

Results of a phase III randomized trial that led to approval by the U.S. Food and Drug Administration of sunitinib for the treatment of patients with progressive well-differentiated neuroendocrine tumors are presented.
Keywords: Sunitinib, Pancreatic neuroendocrine tumor
Abstract
On May 20, 2011, the U.S. Food and Drug Administration (FDA) approved sunitinib malate capsules (Sutent®; Pfizer, Inc., New York) for the treatment of progressive, well-differentiated pancreatic neuroendocrine tumors (pNETs) in patients with unresectable locally advanced or metastatic disease. In a phase III randomized trial, 171 patients received either sunitinib (37.5 mg) or placebo once daily. The progression-free survival (PFS) interval was the primary efficacy endpoint. Secondary endpoints included the overall survival (OS) time, objective response rate (ORR), patient-reported outcomes, and safety.
Based on early results favoring sunitinib, the independent data monitoring committee recommended trial termination prior to the prespecified interim analysis. This premature analysis may have led to an overestimate of the treatment effect. In the FDA analysis of investigator-assessed PFS times, the median values for the sunitinib and placebo arms were 10.2 months and 5.4 months, respectively. The ORRs were 9.3% and 0% in the sunitinib and placebo arms, respectively. The OS data were not mature at the time of approval and were confounded by 69% crossover.
Common adverse reactions in patients receiving sunitinib included diarrhea, nausea, asthenia, fatigue, neutropenia, hypertension, and palmar–plantar erythrodysesthesia syndrome. Two patients on sunitinib died as a result of cardiac failure.
The Oncologic Drugs Advisory Committee voted eight to two that, despite residual uncertainty about the magnitude of the PFS effect because of early trial termination, sunitinib demonstrated a favorable benefit–risk profile in pNET patients. The FDA concurred with the committee's assessment and granted sunitinib regular approval for this rare malignancy with few available therapies.
Introduction
Pancreatic neuroendocrine tumors (pNETs), also known as well-differentiated pancreatic islet cell tumors, are rare, with an estimated incidence in the U.S. of 0.32 per 100,000 adults [1]. Although typically slow growing with low mitotic activity, locally advanced or metastatic pNETs can be aggressive, with an estimated 10-year overall survival rate of 11% for patients with distant disease [1].
Aside from sunitinib, there are three systemic treatments approved by the U.S. Food and Drug Administration (FDA) for patients with pNETs. Streptozocin (Zanosar®; Teva Pharmaceuticals USA, Philadelphia, PA) was approved in 1982 based on response rates in patients with functional and nonfunctional pancreatic islet cell tumors [2]. Studies combining streptozocin with 5-fluorouracil and with doxorubicin have shown response rates in the range of 39%–69% [3, 4]. Octreotide acetate (Sandostatin®; Novartis Pharmaceuticals Corporation, East Hanover, NJ) was approved in 1998 for the symptomatic treatment of diarrhea and flushing episodes associated with carcinoid syndrome and vasoactive intestinal peptide tumors. Recently, everolimus (Afinitor®; Novartis Pharmaceuticals Corporation, East Hanover, NJ) was approved for the treatment of progressive NETs of pancreatic origin in patients with unresectable, locally advanced or metastatic disease based on a progression-free survival (PFS) advantage over placebo.
Sunitinib is a small molecule receptor tyrosine kinase (RTK) inhibitor that blocks signaling of multiple RTKs, including vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), Kit, and Flt-3. Sunitinib is approved by the FDA for the treatment of patients with gastrointestinal stromal tumors after disease progression on or intolerance to imatinib and for the treatment of patients with advanced renal cell carcinoma. The approved dose for these indications is 50 mg orally daily for 4 weeks, followed by 2 weeks off.
Preclinical data suggest that pNETs overexpress VEGF, PDGF, and their cognate receptors. Studies of sunitinib in a RIP-TAG mouse model of pNETs suggested activity [5, 6], and a single-arm, phase II study of sunitinib revealed a 16.7% response rate in a cohort of patients with pNETs [7]. This provided justification for a phase III study comparing sunitinib with placebo in patients with progressive pNETs.
