytometry.
Mononuclear cells from liver, spleen, or mesenteric lymph nodes (MLNs) or the stromal vascular cell fraction (SVC) of AT were isolated using standard procedures. Cell suspensions (2 × 106 cells/sample) from liver, MLNs, spleen, and AT were preincubated in buffer with anti-CD16/32 (Fc “blocking” antibodies [Abs], clone 93) for 15 min at 4°C, then stained with either fluorescent-labeled primary Abs or IgG isotype controls for 30 min at 4°C. The following Abs were used: fluorescein isothiocyanate–conjugated anti-CD11b (clone M1/70); phycoerythrin-conjugated anti-CD86 (clone GL1); peridinin chlorophyll-a protein (PerCP)-conjugated anti-B220 (clone RA3–6B2); PB-conjugated anti-F4/80 (clone BM8); and allophycocyanin-conjugated anti-CD11c (N418). PerCP-conjugated Abs were purchased from BD Biosciences, whereas all other Abs were from eBiosciences (San Diego, CA). Cells were washed gently ×2 in buffer and resuspended in 4% paraformaldehyde (Fisher) before analysis using a FACSCalibur flow cytometer and FACSDiva software (BD Biosciences, San Jose, CA). A minimum of 50,000 cells were analyzed for each sample, and a proportion of up to 1% false-positive events was accepted in the isotype control samples.
Immunomagnetic cell sorting.
For electron microscopic analysis, cell suspensions from liver and AT were further selected using a magnetic cell sorting system (Miltenyi Biotec, Auburn, CA) per the manufacturer’s instructions. Flow cytometry analysis was performed to determine the purity of sorted cells.
Scanning electron microscopy.
Cell suspensions were applied to glass coverslips for 15 min at 37°C then fixed with 2.5% glutaraldehyde, followed by 1% OsO4 (Electron Microscopy Sciences, Hatfield, PA). After drying, samples were mounted onto aluminum stubs (Electron Microscopy Sciences) then sputter coated with 3.5 nm gold/palladium (Auto 108; Cressington, Watford, UK). With the use of a JSM-6330F electron microscope (JEOL, Peabody, MA), at least 100 cells were counted in 5000× fields for each sample, and those with DC morphology (22,23) were scored versus cells lacking these structures.
Transmission electron microscopy.
Pellets of freshly isolated cell suspensions were resuspended in 2.5% glutaraldehyde in PBS, spun at 300 g, and further processed for transmission electron microscopy as described (24). For analysis, two sets of samples were evaluated for cells with DC morphology, using previously described criteria (22,23).
Immunofluorescent microscopy.
Staining and analysis of epididymal fat pads was performed as described previously (25).
Generation and purification of BMDC.
BMDC were prepared as described previously (26). BMDC purity (>90% were CD11b+CD11c+Gr-1−) and immaturity (87% of cells were MHCII−) were confirmed using flow cytometry.
Statistical analysis.
Data are expressed as means ± SEM for 4–12 animals in each group. Data were analyzed using a two-tailed Student’s t test, and a P value of <0.05 was considered significant.
RESULTS
Increases in CD11c+ cells in liver and AT are a marked response to high-fat feeding, and the increase occurs early and is reversible in liver.
We first analyzed the influence of high-fat feeding on the total CD11c+ cell population (Supplementary Fig. 1). Mice rendered obese using an HFD (Fig. 1A–D) had a substantial increase in the absolute number of CD11c+ cells in spleen, AT, and liver, but not MLNs (Fig. 2A and D). When expressed as percentages of the total mononuclear cells or SVC population (Fig. 2B), these increases were significant for liver and AT but not for spleen and MLNs, demonstrating that CD11c+ cells in the former two tissues were specifically affected in obesity. Furthermore, the elevations in CD11c+ cells in AT were predominantly located within the crown-like structures associated with the inflammatory response in obesity (Fig. 2E). It is interesting that increases in liver, but not adipose tissue, CD11c+ cells occurred before substantial alterations in adiposity and glucose tolerance, but in parallel to mild steatosis (Figs. 1B–D and 2C). Notably, other liver immune cells assessed (CD4+ T cells, CD8+ T cells, B cells) did not increase with a short-term diet (data not shown), while NKT cells decreased (20). Furthermore, the dietary influence on CD11c+ cells was reversible in liver (Fig. 2C) demonstrating a direct link between overnutrition and CD11c+ cell alterations, and coincided with a decrease in liver steatosis (Fig. 1D).
FIG. 1.

Characteristics of C57BL/6 male mice exposed to HFD or SCD. Mice were exposed to either the HFD or SCD for 3 or 26 weeks. Additional animals were fed the HFD for 3 weeks, then switched to the SCD for 3 weeks or fed the SCD for the entire 6 weeks. A minimum of 6 animals per group were individually analyzed, and results are presented as means ± SE. Significant differences are indicated (*Compared with the SCD group; #Compared with mice fed the HFD for 3 weeks; P < 0.05). A: Weight was measured weekly in HFD (●) and SCD (○) mice. B: Epididymal and renal adipose depots were excised and weighed after dietary exposure. C: GTT was performed after either 3 or 22 weeks of dietary exposure (◇, mice fed the SCD for 3 weeks; ♦, mice fed the HFD for 3 weeks; □, mice fed the SCD for 22 weeks; ■, mice fed the HFD for 22 weeks). Mice were injected IP with 1.5 g/kg glucose, and blood glucose determined from tail vein samples every 15 min using an Ascencia Elite glucometer (Bayer, Mishawaka, IN), as described previously (20). D: Triglyceride (TG) content of the liver.
