

Abstract
An accurate, predictive understanding of protein-DNA binding specificity is crucial for the successful design and engineering of novel protein-DNA binding complexes. In this review, we summarize recent studies that use atomistic representations of interfaces to predict protein-DNA binding specificity computationally. Although methods with limited structural flexibility have proven successful at recapitulating consensus binding sequences from wild-type complex structures, conformational flexibility is likely important for design and template-based modeling, where non-native conformations need to be sampled and accurately scored. A successful application of such computational modeling techniques in the construction of the TAL-DNA complex structure is discussed. With continued improvements in energy functions, solvation models, and conformational sampling, we are optimistic that reliable and large-scale protein-DNA binding prediction and engineering is a goal within reach.
Introduction
Sequence-specific interactions between proteins and nucleic acids play a central role in a wide variety of cellular processes, including transcriptional and translational regulation, DNA replication, and DNA damage repair. The ability to accurately predict and rationally engineer the specificity of these interactions would have a transformative impact on biology and medicine. Here we review recent progress in molecular modeling of protein-DNA interactions, focusing on algorithms for prediction of DNA-binding specificity. In addition to its intrinsic importance for unraveling regulatory interactions, binding specificity prediction provides critical feedback for structure-based design of protein-DNA interactions: indeed, we suggest that a major barrier to successful redesign of DNA binding specificity is the difficulty in modeling the changes in interface structure and/or specificity induced by designed mutations (Fig. 1). Given the polar nature of protein-DNA interfaces, which often feature specific water-bridged interactions, binding specificity prediction is also a stringent test of electrostatics models and representations of aqueous solvation.
Figure 1. The importance of flexibility in protein-DNA modeling.

(A) Shifts in DNA bases (yellow arrows) are evident in template-based modeling of one zinc finger (the target, shown in red and pink) using another (the template, shown in blue). Interfaces are superimposed based on their protein components. Protein sidechains in the target whose DNA contacts would be disrupted by these DNA shifts are shown in stick representation (example taken from [7]). (B) Superposition of design model (pink) and experimental structure (blue) for the I-MsoI ‘-7C’ design of Ashworth et al. [47] reveals a shift of 2.9 Å in the DNA backbone and a 180° flip of a designed Tryptophan residue. These examples suggest that incorporation of backbone conformational flexibility may be necessary for reliable prediction and design of protein-DNA interactions.
Structure-based prediction of protein-DNA binding specificity
Prediction of the DNA binding specificity of a protein of interest (Fig. 2) typically begins with a structure of the protein bound to a DNA target site. Models of the protein bound to alternate sites (often all single base-pair mutants of the target site) are built from this structure, and relative binding affinities are computed for these sites by comparing the energies of the starting and mutant complexes under a suitable energy function. For benchmarking purposes, these predicted binding affinities may be compared with corresponding experimental values, or a position weight matrix (PWM) model can be constructed from the calculated energies and compared with one estimated from known target sites or high-throughput experimentation [1-3, 4*].
Figure 2. Flowchart of structure-based calculation of the protein-DNA binding specificity.

Step 1. A protein-DNA complex structure, taken from either experiments (X-ray or NMR) or molecular modeling (homology modeling or docking), serves as the input of the method. Here the Zif268 zinc finger protein is shown on the left in cartoon representation as an example (PDB ID: 1AAY). Step 2. The DNA base-pairs in the target site are mutated, perhaps exhaustively to all possible sequences (4L sequences for a site of length L), or more commonly to all single-base mutants (3L sequences). A superposition of four possible base pairs at a given position is shown in stick representation on the left. Step 3. The protein-DNA binding energy change caused by the DNA sequence mutation(s) is evaluated, using either a knowledge-based potential or a molecular mechanics force field. For methods that use molecular mechanics force fields, the binding energy difference is typically evaluated using the thermodynamic cycle shown on the left. Here the protein-DNA binding energy difference caused by the DNA sequence mutation (ΔΔG) is obtained by subtracting the unbound DNA energy (ΔGU) from the bound protein-DNA energy (ΔGB). Step 4. When single base pair mutations are performed, based on the approximation of DNA position independence, the binding energies of the different complexes can be converted into a position weight matrix and represented as a sequence logo [48] to show the binding specificity graphically (such as the logo shown on the left). When the DNA sequence space is exhaustively searched, a collection of high-affinity DNA sequences (such as those with a predicted binding affinity within some cutoff value of the optimal sequence) can also be represented as a sequence logo, although it should be noted that these sequences contain information regarding inter-position correlation that can be represented by higher-order graphical representations [49].
Classification scheme
Here we review a representative subset of recently described approaches to structure-based specificity prediction, organized according to the energy functions and conformational sampling approaches employed. To aid in describing and differentiating these methods, and highlight key issues in structure-based specificity prediction, we provide in Table 1 the answers to the following five questions for each of the discussed methods.
Table 1.