Patients and Methods
A single international, multicenter, double-blinded, randomized, phase III study was submitted to the FDA in support of a new efficacy claim for sunitinib for patients with locally advanced or metastatic well-differentiated pancreatic islet cell tumors.
Patients had to have disease that was not amenable to local therapy with curative intent, that was measurable by the Response Evaluation Criteria in Solid Tumors (RECIST), and that had progressed on a scan in the year prior to screening. In addition, patients had to have adequate organ function and an Eastern Cooperative Oncology Group performance status score of 0 or 1. Patients could be treatment naive or could have received any number of prior treatments, including systemic or liver-directed therapy. Concomitant somatostatin analog use was permitted.
Patients were randomized 1:1 to either sunitinib (37.5 mg oral continuous daily dosing) or placebo. Randomization was balanced by region with no additional stratification factors. Patients were treated until investigator-determined RECIST progression or unacceptable toxicity. At the time of progression, placebo-treated patients were unblinded and offered crossover to sunitinib in an open-label continuation study. Tumor imaging was performed at screening, week 5, week 9, and every 8 weeks thereafter.
The primary endpoint, the PFS interval, was defined as the time from randomization to investigator-determined progression of disease, death for any reason, or radiation therapy. Censoring definitions were standard. Secondary endpoints included the overall survival (OS) time, objective response rate (ORR), and patient-reported outcomes (PRO) using the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire C30.
The study was designed to have 90% power to detect a 50% longer (hazard ratio [HR] of 0.67) median PFS time, 7.6 months versus 5.1 months, in patients randomized to sunitinib. The corresponding planned sample size was 340 patients. The final PFS analysis, planned at 260 events, was to compare PFS outcomes between the two treatments with a two-sided α value of 0.049 after an adjustment for one planned interim analysis at 130 events.
The major change in study conduct occurred with the third protocol amendment, ∼8 months after accrual began, which installed an independent data monitoring committee (IDMC). The IDMC had access to unblinded subject treatment assignment. In addition to monitoring the safety of subjects, the IDMC was to be involved in the conduct and interpretation of the prespecified interim analysis of efficacy at 130 PFS events.
Results
In February 2009, at its third meeting, the IDMC recommended closure of the study based on its review of the preliminary safety and efficacy data after 73 PFS events (28.1% of the planned events) had been observed. The sponsor agreed with the recommendation of the IDMC and notified investigators in March 2009 that the study would be closed and all patients should be offered open-label sunitinib. The final PFS analysis was based on an April 15, 2009 data cutoff, at which time there were 171 patients enrolled and 81 events observed. These 171 randomized patients (sunitinib, n = 86; placebo, n = 85) comprise the intent-to-treat population. Six patients, three in each arm, were randomized but not treated and were excluded from the “as treated” safety population.