FIG. 2.

The influence of HFD on CD11c+ cells. A and B: After 26 weeks of dietary exposure, mononuclear cells were isolated from spleen (Spl), adipose tissue (AT), liver (Liv), and MLN and stained for specific markers, then analyzed by flow cytometry. A: Number of CD11c+ cells in tissues. B: Proportion of CD11c+ cells among total mononuclear cells isolated from spleen, liver, and MLNs and SVC from AT. C: Fold changes in liver CD11c+ cells in 3-week SCD or HFD or 3-week SCD or HFD followed by the SCD for additional 3 weeks (3 + 3 weeks). D: Representative flow cytometry plots of CD11c+ cells (gate 1) isolated from AT and liver of lean and obese animals. For all experiments above, a minimum of 6 animals per group were individually analyzed (n = 6). Results are presented as means ± SE. Significant differences are indicated (*P < 0.05). E: Representative immunofluorescence of epididymal fat pads obtained from lean and obese animals. After fixation in 2% paraformaldehyde, ∼1 mm3 of tissue was labeled in suspension using rat anti-mouse F4/80 (clone 6F12) and hamster anti-mouse CD11c, both at 1:100 dilution (BD Pharminogen). Goat anti-rat Alexa Fluor 488 (1:500; Invitrogen) and goat anti-hamster Cy3 (1:1,000; Jackson ImmunoResearch Laboratories, West Grove, PA) were used as secondary antibodies. Confocal stack tissue reconstructions of 50 µm were taken at 5 µm intervals using an Olympus Fluoview 1000 Microscope (Olympus, Center Valley, PA). FACS, fluorescence-activated cell sorter; FITC, fluorescein isothiocyanate; APC, allophycocyanin. (A high-quality digital representation of this figure is available in the online issue.)
Unlike in AT, triple+ cells in liver do not account for the increase in CD11c+ cells in obesity.
An accumulation of triple+ (CD11b+CD11c+F4/80+) macrophages has previously been described in AT of obese mice (16,17,27), but the influence of obesity on this population in liver is unknown. In AT, the number of CD11b+CD11c+F4/80+ cells was increased fivefold in obese compared with lean mice (3.6 ± 1.2 vs. 0.7 ± 0.2 × 104 cells/g tissue, P < 0.05), similar to previous reports (16,17,27), as did the proportion (Fig. 3A and E). In liver, unlike AT, the proportion and the number (0.5 ± 0.2 vs. 0.8 ± 0.2 × 105 in lean vs. obese animals, respectively) of triple+ cells was unaffected by obesity, demonstrating that increases in CD11c+ cells in liver cannot be accounted for by increases in triple+ macrophages.
FIG. 3.


The influence of obesity on CD11b+CD11c+F4/80+ (triple+) and CD11b+CD11c−F4/80+ (double+) cells in AT and liver. Mononuclear cells from liver and stromal vascular cells from AT were isolated from lean and obese mice, stained for CD11b, CD11c, and F4/80 markers, and analyzed by flow cytometry. Data are presented as mean ± SE (minimum number of 6 animals/group analyzed individually). Significant differences are indicated (*P < 0.05). A: Proportion of triple+ cells (CD11b+CD11c+F4/80+) in AT and liver. B: Proportion of double+ cells (CD11b+CD11c−F4/80+) in AT and liver. C: Proportion of CD11b+ and CD11b+CD11c− cells in AT. D: Proportion of CD11b+ and CD11b+CD11c− cells in liver. MC, mononuclear cells. E: Representative flow cytometry plots of triple+ cells in AT and liver, corresponding to A. F: Representative flow cytometry plots of double+ cells in AT and liver, corresponding to B. G: Representative flow cytometry plots of flow cytometry analysis of CD11b+ (gates 3 and 4) and CD11b+CD11c− cells (gate 3 only) in AT, corresponding to C. H: Representative flow cytometry plots of CD11b+ (gates 3 and 4) and CD11b+CD11c− cells (gate 3 only) in liver, corresponding to D. FACS, fluorescence-activated cell sorter; FITC, fluorescein isothiocyanate; APC, allophycocyanin.
In both tissues, double+ cells (CD11b+CD11c−F4/80+) were unaltered by obesity (Fig. 3B and F), as reported previously for AT (17,27), whereas the total CD11b+ population, i.e., without selecting for F4/80, was highly elevated in AT and liver (Fig. 3C, D, G, and H, respectively). Taken together, these data demonstrate that all obesity-induced increases in CD11b+ cells in AT are accounted for solely by increases in the triple+ population. However, in liver, there is a substantial increase in CD11b+ cells in obesity that are unrelated to changes in the triple+ population.
A substantial proportion of CD11c+ cells in liver and AT in obesity are DC and have a mature phenotype.