Structure-based specificity prediction studies, with answers to the five questions introduced in the text (see “Classification scheme”).
| Citation | Method or software | Energy function | Relaxation of mutants | Unbound DNA | B-P independence | Tested w/templates? | |||
|---|---|---|---|---|---|---|---|---|---|
| intra-DNAa | Proteinb | DNAc | scoringd | samplinge | |||||
| Angarica [20] | DNAPROT | KB | harmonic | N | N | N | N | Y | Y |
| Alamanova [22] | Statistical potential | KB | - | N | N | N | Y | Y | Y |
| Donald [21] | Statistical potential | KB | - | N | N | N | Y | Y | N |
| AlQuraishi [23] | Compressed sensing | KB | - | N | N | N | Y | Y | N |
| Havranek [7] | Rosetta | MM | - | fixbb-rot | N | N | N | N | Y |
| Morozov-stat [24] | Rosetta | MM | harmonic | N | N | N | N | Y | Y |
| Morozov-dyn[24] | Rosetta | MM | harmonic | fixbb-rotmin | min | Y | N | Y | Y |
| Alibes [25*] | FoldX | MM | MM/KB | fixbb | fixbb | N | N | Y | N |
| Siggers [29*] | Tinker | MM | MM | fixbb-rotmin | fixP-rotmin | Y | N | N | Y |
| Rahi [31] | Amber | MM | MM | cst-min | cst-min | Y | N | Y | N |
| Liu [32] | CHARMM | MM | MM | MD | MD | Y | N | Y | N |
| Beierlein [33] | Amber | MM | MM | MD | MD | Y | N | Y | N |
| Seeliger [34*] | Gromacs | MM | MM | MD | MD | Y | N | Y | N |
| Moroni [36] | CHARMM/NAMD | MM | MM | min or MD | min or MD | N | N | N | N |
| Temiz [37*] | Experimental contact energies | KB | - | N | N | N | N | N | Y |
| Zakrzewska [13*,14*] | ADAPT | MM | MM | fixbb-min | min | Y | N | N | N |
| Yanover [39*] | Rosetta | MM | MM | MC | MC | Y | N | N | Y |
The intra-DNA energy function; ‘KB’ denotes knowledge-based potential; ‘MM’ denotes molecular mechanics force field; ‘harmonic’ indicates a model for DNA strain [11] that is quadratic in the base-step (and possibly base-pair) parameters.
Conformational relaxation applied to protein after DNA mutation; ‘N’: none; ‘fixbb’: backbone is fixed; ‘rot’: rotameric sidechain sampling; ‘min’: gradient-based energy minimization; ‘cst’- energetic restraints on the atomic positions; ‘MD’: molecular dynamics relaxation; ‘MC’: Monte Carlo optimization.
Conformational relaxation applied to DNA after mutation; ‘N’, ‘min’, ‘MD’, ‘MC’, ‘cst’: as above; ‘fixbb’: sugar-phosphate backbone is fixed; ‘fixP’: phosphorus atoms are fixed; ‘rot’: small-perturbation DNA rotamers.
Are DNA binding energies position independent? ‘Y’: yes or ‘N’: no.
Does the DNA sequence sampling protocol assume independence between positions (e.g., all single-based mutants of the crystallized target site)? ‘Y’: yes or ‘N’: no.
What type of energy function is employed? Here we focus exclusively on studies that evaluate binding energies using atomically detailed descriptions of complexes. Recent studies can be roughly divided according to whether they use knowledge-based (KB) or molecular mechanics (MM) energy functions. In knowledge-based approaches, the shape of the distance-dependent atomic interaction energy curve is estimated from an experimental dataset such as a collection of protein-DNA complex structures. MM energy functions, on the other hand, are composed of independent, physicochemical energy terms that capture van der Waals and Coulombic interactions, solvation effects, and other atomic interaction energies. While the weights on these terms may be fit to experimental data, the functional forms of the individual terms are more constrained than for KB approaches. For the purposes of this review, we include in the MM category physics-based force fields such as FoldX [5] and Rosetta [6,7] that may incorporate knowledge-based energy components (such as rotamer probability estimates) in addition to standard MM terms.
When building structural models of mutant DNA target sites, what degree of conformational relaxation is allowed after base mutation? DNA mutations are typically performed so as to preserve the plane of the base, the geometry of the glycosidic linkage, and the conformation of the sugar phosphate backbone. If no further conformational relaxation is allowed, this procedure may introduce atomic overlaps and/or unfavorable electrostatic interactions – either internal to the DNA or in the protein-DNA interface – that have the potential to bias the specificity profile toward the DNA site present in the starting structure. When starting from a co-crystal structure of the native protein bound to the high-affinity consensus target site, this effect may enhance performance; in template-based specificity predictions or when assessing the quality of a large-scale specificity redesign, on the other hand, it seems likely that some degree of conformational relaxation is necessary (Fig. 1). For example, sequence-dependent fluctuations in minor groove width, which can contribute to binding specificity for certain families [8], have been successfully modeled using Monte Carlo simulations [9]. For the classification given in Table 1, we consider whether protein and/or DNA backbone flexibility is allowed, and whether local energy minimization or more intensive conformational optimization is used. The latter distinction is motivated by the observation that energy minimization (which involves a search for a nearby local energy minimum) may not sample the discrete rearrangements of interface protein sidechains that are accessible to rotamer-based optimization protocols.