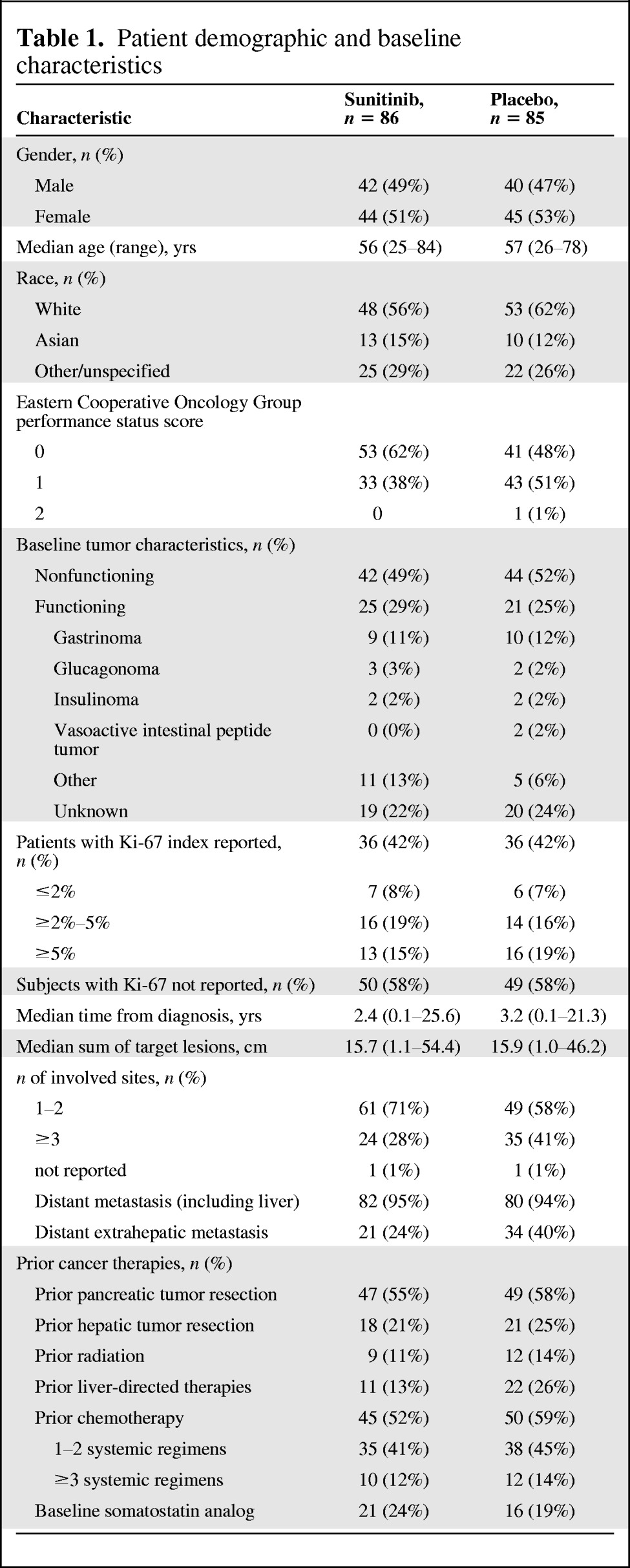
A total of 42 centers in 11 countries enrolled patients into this study. The majority of patients were from Europe (67%), followed by North America (20%). Fourteen patients (8%) were enrolled in the U.S. Table 1 summarizes the baseline patient demographics and disease characteristics. An imbalance in certain baseline characteristics between the treatment groups (such as performance status, number of disease sites, number with distant extrahepatic metastasis, and number with prior liver directed therapies) could have favored the sunitinib arm. However, the results of a post hoc sensitivity analysis of PFS outcomes performed by the FDA adjusting for these potential imbalances were consistent with the primary PFS analysis.
Table 1.
Patient demographic and baseline characteristics
Primary Endpoint
The applicant reported a difference in the median investigator-determined PFS interval of 5.9 months with sunitinib (11.4 months versus 5.5 months; HR, 0.42; 95% confidence interval [CI]; 0.26–0.66; p < .001). However, the FDA conducted an in-depth review of the submitted raw tumor measurements and case report forms and found certain discrepancies, including cases of improper use of RECIST and instances of improper handling of missing data. The FDA analysis of PFS outcomes showed that the difference in the median PFS time was 4.8 months (HR, 0.43; 95% CI, 0.27–0.67; p < .001) (Table 2, Kaplan–Meier curve Fig. 1). A forest plot of PFS times created by the FDA showed that, in all major subgroups analyzed, the PFS outcome tended to favor sunitinib, including in patients with functional and nonfunctional tumors and in patients who did and did not receive concomitant somatostatin analogs.
Table 2.
