

Abstract
Purpose
To identify the underlying genetic defect in four generations of a Chinese family affected with bilateral congenital polymorphic cataracts.
Methods
Family history and clinical data were recorded. The phenotype was documented using slit-lamp photography. Genomic DNA samples were extracted from peripheral blood of family members. Candidate genes were amplified using polymerase chain reaction (PCR) and screened for mutations on both strands using bidirectional sequencing.
Results
Affected individuals exhibited variable opacities in the embryonic nucleus, sutures, and peripheral cortical opacities. The phenotype for this family was identified as polymorphic. Direct sequencing revealed a splice site mutation (c.215+1G>A) at the first base of intron 3 of the crystallin beta A3/A1 (CRYBA3/A1) gene. This mutation co-segregated with all affected individuals in the family and was not found in unaffected family members or in 100 unrelated controls.
Conclusions
Our results identified a recurrent c.215+1G>A mutation in CRYBA3/A1 in a polymorphic congenital cataract family, summarized the variable phenotypes among the patients, which expanded the phenotypic spectrum of congenital cataract in a different ethnic background, and suggested a mechanism that influences cataractogenesis.
Introduction
Congenital cataract, the loss of eye lens transparency, is a significant cause of visual impairment or blindness in childhood. The prevalence of congenital cataracts is 1 to 6 per 10,000 live births, depending on the ascertainment method [1]. Globally, congenital cataracts account for nearly one-tenth of childhood blindness from different causes including infections during embryogenesis, metabolic disorders (galactosemia), and genetic defects [2].Statistical analyses have revealed that about one quarter of congenital cataracts are hereditary [3].Genetically, the majority of isolated congenital cataracts exhibit as autosomal dominant, although autosomal recessive and X-linked inherited forms have also been reported [4].
Over the past few years, remarkable progress has been made toward our understanding of the cataractogenesis process. Currently, there are more than 40 genetic loci to which isolated or primary cataracts have been mapped, and more than 26 genes have been characterized, although this number is constantly increasing [5]. Autosomal dominant congenital cataracts (ADCC) was reportedly caused by mutations in different genes [2]. Approximately half of the mutations are in the crystallin genes and a quarter in connexin genes, with the remainder divided among genes that encode heat shock transcription factor-4 (HSF4), aquaporin-0 (AQP0, MIP), paired-like homeodomain 3 (PITX3), v-maf musculoaponeurotic fibrosarcoma oncogene homolog (MAF), chromatin modifying protein (CHMP4B), lens intrinsic membrane protein 2 (LIM2), beaded filament structural protein-2 (BFSP2), and other genes [2,6]. The crystallin and connexin genes appear to be the most commonly associated with congenital cataract. So, it is suitable to consider these genes as the top candidates for developing congenital cataracts screening strategies.
Congenital cataracts can be classified into several subtypes according to morphology: total, nuclear, cortical, anterior polar, posterior polar, lamellar, cerulean, pulverulent, sutural, coralliform, wedge-shaped, and polymorphic cataracts and other minor subtypes [2].Congenital cataracts are genetically heterogeneous [7]. It is known that different mutations in different genes can cause similar cataract patterns, while the highly variable cataract morphologies within some families suggest that the same mutation in a single gene can lead to different phenotypes [8,9].
In this paper, a four-generation family affected with congenital polymorphic cataracts was investigated in an attempt to identify the genetic defect associated with their cataract phenotype.
Methods
Clinical evaluations and DNA specimens
Four generations of a family suffering with ADCC were recruited from the Eye Center of Affiliated Second Hospital, College of Medicine, Zhejiang University, Hangzhou, China. Informed consent was obtained from all participants in accordance with the Zhejiang Institutional Review Board and the study protocol adhered to the tenets of the Declaration of Helsinki. In total, 11 individuals participated: 7 affected and 4 unaffected (Figure 1). Detailed medical histories were obtained by interviewing all individuals. All participants underwent detailed ophthalmic examinations including visual acuity, slit lamp examination with dilated pupils, ultrasonography, fundus exam, and intraocular pressure measurement. The phenotypes were documented using slit lamp photography (Figure 2). Also,100 unrelated ethnically-matched controls with no family history of congenital cataracts were recruited.
Figure 1.