Is the energy of the unbound DNA site considered? Binding affinities involve energy differences between the bound and unbound states. Evaluating only the energies of the bound complexes risks missing indirect readout mechanisms, which depend on the sequence-dependent deformability of DNA and are determined in part by energetic differences between the bound and unbound state. For example, the greater flexibility of pyrimidine-purine base steps (TpA, CpA, and CpG) is thought to reflect weaker stacking interactions for these steps in unbent B-form DNA [10]. Capturing this effect requires either scoring of unbound models of the DNA sequence, or the explicit inclusion of a knowledge-based scoring term that measures these geometric preferences (such as harmonic base-step potentials derived from crystal structures [11] or molecular dynamics (MD) simulations [12]). Explicit treatment of the unbound state is also critical when there are differences in the internal energy of base pairs or base steps of different sequences, as occurs when intra-DNA interactions are evaluated with an MM potential.
Is independence assumed between DNA base pairs? Positional independence may be a feature of the energy calculations themselves, or may follow from the extent of DNA sequence sampling that is performed to construct a binding specificity model. In the absence of intra-DNA energetics and conformational relaxation of mutant complexes, the binding affinity contribution of a DNA base at one position in the target site is independent of the base identities at other positions. Even when calculated energies are not strictly pairwise independent, however, many studies choose to sample only single base mutants from a consensus target site. The ADAPT method [13*,14*], by contrast, evaluates binding energies for all possible variants of the target site, and thus is able to capture energetic correlations between DNA sequence positions. Although positional independence has been shown to be a reasonably good approximation to binding energetics overall [15], there are clear cases where this assumption breaks down [16], which may be associated with sequence-dependent DNA bending or protein sidechain rearrangements at the interface.
Is the approach tested with homologous templates? We expect that performance evaluations based only on predictions made from structures of native, high-affinity complexes will tend to reward protocols that bias toward the crystal structure sequence, whereas such protocols would be expected to perform less well when predicting binding specificity from the structure of a template with divergent binding preferences or when assessing the quality of a large-scale specificity redesign. Given the limited experimental coverage of protein-DNA complexes, the ability to make accurate predictions starting from structures of homologous protein-DNA complexes [17], or unbound protein structures via protein-DNA docking [18,19], is highly desirable; such applications are, however, likely to require significant backbone and sidechain conformational sampling.
Knowledge-based potentials
Several groups have explored the use of knowledge-based potential energy functions in the context of a static model in which little or no conformational adjustment is performed after base mutation. The DNAPROT method [20] uses a potential energy function that combines Olson’s harmonic model of DNA flexibility [11] with three atomic preference matrices (for hydrogen bonds, water-mediated hydrogen bonds, and hydrophobic interactions) derived from a large database of protein-DNA interfaces. This method performed well at PWM prediction from native structures, and even generated reasonable PWM predictions for two targets whose structures were modeled by homology (with rotamer-based optimization of sidechains at the protein-DNA interface in the context of the template DNA sequence). Donald et al. [21] compared the performance of several statistical potential functions and found that the quasichemical approach outperformed other potentials, including the Amber99 MM force field. Alamanova et al. [22] derived a distance-dependent all-atom statistical potential from protein-DNA crystal structures and successfully applied it to predict PWMs for transcription factors (TFs) in the p53 and NF-κB families. Compressed sensing methods were used by AlQuraishi and McAdams [23] to learn a “de novo” interatomic energy potential from protein-DNA interface structures and binding energies. In this approach, protein-DNA binding interactions are viewed as mesoscopic sensors of an underlying microscopic signal, the atomic interaction potential, which is fit by a regularized logistic regression procedure that enforces sparsity of the components in order to avoid over-training. This approach gave improved results over the quasichemical and DNAPROT methods (as well as the MM approach of Morozov et al. [24], see below) in recovery of crystal structure DNA sequences for the helix-turn-helix superfamily; transferability to other families and applicability of the potential to prediction of specificity from homology models or models where relaxation upon mutation is performed remain to be demonstrated.