U.S. Food and Drug Administration analysis of progression-free survival outcomes
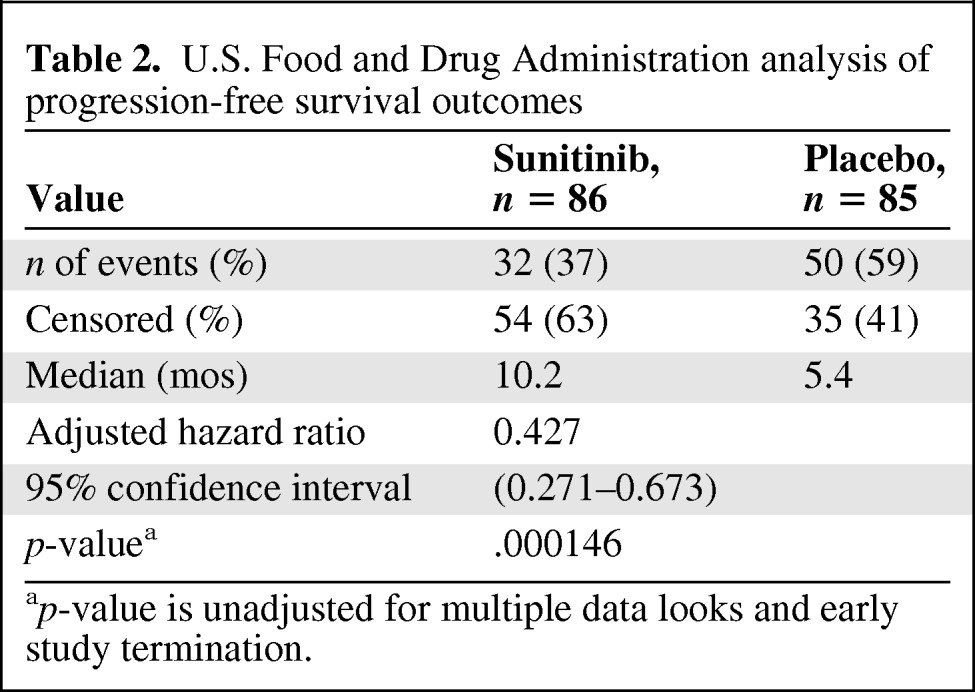
ap-value is unadjusted for multiple data looks and early study termination.
Figure 1.

Progression-free survival (PFS) probabilities in the intent-to-treat population.
Abbreviations: CI, confidence interval; HR, hazard ratio.
As requested by the FDA because of concerns about bias resulting from unblinding, the applicant performed a post hoc blinded independent central radiologic review (BICR) of scans from all patients randomized. The BICR was performed by two radiologists blinded to treatment arm, outside radiology reports, investigator assessments, and adverse events. Discrepancies were adjudicated by a third radiologist.
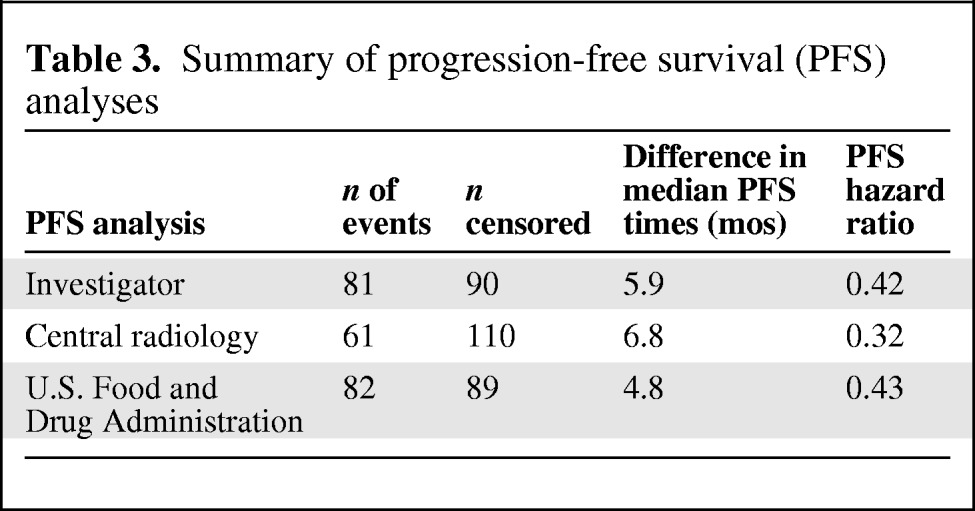
Complete scans were collected for 160 patients (93.6%). The BICR analysis showed a 6.8-month longer median PFS time with sunitinib than with placebo (12.6 months versus 5.8 months; HR, 0.32; 95% CI, 0.18–0.55; p < .0001). The overall concordance rate between the investigator analysis and the BICR for PFS outcomes was 57%. The overall concordance rate between the two central reviewers was 66%. Table 3 provides a summary of the three main PFS analyses.
