

Abstract
1,4-Dihydropyridines are regarded as privileged structures for drug design, i.e. they tend to bind to a wide variety of receptor sites. We have shown that upon appropriate manipulation of the substituent groups on a 1,4-dihydropyridine template, high affinity and selectivity for the A3 subtype of adenosine receptors (‘P1 receptors’) may be attained. In the present study we have begun to extend this approach to P2 receptors which are activated by ATP and other nucleotides. Nicardipine, a representative dihydropyridine, used otherwise as an L-type calcium channel blocker, was shown to be an antagonist at recombinant rat P2X2 (IC50 = 25 μM) and P2X4 (IC50 ~ 220 μM) receptors expressed in Xenopus oocytes. Thus, this class of compounds represents a suitable lead for enhancement of affinity through chemical synthesis. In an attempt to modify the 1,4-dihydropyridine structure with a predicted P2 receptor recognition moiety, we have replaced one of the ester groups with a negatively charged phosphonate group. Several 4-phenyl-5-phosphonato-1,4-dihydropyridine derivatives, MRS 2154 (2,6-dimethyl), MRS 2155 (6-methyl-2-phenyl), and MRS 2156 (2-methyl-6-phenyl), were synthesized through three component condensation reactions. These derivatives were not pure antagonists of the effects of ATP at P2X2 receptors, rather were either inactive (MRS 2156) or potentiated the effects of ATP in a concentration-dependent manner (MRS 2154 in the 0.3–10 μM range and MRS 2155 at >1 μM). Antagonism of the effects of ATP at P2X2 receptor superimposed on the potentiation was also observed at >10 μM (MRS 2154) or 0.3–1 μM (MRS 2155). Thus, while a conventional dihydropyridine, nicardipine, was found to antagonize rat P2X2 receptors ninefold more potently than P2X4 receptors, the effects of novel, anionic 5-phosphonate analogues at the receptor were more complex.
Keywords: Ion channels, Oocytes, Purines, Dihydropyridine derivatives, Potentiator
1. Introduction
Early attempts to find non-nucleotide ligands for P2 receptors identified only weak, non-selective antagonists, often poorly defined chemically (such as Reactive Blue 2) or not specific for P2 receptors (such as suramin) (Jacobson et al., 1997). Synthetic ligands which display high potency and/or selectivity at various subtypes of P2 receptors are currently being developed through library screening and rational design approaches (Williams and Bhagwat, 1996; Fischer, 1999). The cloning of the P2X and P2Y receptors has permitted the use of biophysical and computational methods (North and Barnard, 1997; Moro et al., 1998) to characterize the drug–receptor interactions, thus aiding the drug design process.
Pyridoxal phosphate analogues, of which PPADS (pyridoxal phosphate-6-azophenyl-2′,4′-disulfonic acid) (Lambrecht et al., 1996; Kim et al., 2000) is the prototypical compound, and truncated analogues of suramin (Mateo et al., 1998) have been introduced as principally P2X receptor antagonists. In the pyridoxal phosphate series, the potency at P2X1 receptors and, in some cases, at both P2X1 and P2X3 receptors (Damer et al., 1998) has reached the nanomolar range. In the suramin series, NF023 (8,8′-[carbonylbis(imino-3,1-phenylenecarbonylimino)]bis-(1,3,5-naphthalenetrisulfonic acid) displayed an IC50 value of 0.24 μM at human P2X1 receptors (Mateo et al., 1998). Damer et al. (1998) have described the suramin analogue NF279 (8,8′-[carbonylbis(imino-4,1-phenylenecarbonylimino)]bis(1,3,5-naphthalenetrisulfonic acid) as an antagonist with high affinity and apparent P2X1 receptor-selectivity. At P2Y1 receptors, the most potent and selective antagonists are bisphosphate derivatives of adenine nucleosides (Boyer et al., 1998; Camaiono et al., 1998; Nandanan et al., 2000). MRS 2179 (N6-methyl-2′-deoxyadenosine 3′,5′-bisphosphate) was found to be a competitive antagonist at turkey and human P2Y1 receptors, with a KB value of 100 nM (Boyer et al., 1998). Carbocyclic and acyclic analogues have also been shown to have high affinity at P2Y1 receptors, among which analogues is a ring-constrained carbocyclic bisphosphate derivative, MRS 2279 ((1R,2S,4S,5S)-1-[(phosphato)methyl]-4-(2-chloro-6-aminopurin-9-yl)bicyclo[3.1.0]-hexane-2-phosphate), which has an IC50 value of 52 nM (Nandanan et al., 2000). There are also high affinity antagonists at the yet uncloned P2T receptors, such as ARL 67085 (2-propylthio-D-β,γ-dichloromethylene-ATP), which is in clinical trials as an anti-thrombotic agent (Ingall et al., 1999). Ip5I has been shown to be a potent antagonist of P2X1 receptors (King et al., 1999). 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate (TNP-ATP) is a nucleotide derivative which antagonizes P2X1, P2X3, and P2X2/3 receptors with extremely high affinity (Lewis et al., 1998). The tyrosine derivative KN-62 potently antagonizes P2X7 receptors (Humphreys et al., 1998), although its potency varies among species orthologues of P2X7 receptors.