To test what proportion of the increase in CD11c+ cells observed in response to high-fat feeding in AT and liver are DC, we first analyzed populations of cells that possess a definitive DC marker profile, i.e., CD11b−CD11c+, CD11b−CD11c+B220+, and PDCA-1+ cells (28,29). In both AT and liver of obese mice, significant increases in the number and proportion of CD11b−CD11c+ cells were observed (Figs. 3G and H and 4A and B). Furthermore, PDCA-1+ cells were increased significantly in both tissues (Fig. 4C), with CD11c and B220 being coexpressed on ∼65% of these cells in AT and liver, respectively, suggesting that the majority of these cells were plasmacytoid DC (pDC). Scanning electron microscopy (SEM) analysis of PDCA-1+ fractions from AT and liver revealed a relatively uniform population of cells with round surfaces and short dendritic processes, thereby confirming that these cells were pDC (Fig. 4D).
FIG. 4.

The influence of obesity on CD11b−CD11c+ DCs and pDC (CD11b−CD11c+B220+) in AT and liver. For A and B, mononuclear cells from liver and SVC from AT were isolated from lean and obese mice, stained for appropriate markers, and analyzed by flow cytometry (see also Fig. 3G and H, gate 5, which represents CD11b−CD11c+ cells). A: Number of CD11b−CD11c+ DC in AT and liver. B: Proportion of CD11b−CD11c+ DC among SVC in AT and mononuclear cells in liver. There were a minimum number of 6 animals per group. Results are presented as means ± SE. Significant differences are indicated (*P < 0.05). Results are presented as means ± SE. C: Isolated cells from AT and liver were first separated into PDCA-1+ (plasmacytoid DC antigen-1, ∼65% of PDCA-purified cells were CD11c+, CD11b−, B220+) and PDCA-1− fractions using immunomagnetic beads, labeled for specific markers, and analyzed by flow cytometry. For each individual experiment, immune cells from 2 lean animals (pooled) and 1 obese animal were used. The fold increase of PDCA-1+ cells that were also CD11b−CD11c+B220+ (pDC) in AT and liver of obese vs. lean mice is shown. Results are presented as means ± SD; n = 3 separate experiments (6 lean and 3 obese animals). Significant differences are indicated (*P < 0.05). D: Cell suspensions isolated from liver and AT were separated into PDCA+ and CD11c+PDCA-1− fractions using immunomagnetic beads and PDCA+ cells prepared for SEM. SEM was performed separately on cells from three individual obese mice (n = 3), and cells from 6 lean mice were pooled in groups of 2 (n = 3). PDCA-1+ cells appear as a relatively homogeneous population, with round surface and short dendrites—the morphology typical of freshly isolated pDC.
To determine whether CD11b+CD11c+ DC (conventional DC, cDC) contribute to the increase in the CD11c+ population in obesity, we analyzed the CD11c+PDCA-1− cell population, i.e., a population with pDC removed. SEM analysis of CD11c+PDCA-1− cells from AT and liver revealed a heterogeneous population of DC, macrophages, and rare lymphocytes (Fig. 5A and B). It is important that in AT the proportion of cDC-like cells was increased twofold, from 16% ± 8% in lean mice compared with 30% ± 2.6% (P = 0.043) in the obese mice, and in liver fourfold, from 8 ± 1.7% in lean mice to 33% ± 0.3% (P < 0.0001) in obese mice (data not shown). Transmission electron microscopy of the CD11c+PDCA-1− population revealed the presence of DC, with characteristic dendritic processes and few vacuoles, in both liver and AT (Fig. 5C and D).
FIG. 5.

The influence of obesity on CD11c+PDCA-1− cells in AT and liver. A and B: SVC from AT and mononuclear cells from liver were isolated and the CD11c+PDCA-1− fraction prepared for SEM. Although macrophage morphology is evident (arrowheads), cells displaying the prominent cytoplasmic veils characteristic of DC are also seen (arrows). C and D: SVC from AT and mononuclear cells from liver were isolated and the CD11c+PDCA-1− fraction prepared for transmission electron microscopy as described (15). Both micrographs show the presence of typical dendritic processes on DC, which are absent on macrophages (Mac), and few vacuoles in DC compared with numerous vacuoles in macrophages.
Under noninflammatory steady-state conditions, DC are present mainly as immature cells, which express low levels of CD86 and other costimulatory molecules, whereas the mature state is associated with increased expression of CD86 (29). Thus, to determine the degree of DC maturation, we assessed mean fluorescent intensity (MFI) of CD86 on the CD11c+ population from AT and liver of lean and obese mice. In AT, the number of CD11c+CD86+ cells was elevated in obesity, mirroring the overall increases in these two populations. However, MFI of CD86 expression on these cells was unchanged in obese compared with control animals (Fig. 6A, B, and E). In liver, obesity was associated with both an increased number of CD86+ cells, as well as increased MFI on all CD11c+ populations examined, compared with lean mice (Fig. 6C, D, and F).
FIG. 6.