Table 3.
Summary of progression-free survival (PFS) analyses
Secondary Endpoints
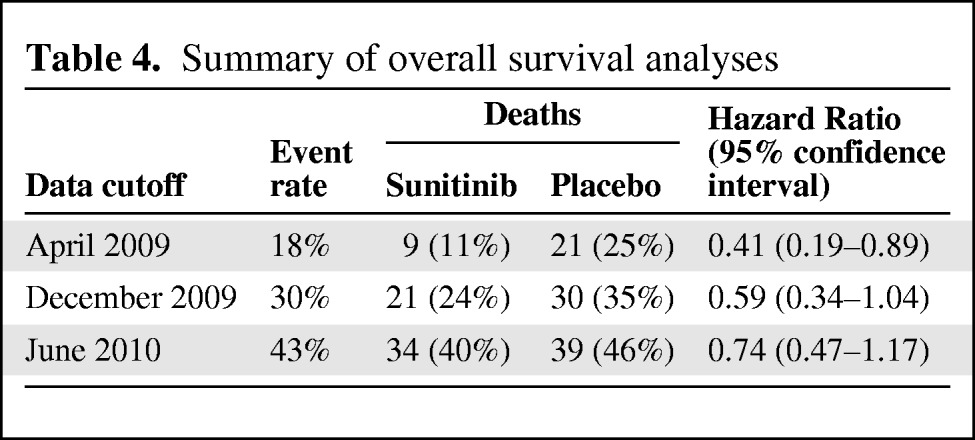
The protocol stipulated that the OS outcome would be analyzed every 2 years for 5 years or until a 95% death rate had occurred. In actuality, at the time of the supplemental new drug application submission, OS analyses had been performed three times: at the April 2009 study termination, in December 2009, and in June 2010 (Table 4). For the initial OS analysis, the 95% CI of the HR did not cross 1.0; however, the event rate of that analysis was low (18%). The subsequent OS analyses are confounded by a 69% crossover rate from placebo to sunitinib.
Table 4.
Summary of overall survival analyses
There were eight partial responses (9.3%) in patients on sunitinib and no objective responses in patients on placebo. With respect to the PRO analysis, the applicant reported no differences between sunitinib and placebo in health-related quality of life measures, functional scales, or symptom scores, except for a higher rate of diarrhea with sunitinib.
Safety data are summarized in Table 5. There were more dose interruptions, dose reductions, discontinuations, total adverse events (AEs), and grade 3–4 AEs in patients on sunitinib. There were more serious AEs and on-study deaths in patients on placebo. Two patients on sunitinib died as a result of cardiac failure. The common adverse reactions are summarized in Table 6. There were certain characteristic sunitinib adverse reactions, such as hair color changes, neutropenia, hypertension, and palmar–plantar erythrodysesthesia syndrome that may have led to inadvertent unblinding.
Table 5.
Safety summary

Abbreviation: NCI-CTC, National Cancer Institute Common Toxicity Criteria.
Table 6.