Molecular mechanics force fields and conformational relaxation
Havranek et al. [7] developed a simple physicochemical model for prediction and design of protein-DNA interactions that featured fixed-backbone, rotameric sampling of interface sidechains together with a frozen DNA approximation. Accurate specificity predictions from native structures were demonstrated, and binding preferences for a set of zinc finger mutants were predicted by homology modeling with good experimental agreement for most targets. Morozov et al. [24] compared specificity prediction approaches with (“dynamic model”) and without (“static model”) conformational relaxation of mutant complexes, and found that the static model gave better performance overall, noting however that this model “…is more likely to fail when DNA conformational change is required to predict novel sites by homology.” The potential function combined MM terms with a harmonic model for DNA strain inspired by Olson et al. [11]; during conformational relaxation of mutant complexes the protein backbone and protein-DNA interface were fixed while protein sidechains and DNA dihedral angles were adjusted by Monte Carlo rotamer optimization and gradient-based minimization. Serrano and co-workers have used the FoldX force field [5] to predict PWMs from native crystal structures using a protocol that features limited conformational rearrangement of mutant complexes, with promising benchmark results [25*]. The FoldX model has been used to predict the effect of protein and DNA mutations on DNA-binding affinities of the Pax6 paired domain [25*], as well as in a variety of structure-based specificity redesign applications [26,27].
Using a molecular mechanics potential based on the Amber [28] force field, Siggers and Honig [29*] found that fixed-backbone, template-based specificity predictions for C2H2 zinc fingers are highly sensitive to the degree of structural divergence between target and template interfaces. Their protocol featured sidechain relaxation of mutant complexes (using a protein conformer library, small-perturbation DNA rotamers, and gradient-based energy minimization) as well as explicit evaluation of unbound DNA energies; it was able to generate highly accurate predictions when given the structure of the target protein (Zif268) or a structurally similar template. Conformational relaxation was found to be important for correctly predicting binding affinity for a series of target sites (as opposed to recapitulating a consensus sequence). Using constrained energy minimization of mutant complexes with the Amber force field and a generalized Born solvation model [30] (and explicit scoring of unbound DNA sites), Rahi et al. [31] successfully recapitulated experimental ΔΔG values for the PurR TF and single amino acid variants, and examined the relative contributions of direct and indirect readout mechanisms to DNA recognition by this TF.
Molecular dynamics simulations and free energy calculations
MD simulations have been widely used to study the energetics of macromolecular binding events, and several groups have used MD simulations and free energy calculations to obtain relative binding affinities of target proteins for alternate DNA sites. In so-called alchemical free energy simulations, the free energy difference between two states is calculated by sampling along an artificial “alchemical” transformation coordinate (λ) that links the two states (e.g., the wild-type and mutant base pairs), using simulations at multiple, fixed values of λ or in which λ is varied continuously between the states. Liu and Bader [32] used the thermodynamic integration (TI) approach and a dual topology representation (in which both states are simultaneously present, with interactions scaled by λ) of the mutated base pair to calculate relative binding affinities for all single base mutations of a TF target site. Calculations for the homeodomain Mat-α2 and bZIP GCN4 showed good agreement with experimental binding data, and analysis of the trajectories suggested an important role for water molecules in sequence-specific recognition. Beierlein et al. [33] investigated the specificity of C-protein:DNA recognition at a highly conserved G:C base pair using TI with a dual topology representation and a soft-core treatment of non-bonding interactions (to avoid singularities at the endpoints of the alchemical transformation). In agreement with experiment, all three base mutations were predicted to destabilize binding. Seeliger et al. [34*] applied both an equilibrium approach (using simulations at fixed λ-values) and a non-equilibrium approach (based on work distributions from many short simulations during which λ is varied between the two states) to compute relative binding affinities of the TF Zif268 for single mutants of its preferred target site. The spacing of intermediate states along the transformation coordinate was chosen so as to maximize phase space overlap between neighboring states. Agreement with experiment was encouraging, with deviations from experimental ΔΔG’s averaging less than 1 kcal/mol. For the non-equilibrium method, insufficient sampling of the endpoint states was found to be a major source of error. Taken together, the results of these alchemical free energy calculations are highly encouraging, although further benchmarking is necessary to assess performance in more challenging applications such as template-based specificity prediction and prediction of binding specificity changes induced by protein mutations. In addition, due to the high computational cost of MD simulations [32,33,35], advances in computational efficiency will likely be necessary for large-scale applications of these methods.
Moroni et al. [36] evaluated the use of MD conformational sampling in endpoint free energy calculations using a variety of implicit solvent models (distance dependent dielectric, solvent accessible surface area-based, and Generalized Born). Energetic contributions from van der Waals and Coulombic interactions and solvation effects were independently weighted to optimize the fit between predicted and experimental ΔΔG values for 52 single base-pair mutations to the lambda repressor target site. The best agreement between prediction and experiment was found when minimizing and scoring mutant complexes using a potential energy function incorporating standard MM terms as well as the Generalized Born plus solvent accessible surface area (MM/GBSA) solvation model, together with a term counting protein-DNA hydrogen bonds. Using MD conformational sampling instead of energy minimization resulted in poorer correlations with experiment, which was attributed by the authors to the presence of large conformational fluctuations in the MD trajectories.