The influence of obesity on maturation of DC in AT and liver. Isolated mononuclear cells from liver and SVC from AT were stained for markers to define subsets of CD11c+ as well as CD86+ cells and analyzed by flow cytometry (n = minimum of 6 animals per group). Results are presented as means ± SE, and significant differences are indicated (*P < 0.05). A: Number of CD11c+CD86+ cells isolated from AT. B: MFI for CD86 on CD11c+CD86+ cells from AT. C: Number of CD11c+CD86+ cells isolated from liver. D: MFI for CD86 on liver CD11c+CD86+ cells. E: Representative flow cytometry plots of CD86+ cells in AT, corresponding to A and B. Gate 1a represents CD11c+CD86+ cells; gate 4a represents CD11b+CD11c+CD86+ cells; and gate 5a represents CD11b−CD11c+CD86+ cells. F: Representative flow cytometry plots of CD86+ cells in liver, corresponding to C and D. Gate 1a represents CD11c+CD86+ cells; gate 4a represents CD11b+CD11c+CD86+ cells; and gate 5a represents CD11b−CD11c+CD86+ cells. FACS, fluorescence-activated cell sorter; APC, allophycocyanin.
DC induce macrophage recruitment to AT and liver, but not spleen.
Given the vital role of DC in the activation and recruitment of macrophages, the data presented above raise the possibility that, in states of overnutrition, DC may play a role in the recruitment of macrophages and triple-positive cells to AT and liver. To address this hypothesis, DC numbers in vivo were manipulated using a number of approaches, and subsequently the effects of altering DC numbers on tissue accumulation of macrophages and T cells were assessed. In response to a single IP injection (0.5 × 106) of BMDC (CD11b+CD11c+Gr-1−) AT and liver, but not spleen or MLNs, exhibited substantial increases in triple+ (CD11b+CD11c+F4/80+) and CD11b+CD11c− cells (Fig. 7A and B), and CD3+CD4+ T cells and CD11b−CD11c+ cells, a DC marker profile (Supplementary Fig. 2). These increases were observed 1 week after the BMDC injection, by which time the original cells could not be detected (30), the increases were far in excess of the original number of BMDC injected, and a bone marrow–derived macrophage injection failed to elicit the same changes (data not shown). Of interest, lipopolysaccharide-treated BMDC and once-weekly BMDC injections for a total of 6 weeks gave a similar immunophenotype in AT to a single BMDC injection, but the effects in liver were lost in the 6-week experiment. Notably, SVC from AT of DC-injected mice produced significantly greater quantities of interferon-γ, granulocyte-macrophage colony-stimulating factor, interleukin-2, and monokine induced by γ-interferon, with no differences in tumor necrosis factor-α or monocyte chemoattractant protein-1 production compared with saline-injected mice (Supplementary Fig. 3). We next assessed the influence of genetic deletion of DC on macrophage numbers. Flt3l−/− mice lacking DC had dramatically reduced numbers of CD11b+CD11c− and CD11b+CD11c+F4/80+ (triple+) cells in AT and liver but not spleen as compared with wild-type animals (Fig. 7C and D). This effect was reversed upon reconstitution of DC in Flt3l−/− mice (Fig. 7E and F) using injections of recombinant Flt3 ligand. Manipulation of DC via Flt3 ligand had a particularly dramatic impact on the triple+ population in AT. In total, these data demonstrate that manipulation of DC by three independent methods altered AT and liver macrophage populations. Finally, we assessed the metabolic effects of high-fat feeding in C57BL/6 Flt3l−/− mice. A 16-week HFD failed to induce obesity, insulin resistance, or liver steatosis in these mice compared with wild-type controls (Fig. 8A, B, D, E, and H). Flt3l−/− mice on a SCD displayed a similar metabolic phenotype to wild-type controls (Fig. 8A and B). Notably, Flt3l−/− mice had elevated caloric intake, increased activity, and elevated metabolic rate (Fig. 8C, F, and G).
FIG. 7.

The effects of gain-and-loss of DC on AT and liver macrophage and triple-positive cell infiltration. A and B: 1 week after IP injection of 0.5–1.0 × 106 CD11c+ BMDC in 200 μL PBS (DC) or PBS alone (PBS), mononuclear cells from liver, spleen, and SVC from AT were isolated from mice fed the SCD, stained for CD11b, CD11c, and F4/80 markers, and analyzed by flow cytometry for CD11b+CD11c− and CD11b+CD11c+F4/80+ (triple+). Results are presented as means ± SE (n = minimum of 6 animals/group). Significant differences are indicated (*P < 0.05). C and D: Wild-type (WT) and Flt3l−/− (Null) mice were fed HFD for 16 weeks before isolation of mononuclear cells from liver, spleen, and SVC from AT for analysis by flow cytometry for CD11b+CD11c− and CD11b+CD11c+F4/80+ (triple+). Results are presented as means ± SE (n = minimum of 5 animals/group). Significant differences are indicated (*P < 0.05). E and F: Flt3l−/− mice were injected IP with either 10 μg human recombinant Flt3 ligand in 100 μL PBS (Flt3L) or PBS alone (PBS) every other day for 2 weeks. Subsequently, mononuclear cells from liver, spleen, and SVC from AT were isolated and stained for analysis by flow cytometry for CD11b+CD11c− and CD11b+CD11c+F4/80+ (triple+). Results are presented as means ± SD (n = 2 animals/group).