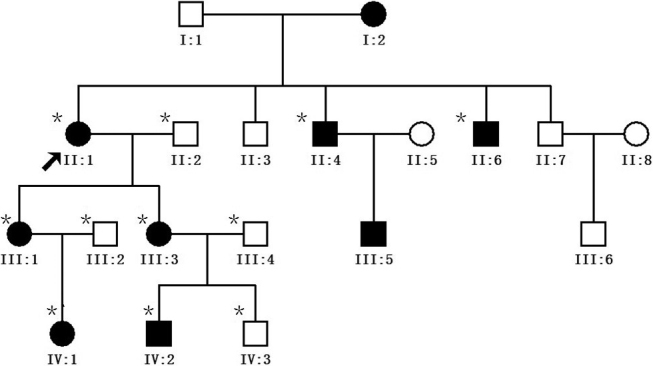
Pedigree of the autosomal dominant congenital cataract mutation. The proband is marked with an arrow. Squares and circles indicate males and females, respectively. Black and white symbols represent affected and unaffected individuals, respectively. The asterisks indicate family members who attend this study.
Figure 2.
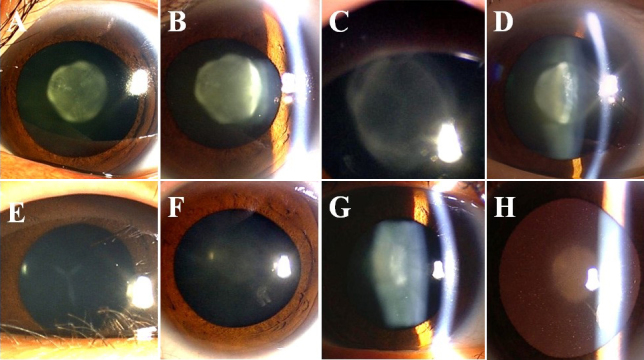
Slit-lamp photograph of family members with congenital cataracts. A, B: The proband (II:1) had nuclear cataract with 'Y' sutural opacities. C, D: The affected member IV:2 showed a different zonular cataract with 'Y' sutural opacities. E: The affected member IV:1 had simple 'Y' sutural opacities. F-H: The affected member III:3 had slight nuclear cataract with curd-like peripheral cortical opacities.
About 2 ml of peripheral blood was collected from the family members and the controls who took part in the study. Blood samples were obtained by venipuncture, collected in Vacutainer tubes (Becton-Dickinson, Franklin Lakes, NJ) containing ethylene diamine tetraacetic acid (EDTA). Leukocyte genomic DNA was extracted using the QIAmp Blood kit (Qiagen, Duesseldorf, Germany).
Mutation analysis
Genomic DNA samples from affected and unaffected members of the family were screened for mutations in crystallin alpha A (CRYAA), crystallin alpha B (CRYAB), crystallin beta A3/A1 (CRYBA3/1), crystallin beta B2 (CRYBB2), crystallin gamma C (CRYGC), crystallin gamma D (CRYGD), gap junction protein, alpha 3 (GJA3), and gap junction protein, alpha 8 (GJA8) genes using direct sequencing. The coding regions of candidate genes were amplified using polymerase chain reaction (PCR) with previously published primer sequences (Table 1) [10-17]. The cycling conditions for PCR were 95 °C pre-activation for 5 min, 10 cycles of touchdown PCR with a 0.5 °C down per 60 °C to 55 °C cycle, followed by 30 cycles with denaturation at 95 °C for 25 s, annealing at 55 °C for 25 s, and extension at 72 °C for 40 s. PCR products were isolated using electrophoresis on 3% agarose gels and sequenced using the BigDye Terminator Cycle sequencing kit V 3.1 (ABI–Applied Biosystems; Sangon Co, China) on an ABI PRISM 3730 Sequence Analyzer (ABI), according to the manufacturer’s instructions. Sequencing results were analyzed using Chromas 1.62 and compared with sequences from NCBI GenBank (CRYAA: 21q22.3; NM_000394, CRYAB: 11q22; NG_009824, CRYBA1: 17q11-q12; NM_005208, CRYBB2: 22q11.2; NM_000496, CRYGC: 2q33-q35; NM_020989, CRYGD: 2q33-q35; NM_006891.3, GJA3: 13q11-q13; NM_021954, and GJA8: 1q21-q25; NM_005267).Direct sequencing was also used to screen the mutation identified in CRYBA1on 100 ethnically-matched controls to confirm the mutation.