Temiz and Camacho [37*] have described a novel approach for predicting the binding affinities of proteins in the C2H2 zinc finger family, one that incorporates MD sampling and homology modeling into an empirical scoring framework based on analysis of protein-DNA hydrogen bonds and atomic desolvation. In this model, hydrogen bond strengths are modulated by a weighting factor that reflects the solvent accessibility of the interacting atoms (computed by explicitly solvating the interface models). Energies computed with this empirical framework show very good agreement with experimental binding data for several zinc finger proteins.
Enhanced sampling of sequences and structures
The ADAPT approach of Lavery and co-workers [38] has recently been extended [13*,14*] to allow large-scale sampling of DNA sequence space with simultaneous conformational relaxation of the DNA target and neighboring protein sidechains. In this approach, the binding site is broken into overlapping five base segments, and binding energies are calculated for all 45 = 1024 possible DNA sequences in each segment. To evaluate the binding energy of a single sequence segment, bound and unbound energy minimizations are performed in which nucleotides within that segment and nearby protein sidechains are flexible while the protein backbone as well as nucleotides in other segments are held fixed. The binding energy for any full-length DNA target site can then be computed by summing segment binding energies, with a weighting scheme that accounts for residue-pair interactions present in multiple overlapping segment minimizations. This approach was applied to the DNA-binding proteins TBP and SRY and, using a grid-computing-enabled approach, to the nucleosome (147 DNA base pairs wrapped around a histone octamer). The use of MD snapshots rather than experimental structures as starting points for the protocol was explored with promising results, and the predicted nucleosome binding energy matrix was used to scan the sequence neighborhood of transcription start sites in the yeast genome, recapitulating features of the experimentally observed nucleosome distribution such as a well-known nucleosome-depleted region upstream of transcription start sites.
To enhance conformational sampling in template-based specificity prediction, a recent study [39*] using the Rosetta software package [6] extended fragment assembly techniques for monomeric protein structure prediction to protein-DNA complexes by adding DNA-duplex and protein-DNA interface fragment replacement moves. In this approach, models of protein-DNA complexes are built in a two-stage procedure consisting of an initial coarse-grained, fragment-assembly simulation followed by an all-atom refinement stage with a high-resolution MM force field. DNA sequence space is explored by the use of Monte Carlo sequence mutation moves in which a randomly selected base pair is mutated and nearby bases and sidechains are re-optimized. Starting from randomized target sites, energy-biased acceptance of these sequence moves focuses sampling in high-affinity regions of sequence space, so that binding models in reasonable agreement with experiment can be extracted from the final, optimized DNA sequences. This approach has been tested on the C2H2 zinc finger family with promising results; transferability to other protein families remains to be assessed.
TAL effector structures and simulations
Transcriptional activator-like (TAL) effectors are bacterial DNA-binding proteins that contain multiple, tandemly-repeated copies of a highly conserved 33-35 amino acid consensus sequence. TAL effectors have been shown to recognize their DNA target sites by a remarkable, modular recognition process in which individual repeat units map to successive DNA base pairs, with the identity of the amino acids at positions 12 and 13 of the repeat unit (termed the repeat-variable diresidue, or RVD) showing a high degree of correlation with the identity of the recognized base pair [40,41]. In a development with relevance to protein-DNA modeling as well as rational engineering of protein-DNA binding specificity, the first structures of TAL effectors were recently solved [42*,43*], revealing the molecular basis for their modular DNA recognition properties (Fig. 3). Notably, the crystal structure of the TAL effector PthXo1 was solved by molecular replacement using structure prediction models of the protein-DNA complex built by the fragment-assembly techniques described above [39*]. In addition, de novo models built prior to the availability of experimental data have also been published [44*], allowing a direct comparison with experiment (Fig. 3B). These studies suggest that protein-DNA interface modeling techniques may play a growing role in experimental structure determination.
Figure 3. TAL effector-DNA interactions.
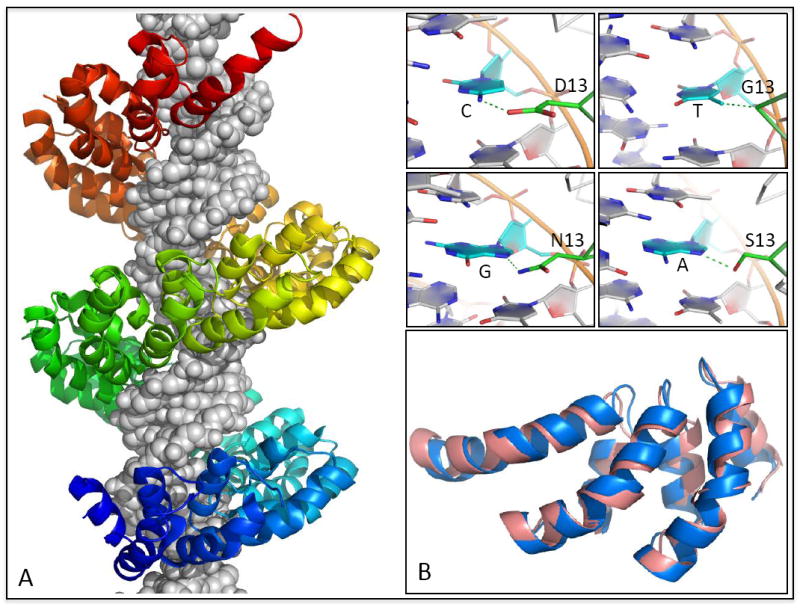
(A) Structure of the 23.5-repeat TAL effector PthXo1 bound to its target site [42*] reveals the mechanistic basis of modular DNA recognition by TAL repeats. Successive repeats form a left-handed helical bundle that wraps around the DNA duplex paralleling the major groove. Inset panels show a subset of the specificity-determining contacts formed between repeat residue 13 and its associated base-pair in the target site. (B) A de novo TAL-effector model [44*] is superimposed onto three repeats of the dHax3 [43*] TAL effector (1.2 Å Cα-RMSD over 102 residues). Whereas structural modeling played a critical role in structure determination for PthXo1, the dHax3 structure was solved by standard techniques and can serve as an independent reference for assessment of the structure predictions.