FIG. 8.

The effects of DC deletion on DIO. A: 7-week-old wild-type (WT) and Flt3l−/− (Null) mice were exposed to either the SCD (○, WT; □, Null) or HFD (●, WT; ■, Null) for 16 weeks and their weight recorded weekly. B–I: WT and Null mice were maintained on the HFD. A minimum of 6 animals per group were analyzed, and data are presented as means ± SE. Significant differences are indicated (*Compared with WT mice on the HFD, P < 0.05). B: Body composition of WT and Null mice was assessed using a Lunar PIXImus densitometer (Lunar, Madison, WI) during the last week of feeding, as described previously (20). C: Caloric intake was assessed for individually caged mice. D: The GTT was performed after 13 weeks of dietary exposure (●, WT mice; △, Null mice). E: The insulin tolerance test was performed 1 week after GTT. Animals were injected IP with 0.75 units/kg of Humulin R (Lilly, Indianapolis, IN) and glucose measured in tail vein blood samples. F: Stereotypical (stereo) and ambulatory (ambul) activity was measured over 24 h by infrared beam interruption (Opto-Varimex mini; Columbus Instruments, Columbus, OH). G: Metabolic rate was analyzed in WT and Null animals during the first week of diet, i.e., before weight gain for 24 h at 23°C by indirect calorimetry using CLAMS (Columbus Instruments). Data were analyzed by repeated-measures ANOVA, followed by Tukey’s post hoc test using the SPSS program. H: Triglyceride (TG) content of the liver. I: Plasma Flt3 ligand level was measured using the Mouse Flt3 Ligand ELISA kit (R&D Systems), as per the manufacturer’s instructions.
DISCUSSION
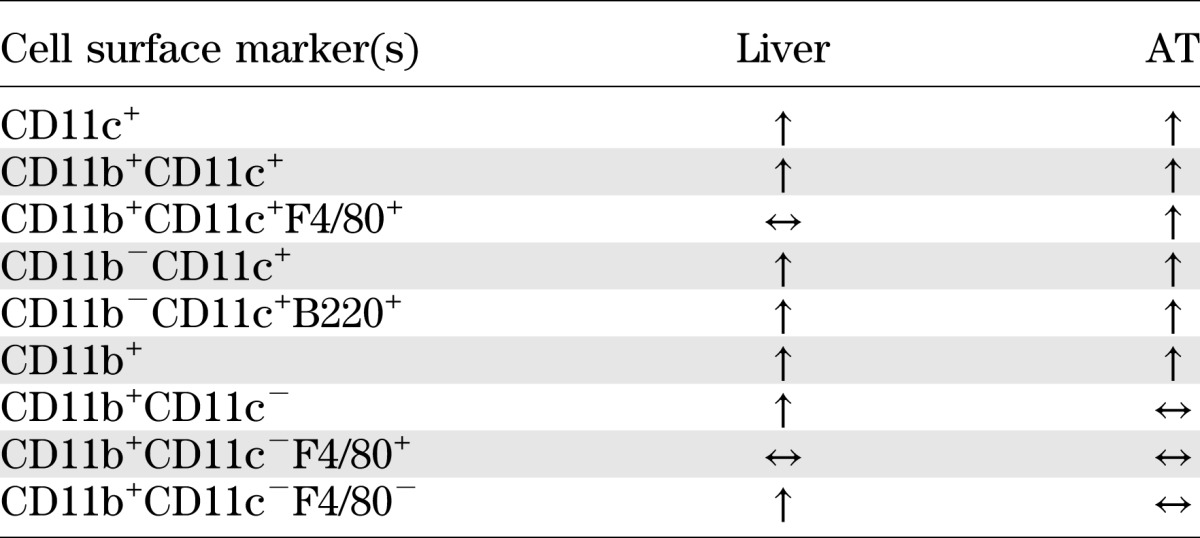
The current study demonstrates that DC represent a significant proportion of the obesity-induced increase in CD11c+ cells in AT and liver. Furthermore, using three independent approaches to manipulate DC number, we present data that strongly implicate a role for DC in mediating obesity-induced increases in AT and liver macrophages. Notably, increases in liver DC occurred rapidly in response to overnutrition and were reversible. There were also clear differences in the CD11c+ populations of liver and AT, most notably the lack of a contribution of triple+ cells to the increase in liver CD11c+ cells in obesity (a summary of immunological changes in response to obesity is presented in Table 1). Finally, mice lacking DC were resistant to the weight gain and metabolic abnormalities of an HFD. Together, these represent the major novel findings of our study.
TABLE 1.