In an effort to find non-nucleotide antagonists of P2 receptors, we have explored the use of 1,4-dihydropyridines (DHPs), used clinically as antagonists of L-type calcium channels, as leads for P2 receptor ligands. DHPs have been shown to be privileged structures for drug design (Triggle, 1985), i.e. they tend to bind to a wide variety of receptor sites, and can be manipulated as a template for the appending groups that are recognized by a given receptor. For P1 receptors, the appropriate manipulation of the substituent groups on a 1,4-DHP ring provided antagonists of high affinity and selectivity for A3 adenosine receptors (Jiang et al., 1997). In the present study, we have begun to extend this approach to P2 receptors, with the expectation that common elements of recognition exist at purine binding sites, whether they be for purine nucleosides (P1) or nucleotides (P2) (Moro et al., 1998). Furthermore, the fact that 1,4-DHPs are already known to block an ion channel, albeit of different structure, increases the expectation that novel interactions may occur at P2 receptor gated ion channels.
Since 1991, our respective laboratories have been collaborating on the synthesis of selective P2 receptor ligands. We credit Prof. Geoffrey Burnstock for fostering this productive relationship, with his encouragement beginning at the International Pharmacology Congress in Sydney, Australia in 1987. Prof. Burnstock’s unrelenting vision of the importance of these initially hypothetical receptors has been the underlying inspiration for our efforts.
2. Materials and methods
2.1. Synthesis
All synthetic reagents were purchased from Aldrich (St. Louis, MO).
1H-NMR spectra were obtained with a Varian Gemini-300 spectrometer using CDCl3 or D2O as a solvent. The chemical shifts are expressed as ppm downfield from tetramethylsilane or as relative ppm from HOD peaks (4.78 ppm). Low-resolution EI (electron impact) mass spectra were carried out with a VG7070F mass spectrometer at 6 kV. High-resolution FAB (fast atom bombardment) mass spectrometry was performed with a JEOL SX102 spectrometer using 6-kV Xe atoms following desorption from a glycerol matrix.
The determinations of purity were performed with a Hewlett-Packard 1090 HPLC system using an SMT OD-5–60 C18 analytical column (250 mm×4.6 mm, Separation Methods Technologies, Newark, DE) with a linear gradient elution of 0.1 M triethylammonium acetate buffer:CH3CN=95:5 to 40:60 for 20 min with a flow rate of 1 ml/min. Peaks were detected by UV absorption using a diode array detector. All phosphonate derivatives showed more than 95% purity in the HPLC system.