Table 1. Polymerase chain reaction primers and product sizes.
| Name | Primer sequence (5′-3′) | Product size (bp) |
|---|---|---|
| CRYBA3/1 | ||
| Exon-1 F | 5′GGCAGAGGGAGAGCAGAGTG 3′ | 207 |
| Exon-1 R | 5′CACTAGGCAGGAGAACTGGG 3′ | |
| Exon-2 F | 5′AGTGAGCAGCAGAGCCAGAA 3′ | 293 |
| Exon-2 R | 5′GGTCAGTCACTGCCTTATGG 3′ | |
| Exon-3 F | 5′AAGCACAGAGTCAGACTGAAGT 3′ | 269 |
| Exon-3 R | 5′CCCCTGTCTGAAGGGACCTG 3′ | |
| Exon-4 F | 5′GTACAGCTCTACTGGGATTG 3′ | 357 |
| Exon-4 R | 5′ACTGATGATAAATAGCATGAACT 3′ | |
| Exon-5 F | 5′GAATGATAGCCATAGCACTAG 3′ | 290 |
| Exon-5 R | 5′TACCGATACGTATGAAATCTGA 3′ | |
| Exon-6 F | 5′CATCTCATACCATTGTGTTGAG 3′ | 295 |
| Exon-6 R | 5′GCAAGGTCTCATGCTTGAGG 3′ | |
| CRYAA | ||
| Exon-1 F | 5′CTTAATGCCTCCATTCTGCT 3′ | 593 |
| Exon-1 R | 5′TGGCTGGTGCCTTACAAA 3′ | |
| Exon-2 F | 5′ CACCTGACCATAGCCAAACAAC 3′ | 512 |
| Exon-2 R | 5′ TCTCCCAGGGTTGAAGGCA 3′ | |
| Exon-3 F | 5′ GGGGCATGAATCCATAAATC 3′ | 487 |
| Exon-3 R | 5′ GGAAGCAAAGGAAGACAGACAC 3′ | |
| CRYAB | ||
| Exon-1 F | 5′ AACCCCTGACATCACCATTC 3′ | 469 |
| Exon-1 R | 5′ GGAGGAAGGCACTAGCAACC 3′ | |
| Exon-2 F | 5′ TGCAGAATAAGACAGCACCTG 3′ | 296 |
| Exon-2 R | 5′ AATGTAGCCAGCCTCCAAAG 3′ | |
| Exon-3 F | 5′ TCTGCCTCTTTCCTCATT 3′ | 473 |
| Exon-3 R | 5′ CCTTGGAGCCCTCTAAAT 3′ | |
| CRYBB2 | ||
| Exon-2 F | 5′ TGCTCTCTTTCTTTGAGTAGACCTC 3′ | 385 |
| Exon-2 R | 5′CCCATTTTACAGAAGGGCAAC 3′ | |
| Exon-3 F | 5′ ACCCTTCAGCATCCTTTG G 3′ | 314 |
| Exon-3 R | 5′ GCAGACAGGAGCAAGGGTAG 3′ | |
| Exon-4 F | 5′ GCTTGGAGTGGAACTGACCTG 3′ | 244 |
| Exon-4 R | 5′ GGCAGAGAGAGAAAGTAGGATGATG 3′ | |
| Exon-5 F | 5′ GCCCCCTCACCCATACTC 3′ | 242 |
| Exon-5 R | 5′ CCCCAGAGTCTCAGTTTCCTG 3′ | |
| Exon-6 F | 5′ CCTAGTGGCTTATGGATGCTC 3′ | 347 |
| Exon-6 R | 5′ TCTTCACTTGGAGGTCTGGAG 3′ | |
| CRYGC | ||
| Exon-1.2 F | 5′ TGCATAAAATCCCCTTACCGCTGA 3′ | 524 |
| Exon-1.2 R | 5′ ACTCTGGCGGCATGATGGAAATC 3′ | |
| Exon-3 F | 5′AGACTCATTTGCTTTTTTCCATCCTTCTTTC 3′ | 407 |
| Exon-3 R | 5′GAAAGAATGACAGAAGTCAGCAATTGCC 3′ | |
| CRYGD | ||
| Exon-1.2 F | 5′ CCTCGCCTTGTCCCGC 3′ | 340 |
| Exon-1.2 R | 5′ TTAACTTTTGCTTGAAACCATCCA 3′ | |
| Exon-3 F | 5′ TGCTTTTCTTCTCTTTTTATTTCTGGGTCC 3′ | 400 |
| Exon-3 R | 5′AGTAAAGAAAGACACAAGCAAATCAGTGCC 3′ | |
| GJA3 | ||
| Exon-1–1 F | 5′ CTCTTCTGGCTCTGGCTTCC 3′ | 741 |
| Exon-1–1R | 5′ CACCTCGAACAGCGTCTTGA 3′ | |
| Exon-1–2 F | 5′ CTTCCCCATCTCCCACATCC 3′ | 749 |
| Exon-1–2 R | 5′ GGTGGCCGTTGTAGAGCTTG 3′ | |
| Exon-1–3 F | 5′ TCCGCCAAGCTCTACAACG 3′ | 535 |