Conclusions
Atomistic molecular modeling represents one promising avenue for the rational prediction and reengineering of protein interactions. Success depends on potential energy functions that are able to accurately balance the contributions of electrostatic, hydrogen-bonding, and non-polar interactions in the presence of solvent and ions, as well as conformational sampling algorithms that can recapitulate the structural rearrangements that accompany sequence changes in binding proteins or their partners. Given the increasing availability of standardized, high-throughput experimental datasets on protein-DNA interactions [1-3], the many structural templates for these interactions in the PDB [45,46], and the largely polar (and hence challenging) nature of protein-DNA interfaces, prediction of protein-DNA binding specificity is emerging as an excellent in silico test of interface modeling approaches. In this review, we have described a representative collection of recent approaches to structure-based prediction of protein-DNA interactions, all of which depend on atomically detailed structural models of the binding interface. We conclude that the majority of these approaches give good results when predicting consensus sequences from native, high-affinity structures. In the context of this somewhat artificial test, conservative approaches with little or no conformational flexibility appear to give the best results [21,23,24,25*]. In template-based specificity, and more generally when modeling interactions not directly visualized by the input structure, there is evidence that conformational flexibility improves prediction accuracy [20,29*,39*], although the lack of comprehensive benchmarks that demand extensive sampling hinders comparison of different approaches. Moving beyond the native-recapitulation tests that dominate these studies should advance method development in this area. Looking ahead, we identify as promising research directions the accurate modeling of interfacial waters, the incorporation of cofactor binding influences by explicit modeling of multi-protein complexes, and the further development and application of backbone sampling techniques in prediction and design.
Highlights.
We review structure-based approaches for protein-DNA binding specificity prediction.
Interface flexibility is necessary for accurate template-based predictions.
Assessments on native complexes are likely biased toward conservative approaches.
Applications to structure determination and protein design are discussed.
Acknowledgments
We gratefully acknowledge the contributions of the authors whose publications we have reviewed, and apologize for any inaccuracies and omissions. We thank Justin Ashworth, Jim Havranek, and Alex Morozov for stimulating discussions and for their insightful comments on earlier versions of this article. This work was supported by a grant from the National Institutes of Health (R01GM088277).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Limin Angela Liu, Email: lliu2@fhcrc.org.
Philip Bradley, Email: pbradley@fhcrc.org.
References
Papers of particular interest, published within the period of review, have been highlighted as:
-
*
of special interest
- 1.Zykovich A, Korf I, Segal DJ. Bind-n-Seq: high-throughput analysis of in vitro protein-DNA interactions using massively parallel sequencing. Nucleic Acids Research. 2009;37 doi: 10.1093/nar/gkp802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robasky K, Bulyk ML. UniPROBE, update 2011: expanded content and search tools in the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Research. 2011;39:D124–D128. doi: 10.1093/nar/gkq992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nutiu R, Friedman RC, Luo S, Khrebtukova I, Silva D, Li R, Zhang L, Schroth GP, Burge CB. Direct measurement of DNA affinity landscapes on a high-throughput sequencing instrument. Nature Biotechnology. 2011;29:659–U146. doi: 10.1038/nbt.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *4.Stormo GD, Zhao Y. Determining the specificity of protein-DNA interactions. Nature Reviews Genetics. 2010;11:751–760. doi: 10.1038/nrg2845. A comprehensive review of state-of-the-art experimental techniques for determining protein-DNA binding specificity; both in vivo and in vitro methods are discussed. [DOI] [PubMed] [Google Scholar]
- 5.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 6.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Havranek JJ, Duarte CM, Baker D. A simple physical model for the prediction and design of protein-DNA interactions. J Mol Biol. 2004;344:59–70. doi: 10.1016/j.jmb.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 8.Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B. The role of DNA shape in protein-DNA recognition. Nature. 2009;461:1248–U1281. doi: 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rohs R, Sklenar H, Shakked Z. Structural and energetic origins of sequence-specific DNA bending: Monte Carlo simulations of papillomavirus E2-DNA binding sites. Structure. 2005;13:1499–1509. doi: 10.1016/j.str.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 10.Dickerson RE. DNA bending: the prevalence of kinkiness and the virtues of normality. Nucleic Acids Res. 1998;26:1906–1926. doi: 10.1093/nar/26.8.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olson WK, Gorin AA, Lu XJ, Hock LM, Zhurkin VB. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc Natl Acad Sci U S A. 1998;95:11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lankas F, Sponer J, Langowski J, Cheatham TE., 3rd DNA basepair step deformability inferred from molecular dynamics simulations. Biophys J. 2003;85:2872–2883. doi: 10.1016/S0006-3495(03)74710-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *13.Deremble C, Lavery R, Zakrzewska K. Protein-DNA recognition: Breaking the combinatorial barrier. Computer Physics Communications. 2008;179:112–119. [Google Scholar]