Summary of changes in the proportion of various immune cell populations in liver and AT of diet-induced obese mice compared with lean mice
We used a number of DC-specific characteristics, in combination and separately, to identify/characterize an obesity-induced elevation in DC in liver and AT. First, our flow cytometry approach included an analysis of CD11c+ cells that were CD11b−. Any increase in this population could not be macrophages since these cells lack the CD11b marker. Second, we analyzed cells that were CD11c+CD11b− and either B220+ (by flow cytometry) or PDCA-1+ (by flow cytometry and SEM), a combination of markers used to identify pDC (29). Finally, after depleting the CD11c+ population of PDCA-1+ cells to remove CD11b−CD11c+, we made use of the fact that cDC possess specific morphological features that can be visualized by electron microscopy (typically defined as prominent membrane veils and dendrites) (22) to determine the proportion of the CD11b+CD11c+ population that was DC like. With the use of these approaches, various subpopulations of DC were identified, specifically conventional (cDC - CD11b+CD11c+), nonconventional (CD11b−CD11c+), and plasmacytoid (pDC - CD11b−CD11c+B220+) that were increased in liver and AT of obese mice. To our knowledge, this is the first definitive report of DC in AT of mice. Very little is known about DC function in AT, but it is plausible to suggest that their role is similar to that in liver, i.e., the regulation of both innate and adaptive immunity and immune tolerance (29,31).
Because DC have been shown to play an important role in the activation and recruitment of macrophages at sites of immune system activation not related to high-fat feeding (4), we hypothesized that DC could also influence the pattern of immune cell composition of AT and liver. To address this question, we first injected lean mice with DC derived from BM, a strategy that has previously been used to alter immune response (32), notably in the NOD mouse (33) and in transplant models (34). A single injection of DC was sufficient, 1 week after the injection, to increase the number of both CD11b+CD11c− and triple+ cells in AT and liver. We next analyzed the Flt3l−/− (DC deficient) mice (35) for the presence of CD11b+CD11c− and triple+ cells in AT and liver. Our data demonstrate that a lack of DC reduces accumulation of CD11b+CD11c− and triple+ cells in AT and liver. The effects on CD11b+CD11c− cells were more pronounced in liver, whereas the effect on triple+ cells was particularly striking in AT. It is interesting that our data demonstrate that in DIO CD11b+CD11c− cells accumulate mainly in liver, whereas triple+ cells predominantly infiltrate AT. Our final approach was to reconstitute DC in Flt3l−/− mice by administration of recombinant Flt3 ligand, the nonredundant hematopoetic growth factor required for DC development (35). Notably, this approach reversed the effect of DC deficiency on immune cell populations in Flt3l−/− mice. In total, these data suggest that DC play a role in promoting infiltration of immune cells, including macrophages, in AT and liver.
Our study revealed substantial obesity-induced increases in CD11c+ cells in AT, liver, and spleen. Although the changes in CD11c+ populations seen in AT and spleen are in agreement with earlier studies, the increase in CD11c+ cells in liver of obese mice has not been reported previously. Nguyen et al. (17) demonstrated increased CD11c expression in skeletal muscle of high-fat–fed mice, whereas Macia et al. (36) reported an increased incidence of CD11c+ cells in spleens and skin of ob/ob mice. Combined, these data indicate that the influence of obesity on CD11c+ cells may be a more generalized phenomenon than previously recognized. Notably, in healthy Pima Indians, the expression of CD11c in AT has been associated with the degree of adiposity and negatively correlated with whole-body insulin sensitivity (37), indicating that the relationship between obesity and CD11c+ cells also extends to humans.
Although the HFD had a similar effect on the proportion of CD11c+ populations in liver and AT, further analysis revealed significant differences between the two tissues. Triple+ cells contributed substantially to alterations of CD11c+ cells in AT of obese animals, in agreement with previous reports (17,27). However, very few liver CD11c+ cells were triple+, and the HFD had no effect on this population. It is interesting that a similar observation was made in a mouse model of liver steatosis (38). In addition, CD11b+CD11c− cells were increased in liver but not AT of obese animals, with most of these cells in the liver being F4/80−. These data suggest differing phenotypes of liver-resident CD11c+ cells compared with AT, in agreement with the prevailing view that immune cells, such as macrophages and DC, may be organ- and tissue-specific, and stress the importance of analyzing multiple cell markers when evaluating immune cell responses to obesity.
DC reside in liver as “immature” antigen-presenting cells, characterized by expression of low levels of CD86 and other costimulatory molecules (29,39). The increased expression of CD86 observed on liver DC of obese mice suggests the promotion by obesity/overnutrition of their phenotypic maturation. This process is also considered essential for the initiation of acquired immune response by DC (29). It is interesting that the expression of CD86 was unchanged in DC from AT of obese mice, representing another differential effect of the HFD on DC in AT and liver. In addition, CD86 expression was increased specifically in liver CD11b−CD11c+ cells after just 3 weeks of high-fat feeding. Therefore, the proliferation and maturation of CD11c+ cells and, more specifically, CD11b-CD11c+ cells in the liver, most likely represents one of the early steps in diet-induced immune system alterations.