| Exon-1–3 R | 5′ GAAACCTGATCTCTCCTCCAT 3′ | |
| GJA8 | ||
| Exon-2–1 F | 5′ CAGATATTGACTCAGGGTTG 3′ | 542 |
| Exon-2–1R | 5′ GATGATGTGGCAGATGTAGG 3′ | |
| Exon-2–2 F | 5′ GGCAGCAAAGGCACTAAG 3′ | 465 |
| Exon-2–2 R | 5′ CTCCACCATCCCAACCTC 3′ | |
| Exon-2–3 F | 5′ ATCGTTTCCCACTATTTCC 3′ | 492 |
| Exon-2–3 R | 5′ GGCGTCACTTCATACGGTTA 3′ | |
Results
Clinical evaluations
The cataract exhibited an autosomal dominant inheritance pattern in the family (Figure 1).Three of the seven patients had undergone lens surgery. All affected patients had bilateral lens opacification, but the degree of lens opacities was highly variable (Figure 2).The proband (II:1), who was a 59-year-old woman, had nuclear cataract with ‘Y’ sutural opacities (Figure 2A,B).The affected member III:3 (Figure 2F-H), who was the daughter of the proband, had slight nuclear cataract with curd-like peripheral cortical opacities, while her son (IV:2; Figure 2C,D) showed a different zonular cataract with ‘Y’ sutural opacities. The affected member IV:1(Figure 2E) had a simple ‘Y’ sutural opacity. The clinical evaluation of the affected individuals is provided in Table 2. Prior to surgery, the affected members had visual acuity ranging from 0.05 to 0.8. After surgery, all patients achieved a best-corrected visual acuity of 0.8 to 1.0. There was no family history of other ocular or systemic abnormalities.
Table 2. Clinical features of affected individuals.
| Affected individual | Gender | Age | Surgery age | Phenotype |
|---|---|---|---|---|
| II:1 | Female | 59 | 59 | Nuclear cataract with ‘Y’ sutural opacities |
| II:4 | Male | 55 | 43 | IOL, after cataract surgery |
| II:6 | Male | 53 | 41 | IOL, after cataract surgery |
| III:1 | Female | 34 | 29 | IOL, after cataract surgery |
| III:3 | Female | 33 | No surgery | Nuclear cataract with curd-like peripheral cortical opacities |
| IV:1 | Female | 6 | No surgery | ‘Y’ sutural opacities |
| IV:2 | Male | 8 | 8 | zonular cataract with ‘Y’ sutural opacities and peripheral cortical opacities |
Mutation screening
Through bidirectional sequencing of the coding regions of the candidate genes, we identified a c.215+1G>A substitution in the donor splice site of intron 3 in CRYBA3/A1 in all affected individuals (Figure 3) that co-segregated with all affected individuals, whereas this heterozygous mutation was not present in the unaffected family members, nor in 100 unrelated Chinese without cataracts who served as controls.
Figure 3.