- *14.Zakrzewska K, Bouvier B, Michon A, Blanchet C, Lavery R. Protein-DNA binding specificity: a grid-enabled computational approach applied to single and multiple protein assemblies. Physical Chemistry Chemical Physics. 2009;11:10712–10721. doi: 10.1039/b910888m. These studies applied a “divide-and-conquer” approach that separated the DNA sequence into overlapping pentanucleotides and allowed exhaustive sequence search within the pentanucleotides. The exponential computational cost of sequence sampling was thus drastically reduced. [DOI] [PubMed] [Google Scholar]
- 15.Benos PV, Bulyk ML, Stormo GD. Additivity in protein-DNA interactions: how good an approximation is it? Nucleic Acids Res. 2002;30:4442–4451. doi: 10.1093/nar/gkf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Man TK, Stormo GD. Non-independence of Mnt repressor-operator interaction determined by a new quantitative multiple fluorescence relative affinity (QuMFRA) assay. Nucleic Acids Res. 2001;29:2471–2478. doi: 10.1093/nar/29.12.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao M, Skolnick J. A threading-based method for the prediction of DNA-binding proteins with application to the human genome. PLoS Comput Biol. 2009;5:e1000567. doi: 10.1371/journal.pcbi.1000567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Dijk M, Bonvin AMJJ. Pushing the limits of what is achievable in protein-DNA docking: benchmarking HADDOCK’s performance. Nucleic Acids Research. 2010;38:5634–5647. doi: 10.1093/nar/gkq222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banitt I, Wolfson HJ. ParaDock: a flexible non-specific DNA--rigid protein docking algorithm. Nucleic Acids Res. 2011;39:e135. doi: 10.1093/nar/gkr620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Angarica VE, Perez AG, Vasconcelos AT, Collado-Vides J, Contreras-Moreira B. Prediction of TF target sites based on atomistic models of protein-DNA complexes. Bmc Bioinformatics. 2008;9:436. doi: 10.1186/1471-2105-9-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donald JE, Chen WW, Shakhnovich EI. Energetics of protein-DNA interactions. Nucleic Acids Res. 2007;35:1039–1047. doi: 10.1093/nar/gkl1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alamanova D, Stegmaier P, Kel A. Creating PWMs of transcription factors using 3D structure-based computation of protein-DNA free binding energies. Bmc Bioinformatics. 2010;11 doi: 10.1186/1471-2105-11-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.AlQuraishi M, McAdams HH. Direct inference of protein-DNA interactions using compressed sensing methods. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:14819–14824. doi: 10.1073/pnas.1106460108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morozov AV, Havranek JJ, Baker D, Siggia ED. Protein-DNA binding specificity predictions with structural models. Nucleic Acids Res. 2005;33:5781–5798. doi: 10.1093/nar/gki875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *25.Alibes A, Nadra AD, De Masi F, Bulyk ML, Serrano L, Stricher F. Using protein design algorithms to understand the molecular basis of disease caused by protein-DNA interactions: the Pax6 example. Nucleic Acids Research. 2010;38:7422–7431. doi: 10.1093/nar/gkq683. After benchmarking its performance in protein-DNA binding specificity prediction, the authors apply the FoldX method to predict changes in stability and DNA-binding specificity induced by missense mutations in the transcription factor Pax6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alibes A, Serrano L, Nadra AD. Structure-based DNA-binding prediction and design. Methods Mol Biol. 2010;649:77–88. doi: 10.1007/978-1-60761-753-2_4. [DOI] [PubMed] [Google Scholar]
- 27.Redondo P, Prieto J, Munoz IG, Alibes A, Stricher F, Serrano L, Cabaniols JP, Daboussi F, Arnould S, Perez C, et al. Molecular basis of xeroderma pigmentosum group C DNA recognition by engineered meganucleases. Nature. 2008;456:107–111. doi: 10.1038/nature07343. [DOI] [PubMed] [Google Scholar]