Adverse reactions occurring in ≥15% of patients in the sunitinib group and occurring more frequently (>5%) with sunitinib than with placebo
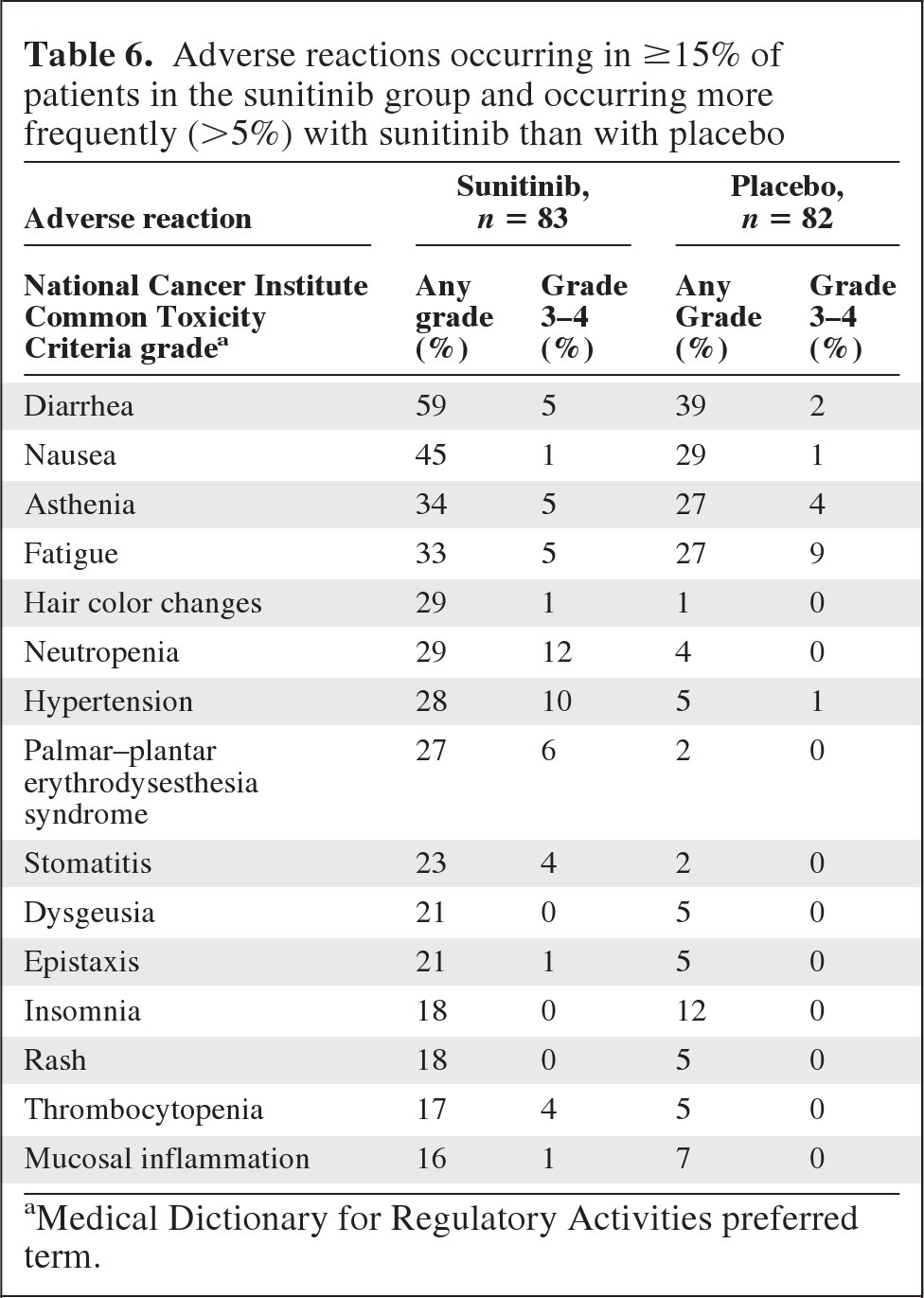
aMedical Dictionary for Regulatory Activities preferred term.
Discussion
The phase III study sought to determine whether or not sunitinib produces a longer PFS interval than with placebo in patients with well-differentiated, progressive, locally advanced or metastatic pNETs. The applicant's justification for an intermediate endpoint included the rarity and long natural history of pNETs, which would limit the feasibility of using the OS time as the primary endpoint.