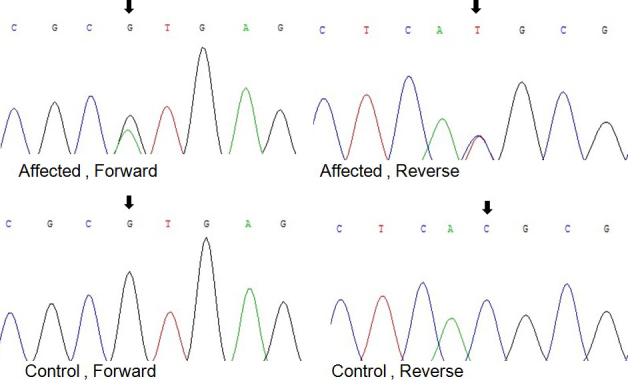
Forward and reverse sequence analyses of the affected and unaffected individuals in the ADCC Chinese family, showing a c.215+1G>A mutation of CRYBA3/A1 (black arrows).
Discussion
In this study, we identified a splice site mutation within CRYBA3/A1 in a four-generation Chinese pedigree with autosomal dominant polymorphic cataract.
Crystallins are known to constitute about 90% of the water-soluble proteins of the lens and contribute to transparency and refractive properties by forming a uniform concentration gradient in the lens. A mutation in the crystallin gene may alter crystallin stability, solubility, or ability to oligomerize and may precipitate from solution, resulting in lens opacity. So, they are considered to be good candidate genes for congenital cataract [18].The vertebrate crystallins are divided into two families: α-crystallins and the β- and γ-crystallin families [19,20]. The β- and γ-crystallins share a commonly features anti-parallel β-sheets in the proteins, referred to as the “Greek key motif.” All vertebrate lens β-crystallins consist of two domains and each one folds into two similar “Greek key motifs,” with each “Greek key motif” comprised of four consecutive anti-parallel β-strands [21].
The CRYBA3/A1 gene uses an alternative translation initiation site to encode both the βA3- and βA1-crystallins.The βA3-crystallins are longer than the βA1-crystallins by the addition of 17 amino acids at the 5′-terminal end [22]. An intermediate form of the βA3-crystallin gene has an N-terminal arm shortened by 8 amino acids [23]. The βA1-crystallin aggregates ranged from dimers to octamers and further complexity is related to temporal and spatial regulation of expression as well as posttranslational modifications [24].
The CRYBA3/A1 gene consists of six exons: the first two exons encode the N-terminal arm, and the subsequent four exons are responsible for the Greek key motifs [25]. So far, four mutations within the CRYBA3/A1 gene was reportedly associated with congenital cataract in different families (Table 3).One is the c.215+1G>A mutation which we reported here, another is the c.215+1G>C [26], the third type is c.215+1G>T [27],and the fourth is a 3-bp deletion at positions 279–281 (c.279_281del) in exon 4, which causes an in-frame deletion of a glycine residue at position 91 (p.Gly91del) [28-31].
Table 3. Previous CRYBA3/BA1 gene mutations associated with congenital cataracts.
| Bp exchange | Aa exchange | Biologic consequence | Origin of family | Reference |
|---|---|---|---|---|
| c.215+1G>A | Splice site mutation | zonular lamellar opacities cataract and floriform | Indian | [6] |
| c.215+1G>C | Splice site mutation | pulverulent, star-shaped, shieldlike and radial cataract | Brazilian | [26] |
| c.215+1G>T | Splice site mutation | Y-suture, nucleus and cortical cataract | Chinese | [27] |
| c.215+1G>A | Splice site mutation | Y-sutural,mild nucleus and cortical dot cataract | Australian | [32] |
| c.215+1G>A | Splice site mutation | progressive childhood nucleus and peripheral cortex cataract | Chinese | [33] |
| c.215+1G>A | Splice site mutation | posterior polar cataract | Chinese | [34] |
| c.215+1G>A | Splice site mutation | zonular cataract with sutural opacity | Indian | [35] |
| c.279_281del | p.Gly91del | nuclear cataract | Chinese | [28] |
| c.279_281del | p.Gly91del | pulverulent nuclear congenital cataracts | Chinese | [29] |
| c.279_281del | p.Gly91del | pulverulent lamellar congenital cataracts | Chinese | [29] |
| c.279_281del | p.Gly91del | nuclear cataract | Swiss | [30] |
| c.279_281del | p.Gly91del | lamellar cataract | Britain | [31] |