Issues arising from this study are understanding the role of DC in overnutrition-induced obesity and metabolic dysregulation, what signals initiate DC activation and trafficking into AT and liver, and why DC traffic to these tissues. Presently, only speculative answers to these questions can be given. Our data demonstrate that genetic deletion of DC protects against the development of DIO and attendant metabolic abnormalities. This protection occurred despite an increase in caloric intake and was associated with increased metabolic rate and activity. It is likely that the resistance of Flt3l−/− mice to DIO results from a combinatorial effect of DC deletion from multiple tissues since there is now clear evidence that immune alterations in obesity are not restricted to adipose tissue, having also been described in liver, the central nervous system, skeletal muscle, and pancreas (15). The Flt3l−/− phenotype most closely resembles that of mice lacking mast cells, which are also protected against DIO, with attendant elevated caloric intake and increased metabolic rate (40). Work also related to the current study demonstrated that targeted deletion of all CD11c+ cells in obese animals improves insulin resistance (41). Another relevant study demonstrated that mice that do not express CD11c have improved glucose tolerance and an improved homeostasis model assessment-insulin resistance (42). However, these mice did gain weight on an HFD, but whether a lack of expression of CD11c alters the DC population was not addressed in this study. If these studies are put in the context of the broader literature in this field, the evidence suggests that the manipulation of specific elements of the immune system can alter the fundamental physiological processes of metabolic rate, fat deposition, carbohydrate/lipid metabolism, and insulin action in such a way as to contribute to the determination of metabolic health and body composition. An ongoing challenge is to identify the mechanisms that underlie these effects.
A candidate DC activating signal is free fatty acids (FFAs), since saturated FFAs activate CD11c+ cells via TLR2 and TLR4 and JNK signaling pathways, each of which have been implicated in obesity-induced insulin resistance (17). Notably, plasma FFAs are elevated in obesity, and there is an increased release of FFA from AT and FFA delivery to liver. The recent description of T cell (43,44) and B cell (45) roles in the inflammation and metabolic abnormalities of obesity, and the provocative demonstration that IgG from obese mice induce metabolic dysregulation in lean mice (45), demand that an antigen-mediated process be considered a candidate mechanism for DC activation in the current study. However, this hypothesis calls for the existence of an obesity and/or overnutrition-derived antigen that is derived from and/or enriched in AT and liver (and also potentially other tissues). To date, the existence of such an antigen remains speculative. Finally, indirect mechanisms, notably the cellular stress responses to diets rich in saturated fatty acids and sucrose, and tissue hypoxia (at least for adipose tissue), must also be considered as potential initiating events in the activation and recruitment of DC.
ACKNOWLEDGMENTS
This work was supported by K08-DK-067272 (M.S.-R.), National Institutes of Health (NIH) Grants R01-DK-058855 and R01-DK-072162 (R.M.O.), NIH Grant F30-DK-085865 (B.S.M.), and P01-AI-81678 (A.W.T.). M.S.T. was supported by a training grant from NIH 5T32-CA-82084-10. Electron microscopy and tissue immunofluorescence was performed at the University of Pittsburgh Center for Biologic Imaging.
No potential conflicts of interest relevant to this article were reported.
M.S.-R., X.Y., M.S.T., B.S.M., D.B.S., T.L.S., I.J.S., N.D., P.A.M., and R.M.O. researched data. R.M.O. and M.S.-R. wrote the manuscript. D.K.S., P.A.M., and A.W.T. reviewed and edited the manuscript and contributed to the discussion. R.M.O. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-1523/-/DC1.
REFERENCES
- 1.Wu L, Liu YJ. Development of dendritic-cell lineages. Immunity 2007;26:741–750 [DOI] [PubMed] [Google Scholar]
- 2.Domínguez PM, Ardavín C. Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol Rev 2010;234:90–104 [DOI] [PubMed] [Google Scholar]
- 3.Joffre O, Nolte MA, Spörri R, Reis e Sousa C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev 2009;227:234–247 [DOI] [PubMed] [Google Scholar]
- 4.Heymann F, Meyer-Schwesinger C, Hamilton-Williams EE, et al. Kidney dendritic cell activation is required for progression of renal disease in a mouse model of glomerular injury. J Clin Invest 2009;119:1286–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998;392:245–252 [DOI] [PubMed] [Google Scholar]
- 6.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol 2003;21:685–711 [DOI] [PubMed] [Google Scholar]
- 7.Huang W, Metlakunta A, Dedousis N, et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010;59:347–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003;112:1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003;112:1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cancello R, Tordjman J, Poitou C, et al. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. Diabetes 2006;55:1554–1561 [DOI] [PubMed] [Google Scholar]
- 11.Kang K, Reilly SM, Karabacak V, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab 2008;7:485–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab 2008;7:496–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrante AW., Jr Obesity-induced inflammation: a metabolic dialogue in the language of inflammation. J Intern Med 2007;262:408–414 [DOI] [PubMed] [Google Scholar]
- 14.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 2010;72:219–246 [DOI] [PubMed] [Google Scholar]
- 15.Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med 2012;18:363–374 [DOI] [PubMed] [Google Scholar]