In considering approval of this application, several issues were identified. One issue was that pNETs are relatively indolent cancers, and some patients with slowly growing, asymptomatic disease do not require immediate therapy. A key eligibility criterion was progression within the prior year, which may have selected for patients in greater need of therapy. The applicant did not collect data on the PFS interval seen with the prior therapy and prior to randomization, which may have provided more information on the pace of tumor growth. Nevertheless, the relatively brisk median progression time in the placebo arm of 5–6 months and the estimated median survival time of 2–3 years indicate that the tumor was aggressive for many patients.
A major issue in the first cycle of this review was potential unblinding resulting from characteristic and unusual sunitinib toxicities, such as hair color changes and palmar–plantar erythrodysesthesia syndrome, which could lead to bias in investigator determination of the PFS interval. Therefore, the FDA requested the BICR. Despite higher rates of censoring, the central review was consistent with the investigator and FDA PFS analyses. This provided reassurance that there was no systematic bias on the part of the investigators favoring sunitinib. In oncology pivotal trials, there is an ∼35%–55% discordance rate between investigator and central PFS analyses [8], and the 43% discordance rate observed in this trial was consistent with the historical data. Whether or not real-time PFS analyses can improve discordance rates and are feasible in oncology trials remains an important question [8].
The central issue surrounding this application was potential overestimation of the magnitude of the PFS effect resulting from premature trial termination and unplanned early efficacy data looks by the IDMC. Unplanned efficacy looks give a trial multiple chances to “win” and, thus, increase the risk of observing a spurious positive effect and committing a type I error. Trial designs allowing interim monitoring of efficacy data (group sequential designs) control the type I error through appropriate alpha allocation. However, irrespective of type I error control, the earlier a trial is stopped for efficacy, the more likely the observed magnitude of treatment effect is an overestimate [9]. In essence, if a trial is stopped early for efficacy, we know that the treatment has some effect; however, the magnitude of the observed treatment effect is likely an overestimate and therefore uncertain. This uncertainty is particularly problematic with a more subjective endpoint such as the PFS time.
Our concerns about potential overestimation of the PFS interval leading to uncertainty in the benefit–risk assessment were discussed at the Oncologic Drugs Advisory Committee (ODAC) meeting in April 2011. Many of the committee members shared the FDA review team's concerns. Ultimately, the committee voted eight to two that the totality of the data supported a favorable benefit–risk assessment for sunitinib in the treatment of patients with pNETs. In its final assessment, the FDA considered the comments and vote of the ODAC, the consistency of the PFS data across all analyses and subgroups, the higher response rate than with placebo, the early imbalance of deaths in the placebo arm, and the rarity of the tumor and paucity of available therapies to conclude that the benefit–risk assessment was favorable.
However, as discussed at the ODAC meeting, early and unplanned looks at PFS or OS data for evaluating the efficacy of an experimental treatment are strongly discouraged. Early and unplanned looks at the data are particularly problematic with more subjective endpoints such as the PFS interval, for which a precise estimation of the treatment effect is critical to determining the benefit–risk profile of a drug. The FDA typically recommends careful deliberation about when to conduct interim analyses for efficacy because of the greater potential for overestimating the treatment effect magnitude at earlier analysis times.
Another issue with this application was the interpretation of the OS results. It is noteworthy that the manuscript detailing the pivotal trial used the April 2009 analysis with an 18% event rate to conclude that sunitinib leads to longer survival times in patients with advanced pNETs [10]. We believe that this premature analysis is not statistically robust and no firm conclusions about OS outcomes can be made. Furthermore, with a 69% rate of crossover from placebo to sunitinib, future interpretation of OS data will be challenging. To avoid this confounding of OS results, future trials could consider eliminating crossover to experimental therapy not previously shown to be effective in later-line settings, as was recently suggested by Korn et al. [11]. However, the FDA acknowledges the potential accrual and ethical challenges with trials without crossover, particularly placebo-controlled trials. Therefore, investigators will need to balance the desire to avoid crossover confounding of OS results with ethical considerations regarding denial of potentially effective (albeit unproven) therapies to patients with serious and life-threatening conditions.