Previously, five geographically distinct families have been reported to possess the c.215+1G>A mutation, which is associated with diverse phenotypes including zonular, lamellar, nuclear, cortical,sutural, and posterior polar cataract [6,32-35].Diverse cataract phenotypes caused by exactly the same mutation within CRYBA3/A1 in different ethnic backgrounds suggest that ethic background including environmental factors or, more likely, other genetic modifiers may influence the expression and function of this gene in lens development and cataract formation. In the family we studied, the phenotypes show considerable variation in morphology, and the severity of the disease ranged from requiring surgery to unawareness of the affliction before this study. Of the four patients who had pictures of their affected eyes taken,II:1 (nuclear cataract with ‘Y’ sutural) and IV:2 (zonular cataract with ‘Y’ sutural), are more severe than III:3 (mild nuclear cataract) and IV:1 (simple ‘Y’ sutural cataract). In addition, after a 5-year followed up of this family, we found the opacities of lens in the affected individuals are not progressive. So, the phenotype of this family was identified as polymorphic.Splice-site mutation is a genetic mutation that inserts or deletes several nucleotides at the splice junction during mRNA processing. It was reported to contribute to exon skipping, activation of cryptic splice sites, creation of pseudo-exon within an intron, or intron retention, which commonly results in exon skipping [36].As speculated by Kannabiran et al. [35], the c.215+1G>A mutation (position 474) would result in skipping of a donor splice junction, recruitment of a cryptic splice site (position 460),or possibly both. All possibilities would cause improper folding of the first Greek key motif, which leads to structural instability of βA1/A3-crystallin and subsequent cataract formation.
Conclusions
In conclusion, we have identified a polymorphic form of congenital cataracts associated with a c.215+1G>A mutation of the CRYBA1/A3 gene in a Chinese family. This mutation supports the role of the CRYBA3/A1 gene in human cataract formation and provides additional evidence for the genetic heterogeneity of congenital cataracts in a different ethnic background.
Acknowledgments
We thank the family for participating in this study. This research is supported by the Innovative Research Team Program of Zhejiang University (2012FZA7018), Zhejiang Key Innovative Research Team Project of China (2009R50039), and Zhejiang Key Lab Fund of China (2011E10006).
References
- 1.Holmes JM, Leske DA, Burke JP, Hodge DO. Birth prevalence of visually significant infantile cataract in a defined U.S. population. Ophthalmic Epidemiol. 2003;10:67–74. doi: 10.1076/opep.10.2.67.13894. [DOI] [PubMed] [Google Scholar]
- 2.Reddy MA, Francis PJ, Berry V, Bhattacharya SS, Moore AT. Molecular genetic basis of inherited cataract and associated phenotypes. Surv Ophthalmol. 2004;49:300–15. doi: 10.1016/j.survophthal.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Amaya L, Taylor D, Russell-Eggitt I, Nischal KK, Lengyel D. The morphology and natural history of childhood cataracts. Surv Ophthalmol. 2003;48:125–44. doi: 10.1016/s0039-6257(02)00462-9. [DOI] [PubMed] [Google Scholar]
- 4.Vanita, Singh JR, Singh D. Genetic and segregation analysis of congenital cataract in the Indian population. Clin Genet. 1999;56:389–93. doi: 10.1034/j.1399-0004.1999.560507.x. [DOI] [PubMed] [Google Scholar]
- 5.Shiels A, Hejtmancik JF. Genetic origins of cataract. Arch Ophthalmol. 2007;125:165–73. doi: 10.1001/archopht.125.2.165. [DOI] [PubMed] [Google Scholar]