- 28.Cheatham TE, 3rd, Cieplak P, Kollman PA. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J Biomol Struct Dyn. 1999;16:845–862. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- *29.Siggers TW, Honig B. Structure-based prediction of C2H2 zinc-finger binding specificity: sensitivity to docking geometry. Nucleic Acids Research. 2007;35:1085–1097. doi: 10.1093/nar/gkl1155. Success in template-based binding specificity predictions for the zinc finger transcription factor Zif268 is shown to depend strongly on the degree of structural similarity between the target and template binding interfaces. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 31.Rahi S, Virnau P, Mirny LA, Kardar M. Predicting transcription factor specificity with all-atom models. Nucleic Acids Res. 2008;36:6209–6217. doi: 10.1093/nar/gkn589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu LA, Bader JS. Ab initio prediction of transcription factor binding sites. Pac Symp Biocomput. 2007:484–495. [PubMed] [Google Scholar]
- 33.Beierlein FR, Kneale GG, Clark T. Predicting the Effects of Basepair Mutations in DNA-Protein Complexes by Thermodynamic Integration. Biophysical Journal. 2011;101:1130–1138. doi: 10.1016/j.bpj.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *34.Seeliger D, Buelens FP, Goette M, de Groot BL, Grubmuller H. Towards computional specificity screening of DNA-binding proteins. Nucleic Acids Research. 2011;39:8281–8290. doi: 10.1093/nar/gkr531. This study tested both equilibrium and non-equilibrium sampling for free energy calculations of protein-DNA complexes. Among all MD based approaches, this method achieved the highest accuracy in binding energy calculations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu LA, Bader JS. Structure-based ab initio prediction of transcription factor-binding sites. Methods Mol Biol. 2009;541:23–41. doi: 10.1007/978-1-59745-243-4_2. [DOI] [PubMed] [Google Scholar]
- 36.Moroni E, Caselle M, Fogolari F. Identification of DNA-binding protein target sequences by physical effective energy functions: free energy analysis of lambda repressor-DNA complexes. BMC Struct Biol. 2007;7:61. doi: 10.1186/1472-6807-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *37.Temiz NA, Camacho CJ. Experimentally based contact energies decode interactions responsible for protein-DNA affinity and the role of molecular waters at the binding interface. Nucleic Acids Research. 2009;37:4076–4088. doi: 10.1093/nar/gkp289. An empirical binding model based on hydrogen bonding energies and desolvation penalties derived from experiment predicts binding affinities for zinc finger mutants with good accuracy. Hydrogen bond strengths are scaled by a measure of solvent accessibility. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lafontaine I, Lavery R. ADAPT: a molecular mechanics approach for studying the structural properties of long DNA sequences. Biopolymers. 2000;56:292–310. doi: 10.1002/1097-0282(2000)56:4<292::AID-BIP10028>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- *39.Yanover C, Bradley P. Extensive protein and DNA backbone sampling improves structure-based specificity prediction for C(2)H(2) zinc fingers. Nucleic Acids Research. 2011;39:4564–4576. doi: 10.1093/nar/gkr048. The fragment-replacement conformational sampling strategy is extended to protein-DNA complexes and combined with large-scale exploration of DNA sequence space to generate specificity predictions for zinc finger transcription factors. Extensive backbone sampling is found to improve template-based predictions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moscou MJ, Bogdanove AJ. A Simple Cipher Governs DNA Recognition by TAL Effectors. Science. 2009;326:1501–1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 41.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Breaking the Code of DNA Binding Specificity of TAL-Type III Effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- *42.Mak AN, Bradley P, Cernadas RA, Bogdanove AJ, Stoddard BL. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science. 2012;335:716–719. doi: 10.1126/science.1216211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *43.Deng D, Yan C, Pan X, Mahfouz M, Wang J, Zhu J-K, Shi Y, Yan N. Structural Basis for Sequence-Specific Recognition of DNA by TAL Effectors. Science. 2012 doi: 10.1126/science.1215670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *44.Bradley P. Structural modeling of TAL effector-DNA interactions. Protein Sci. 2012 doi: 10.1002/pro.2034. These papers present the first 3D structural models of TAL proteins, whose modular and highly specific DNA-binding properties make them optimal tools for sequence-specific DNA targeting. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Contreras-Moreira B. 3D-footprint: a database for the structural analysis of protein-DNA complexes. Nucleic Acids Res. 2010;38:D91–97. doi: 10.1093/nar/gkp781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Norambuena T, Melo F. The Protein-DNA Interface database. Bmc Bioinformatics. 2010;11 doi: 10.1186/1471-2105-11-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ashworth J, Taylor GK, Havranek JJ, Quadri SA, Stoddard BL, Baker D. Computational reprogramming of homing endonuclease specificity at multiple adjacent base pairs. Nucleic Acids Research. 2010;38:5601–5608. doi: 10.1093/nar/gkq283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, et al. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell. 2010;18:662–674. doi: 10.1016/j.devcel.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]