In summary, the results from the phase III study demonstrated that sunitinib has a favorable benefit–risk profile in patients with progressive, well-differentiated, locally advanced, or metastatic pNETs, which justified approval. This provides a new treatment option for this rare patient population. Future directions for treatment may include exploring combination mammalian target of rapamycin and VEGF–PDGF inhibition or inhibition of novel molecular targets as our genetic understanding of pNETs expands [12].
Acknowledgments
The views expressed are the result of independent work and do not necessarily represent the views and findings of the U.S. Food and Drug Administration. The authors acknowledge Harry B. Smith, CDER/FDA, for his contribution to editing the manuscript.
Author Contributions
Conception/Design: Gideon M. Blumenthal, Patricia Cortazar, Jenny J. Zhang, Rajeshwari Sridhara, Anthony Murgo, Robert Justice, Richard Pazdur
Provision of study material or patients: Gideon M. Blumenthal, Patricia Cortazar
Collection and/or assembly of data: Gideon M. Blumenthal, Patricia Cortazar, Jenny J. Zhang, Shenghui Tang
Data analysis and interpretation: Gideon M. Blumenthal, Patricia Cortazar, Jenny J. Zhang, Shenghui Tang, Rajeshwari Sridhara, Anthony Murgo, Robert Justice, Richard Pazdur
Manuscript writing: Gideon M. Blumenthal, Patricia Cortazar, Jenny J. Zhang, Robert Justice
Final approval of manuscript: Gideon M. Blumenthal, Patricia Cortazar, Rajeshwari Sridhara, Anthony Murgo, Robert Justice, Richard Pazdur
References
- 1.Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063–3072. doi: 10.1200/JCO.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 2.Broder LE, Carter SK. Pancreatic islet cell carcinoma. II. Results of therapy with streptozotocin in 52 patients. Ann Intern Med. 1973;79:108–118. doi: 10.7326/0003-4819-79-1-108. [DOI] [PubMed] [Google Scholar]
- 3.Moertel CG, Hanley JA, Johnson LA. Streptozocin alone compared with streptozocin plus fluorouracil in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1980;303:1189–1194. doi: 10.1056/NEJM198011203032101. [DOI] [PubMed] [Google Scholar]
- 4.Kouvaraki MA, Ajani JA, Wolff R, et al. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol. 2004;22:4762–4771. doi: 10.1200/JCO.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 5.Bergers G, Song S, Meyer-Morse N, et al. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pietras K, Hanahan D. A multitargeted, metronomic, and maximum-tolerated dose “chemo-switch” regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer. J Clin Oncol. 2005;23:939–952. doi: 10.1200/JCO.2005.07.093. [DOI] [PubMed] [Google Scholar]
- 7.Kulke MH, Lenz HJ, Meropol NJ, et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. 2008;26:3403–3410. doi: 10.1200/JCO.2007.15.9020. [DOI] [PubMed] [Google Scholar]
- 8.Dodd LE, Korn EL, Freidlin B, et al. Blinded independent central review of progression-free survival in phase III clinical trials: Important design element or unnecessary expense? J Clin Oncol. 2008;26:3791–3796. doi: 10.1200/JCO.2008.16.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellenberg SS, DeMets DL, Fleming TR. Bias and trials stopped early for benefit. JAMA. 2010;304:158. doi: 10.1001/jama.2010.933. [DOI] [PubMed] [Google Scholar]
- 10.Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501–513. doi: 10.1056/NEJMoa1003825. [DOI] [PubMed] [Google Scholar]
- 11.Korn EL, Freidlin B, Abrams JS. Overall Survival as the outcome for randomized clinical trials with effective subsequent therapies. J Clin Oncol. 2011;29:2439–2342. doi: 10.1200/JCO.2011.34.6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiao Y, Edil BH, de Wilde RF, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]