- 16.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007;117:175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen MT, Favelyukis S, Nguyen AK, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem 2007;282:35279–35292 [DOI] [PubMed] [Google Scholar]
- 18.Crowley M, Inaba K, Witmer-Pack M, Steinman RM. The cell surface of mouse dendritic cells: FACS analyses of dendritic cells from different tissues including thymus. Cell Immunol 1989;118:108–125 [DOI] [PubMed] [Google Scholar]
- 19.Metlay JP, Witmer-Pack MD, Agger R, Crowley MT, Lawless D, Steinman RM. The distinct leukocyte integrins of mouse spleen dendritic cells as identified with new hamster monoclonal antibodies. J Exp Med 1990;171:1753–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mantell BS, Stefanovic-Racic M, Yang X, Dedousis N, Sipula IJ, O’Doherty RM. Mice lacking NKT cells but with a complete complement of CD8+ T-cells are not protected against the metabolic abnormalities of diet-induced obesity. PLoS ONE 2011;6:e19831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stefanovic-Racic M, Perdomo G, Mantell BS, Sipula IJ, Brown NF, O’Doherty RM. A moderate increase in carnitine palmitoyltransferase 1a activity is sufficient to substantially reduce hepatic triglyceride levels. Am J Physiol Endocrinol Metab 2008;294:E969–E977 [DOI] [PubMed] [Google Scholar]
- 22.Lu L, Woo J, Rao AS, et al. Propagation of dendritic cell progenitors from normal mouse liver using granulocyte/macrophage colony-stimulating factor and their maturational development in the presence of type-1 collagen. J Exp Med 1994;179:1823–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woo J, Lu L, Rao AS, et al. Isolation, phenotype, and allostimulatory activity of mouse liver dendritic cells. Transplantation 1994;58:484–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wack KE, Ross MA, Zegarra V, Sysko LR, Watkins SC, Stolz DB. Sinusoidal ultrastructure evaluated during the revascularization of regenerating rat liver. Hepatology 2001;33:363–378 [DOI] [PubMed] [Google Scholar]
- 25.Tomiyama K, Murase N, Stolz DB, et al. Characterization of transplanted green fluorescent protein+ bone marrow cells into adipose tissue. Stem Cells 2008;26:330–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner MS, Kane LP, Morel PA. Dominant role of antigen dose in CD4+Foxp3+ regulatory T cell induction and expansion. J Immunol 2009;183:4895–4903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brake DK, Smith EO, Mersmann H, Smith CW, Robker RL. ICAM-1 expression in adipose tissue: effects of diet-induced obesity in mice. Am J Physiol Cell Physiol 2006;291:C1232–C1239 [DOI] [PubMed] [Google Scholar]
- 28.Blasius AL, Giurisato E, Cella M, Schreiber RD, Shaw AS, Colonna M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J Immunol 2006;177:3260–3265 [DOI] [PubMed] [Google Scholar]
- 29.Sumpter TL, Abe M, Tokita D, Thomson AW. Dendritic cells, the liver, and transplantation. Hepatology 2007;46:2021–2031 [DOI] [PubMed] [Google Scholar]
- 30.Inaba K, Turley S, Yamaide F, et al. Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J Exp Med 1998;188:2163–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shu SA, Lian ZX, Chuang YH, et al. The role of CD11c+ hepatic dendritic cells in the induction of innate immune responses. Clin Exp Immunol 2007;149:335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fields RC, Shimizu K, Mulé JJ. Murine dendritic cells pulsed with whole tumor lysates mediate potent antitumor immune responses in vitro and in vivo. Proc Natl Acad Sci USA 1998;95:9482–9487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feili-Hariri M, Dong X, Alber SM, Watkins SC, Salter RD, Morel PA. Immunotherapy of NOD mice with bone marrow-derived dendritic cells. Diabetes 1999;48:2300–2308 [DOI] [PubMed] [Google Scholar]
- 34.Thomson AW, Lu L, Murase N, Demetris AJ, Rao AS, Starzl TE. Microchimerism, dendritic cell progenitors and transplantation tolerance. Stem Cells 1995;13:622–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKenna HJ, Stocking KL, Miller RE, et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood 2000;95:3489–3497 [PubMed] [Google Scholar]
- 36.Macia L, Delacre M, Abboud G, et al. Impairment of dendritic cell functionality and steady-state number in obese mice. J Immunol 2006;177:5997–6006 [DOI] [PubMed] [Google Scholar]
- 37.Ortega Martinez de Victoria E, Xu X, Koska J, et al. Macrophage content in subcutaneous adipose tissue: associations with adiposity, age, inflammatory markers, and whole-body insulin action in healthy Pima Indians. Diabetes 2009;58:385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lloyd CM, Phillips AR, Cooper GJ, Dunbar PR. Three-colour fluorescence immunohistochemistry reveals the diversity of cells staining for macrophage markers in murine spleen and liver. J Immunol Methods 2008;334:70–81 [DOI] [PubMed] [Google Scholar]
- 39.Jomantaite I, Dikopoulos N, Kröger A, et al. Hepatic dendritic cell subsets in the mouse. Eur J Immunol 2004;34:355–365 [DOI] [PubMed] [Google Scholar]
- 40.Liu J, Divoux A, Sun J, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med 2009;15:940–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab 2008;8:301–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu H, Perrard XD, Wang Q, et al. CD11c expression in adipose tissue and blood and its role in diet-induced obesity. Arterioscler Thromb Vasc Biol 2010;30:186–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 2009;15:914–920 [DOI] [PubMed] [Google Scholar]
- 44.Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 2009;15:930–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winer DA, Winer S, Shen L, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 2011;17:610–617 [DOI] [PMC free article] [PubMed] [Google Scholar]