- 6.Devi RR, Yao W, Vijayalakshmi P, Sergeev YV, Sundaresan P, Hejtmancik JF. Crystallin gene mutations in Indian families with inherited pediatric cataract. Mol Vis. 2008;14:1157–70. [PMC free article] [PubMed] [Google Scholar]
- 7.Scott MH, Hejtmancik JF, Wozencraft LA, Reuter LM, Parks MM, Kaiser-Kupfer MI. Autosomal dominant congenital cataract. Interocular phenotypic variability. Ophthalmology. 1994;101:866–71. doi: 10.1016/s0161-6420(94)31246-2. [DOI] [PubMed] [Google Scholar]
- 8.Gill D, Klose R, Munier FL, McFadden M, Priston M, Billingsley G, Ducrey N, Schorderet DF, Héon E. Genetic heterogeneity of the Coppock-like cataract: a mutation in CRYBB2 on chromosome 22q11.2. Invest Ophthalmol Vis Sci. 2000;41:159–65. [PubMed] [Google Scholar]
- 9.Héon E, Priston M, Schorderet DF, Billingsley GD, Girard PO, Lubsen N, Munier FL. The gamma-crystallins and human cataracts: a puzzle made clearer. Am J Hum Genet. 1999;65:1261–7. doi: 10.1086/302619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vanita V, Singh JR, Hejtmancik JF, Nuernberg P, Hennies HC, Singh D, Sperling K. A novel fan-shaped cataract-microcornea syndrome caused by a mutation of CRYAA in an Indian family. Mol Vis. 2006;12:518–22. [PubMed] [Google Scholar]
- 11.Lu S, Zhao C, Jiao H, Kere J, Tang X, Zhao F, Zhang X, Zhao K, Larsson C. Two Chinese families with pulverulent congenital cataracts and Delta G91 CRYBA1 mutations. Mol Vis. 2007;13:1154–60. [PubMed] [Google Scholar]
- 12.Litt M, Carrero-Valenzuela R, LaMorticella DM, Schultz DW, Mitchell TN, Kramer P, Maumenee IH. Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human beta-crystallin gene CRYBB2. Hum Mol Genet. 1997;6:665–8. doi: 10.1093/hmg/6.5.665. [DOI] [PubMed] [Google Scholar]
- 13.Zhang LY, Yam GH, Fan DS, Tam PO, Lam DS, Pang CP. A novel deletion variant of gamma D-crystallin responsible for congenital nuclear cataract. Mol Vis. 2007;13:2096–104. [PubMed] [Google Scholar]
- 14.Hansen L, Yao W, Eiberg H, Funding M, Riise R, Kjaer KW, Hejtmancik JF, Rosenberg T. The congenital “ant-egg” cataract phenotype is caused by a missense mutation in connexin46. Mol Vis. 2006;12:1033–9. [PubMed] [Google Scholar]
- 15.Schmidt W, Klopp N, Illig T, Graw J. A novel GJA8 mutation causing a recessive triangular cataract. Mol Vis. 2008;14:851–6. [PMC free article] [PubMed] [Google Scholar]
- 16.Brémond-Gignac D, Bitoun P, Reis LM, Copin H, Murray JC, Semina EV. Identification of dominant FOXE3 and PAX6 mutations in patients with congenital cataract and aniridia. Mol Vis. 2010;16:1705–11. [PMC free article] [PubMed] [Google Scholar]
- 17.Shiels A, Bennett TM, Knopf HL, Yamada K, Yoshiura K, Niikawa N, Shim S, Hanson PI. CHMP4B, a novel gene for autosomal dominant cataracts linked to chromosome 20q. Am J Hum Genet. 2007;81:596–606. doi: 10.1086/519980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hejtmancik JF. Congenital cataracts and their molecular genetics. Semin Cell Dev Biol. 2008;19:134–49. doi: 10.1016/j.semcdb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graw J. Cataract mutations and lens development. Prog Retin Eye Res. 1999;18:235–67. doi: 10.1016/s1350-9462(98)00018-4. [DOI] [PubMed] [Google Scholar]
- 20.Andley UP. Crystallins in the eye: Function and pathology. Prog Retin Eye Res. 2007;26:78–98. doi: 10.1016/j.preteyeres.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 21.Graw J. Genetics of crystallins: cataract and beyond. Exp Eye Res. 2009;88:173–89. doi: 10.1016/j.exer.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 22.Quax-Jeuken Y, Janssen C, Quax W, van den Heuvel R, Bloemendal H. Bovine beta-crystallin complementary DNA clones. Alternating proline/alanine sequence of beta B1 subunit originates from a repetitive DNA sequence. J Mol Biol. 1984;180:457–72. doi: 10.1016/0022-2836(84)90022-6. [DOI] [PubMed] [Google Scholar]
- 23.Hope JN, Chen HC, Hejtmancik JF. Beta A3/A1-crystallin association: role of the N-terminal arm. Protein Eng. 1994;7:445–51. doi: 10.1093/protein/7.3.445. [DOI] [PubMed] [Google Scholar]
- 24.Werten PJ, Carver JA, Jaenicke R, de Jong WW. The elusive role of the N-terminal extension of beta A3- and beta A1-crystallin. Protein Eng. 1996;9:1021–8. doi: 10.1093/protein/9.11.1021. [DOI] [PubMed] [Google Scholar]
- 25.Hogg D, Tsui LC, Gorin M, Breitman ML. Characterization of the human beta-crystallin gene Hu beta A3/A1 reveals ancestral relationships among the beta gamma-crystallin superfamily. J Biol Chem. 1986;261:12420–7. [PubMed] [Google Scholar]
- 26.Bateman JB, Geyer DD, Flodman P, Johannes M, Sikela J, Walter N, Moreira AT, Clancy K, Spence MA. A new beta A1-crystallin splice junction mutation in autosomal dominant cataract. Invest Ophthalmol Vis Sci. 2000;41:3278–85. [PubMed] [Google Scholar]
- 27.Yang Z, Li Q, Ma Z, Guo Y, Zhu S, Ma X. AG→T splice site mutation of CRYBA1/A3 associated with autosomal dominant suture cataracts in a Chinese family. Mol Vis. 2011;17:2065–71. [PMC free article] [PubMed] [Google Scholar]
- 28.Qi Y, Jia H, Huang S, Lin H, Gu J, Su H, Zhang T, Gao Y, Qu L, Li D, Li Y. A deletion mutation in the betaA1/A3 crystallin gene (CRYBA1/A3) is associated with autosomal dominant congenital nuclear cataract in a Chinese family. Hum Genet. 2004;114:192–7. doi: 10.1007/s00439-003-1049-7. [DOI] [PubMed] [Google Scholar]
- 29.Lu S, Zhao C, Jiao H, Kere J, Tang X, Zhao F, Zhang X, Zhao K, Larsson C. Two Chinese families with pulverulent congenital cataracts and deltaG91 CRYBA1 mutations. Mol Vis. 2007;13:1154–60. [PubMed] [Google Scholar]
- 30.Ferrini W, Schorderet DF, Othenin-Girard P, Uffer S, Héon E, Munier FL. CRYBA3/A1 gene mutation associated with suture-sparing autosomal dominant congenital nuclear cataract: A novel phenotype. Invest Ophthalmol Vis Sci. 2004;45:1436–41. doi: 10.1167/iovs.03-0760. [DOI] [PubMed] [Google Scholar]
- 31.Reddy MA, Bateman OA, Chakarova C, Ferris J, Berry V, Lomas E, Sarra R, Smith MA, Moore AT, Bhattacharya SS, Slingsby C. Characterization of the G91del CRYBA1/3-crystallin protein: a cause of human inherited cataract. Hum Mol Genet. 2004;13:945–53. doi: 10.1093/hmg/ddh110. [DOI] [PubMed] [Google Scholar]
- 32.Burdon KP, Wirth MG, Mackey DA, Russell-Eggitt IM, Craig JE, Elder JE, Dickinson JL, Sale MM. Investigation of crystallin genes in familial cataract, and report of two disease associated mutations. Br J Ophthalmol. 2004;88:79–83. doi: 10.1136/bjo.88.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu Y, Shentu X, Wang W, Li J, Jin C, Yao K. A Chinese family with progressive childhood cataracts and IVS3+1G>A CRYBA3/A1 mutations. Mol Vis. 2010;16:2347–53. [PMC free article] [PubMed] [Google Scholar]
- 34.Gu Z, Ji B, Wan C, He G, Zhang J, Zhang M, Feng G, He L, Gao L. A splice site mutation in CRYBA1/A3 causing autosomal dominant posterior polar cataract in a Chinese pedigree. Mol Vis. 2010;16:154–60. [PMC free article] [PubMed] [Google Scholar]
- 35.Kannabiran C, Rogan PK, Olmos L, Basti S, Rao GN, Kaiser-Kupfer M, Hejtmancik JF. Autosomal dominant zonular cataract with sutural opacities is associated with a splice mutation in the betaA3/A1-crystallin gene. Mol Vis. 1998;4:21. [PubMed] [Google Scholar]
- 36.Nakai K, Sakamoto H. Construction of a novel database containing aberrant splicing mutations of mammalian genes. Gene. 1994;141:171–7. doi: 10.1016/0378-1119(94)90567-3. [DOI] [PubMed] [Google Scholar]