2.1.1. Dimethyl 2,6-dimethyl-3-(ethoxycarbonyl)-4-phenyl-1,4-(±)-dihydropyridine-5-phosphonate (4a)
A solution of ethyl 3-aminocrotonate (1a, 63.2 μl, 0.5 mmol), benzaldehyde (2, 50.5 μl, 0.5 mmol) and dimethyl-(2-oxopropyl)phosphonate (3, 72.7 μl, 0.5 mmol) in 1 ml of EtOH was heated in a sealed tube at 90°C for 24 h (Morita et al., 1987). After evaporation, the residue was purified by preparative thin layer chromatography (CHCl3:MeOH=30:1) to afford 54 mg of 4a (30%). 1H-NMR (CDCl3) 1.23 (3H, t, J=6.8 Hz, 3–CH3), 2.26 (3H, d, J=1.9 Hz, 6–CH3), 2.31 (3H, s, 2–CH3), 3.22 (3H, d, J=11.7 Hz, –OCH3), 3.50 (3H, d, J=11.7 Hz, –OCH3), 4.03–4.14 (2H, m, 3–OCH2–), 4.65 (1H, d, J=10.8 Hz, 4–CH–), 6.91 (1H, d, J=4.9 Hz, NH), 7.10–7.32 (5H, m, 4–Ph). MS (EI): 365 (M+). Anal. calcd. for C18H24NO5P; C 59.17, H 6.62, N 3.83, found: C 59.11, H 6.57, N 3.75.
2.1.2. Dimethyl 2,4-diphenyl-3-(ethoxycarbonyl)-6-methyl-1,4-(±)-dihydropyridine-5-phosphonate (4b)
Following the same procedure for the preparation of 4a, with ethyl 1-aminocinnamate (95.6 μl, 0.5 mmol), 45 mg of 4b was obtained (21%). 1H-NMR (CDCl3) 0.85 (3H, t, J=6.8 Hz, 3–CH3), 2.30 (3H, d, J=1.9 Hz, 6–CH3), 3.20 (3H, d, J=11.7 Hz, –OCH3), 3.54 (3H, d, J=11.7 Hz,–OCH3), 4.78–4.87 (2H, m, 3–OCH2–), 4.74 (1H, d, J=10.8 Hz, 4–CH–), 6.26 (1H, d, J=4.9 Hz, NH), 7.18–7.45 (10H, m, 4–Ph, 2–Ph). MS (EI): 427 (M+). Anal. calcd. for C23H26NO5P; C 64.63, H 6.13, N 3.27, found: C 64.74, H 6.12, N 3.30.
2.1.3. 2,6-Dimethyl-3-(ethoxycarbonyl)-4-phenyl-1,4-(±)-dihydropyridine-5-phosphonate diammonium salt (5a)
To a solution of 4a (0.04 g, 0.109 mmol) in 1 ml of anhydrous CH3CN was added trimethylsilyl bromide (56 μl, 0.411 mmol) at 25°C under N2 atmosphere. The mixture was stirred for 18 h and the solvent was removed by N2 stream. The residue was partitioned between ether and 0.1 M ammonium bicarbonate solution. The water fraction was purified by an ion-exchange column chromatography using Sephadex-DEAE A-25 resin with a linear gradient of 0.5 M ammonium bicarbonate (0% to 100%) as the mobile phase, and UV and HPLC were used to monitor the elution to give 39 mg of 5a (96%). 1H-NMR (D2O) 1.20 (3H, t, J=6.8 Hz, 3–CH3), 2.12 (3H, d, J=1.9 Hz, 6–CH3), 2.18 (3H, s, 2–CH3), 4.06 (2H, q, J=6.8 Hz, 3–OCH2–), 4.69 (1H, d, J=10.8 Hz, 4–CH–), 7.14–7.35 (5H, m, 4–Ph). HRMS (FAB+) calcd. 338.1157, found 338.1171. The HPLC retention time was 5.04 min.
2.1.4. 2,4-Diphenyl-3-(ethoxycarbonyl)-6-methyl-1,4-(±)-dihydropyridine-5-phosphonate diammonium salt (5b)
Following the same procedure for the preparation of 5a, starting from 4b (0.02 g, 0.468 mmol), 0.01 g of 5b was obtained (54%). 1H NMR (D2O) 1.02 (3H, t, J=6.8 Hz, 3–CH3), 2.41 (3H, d, J=1.9 Hz, 6–CH3), 4.00 (2H, q, J=6.8 Hz, 3–OCH2–), 5.01 (1H, d, J=10.8 Hz, 4–CH–), 7.42–7.74 (10H, m, 4–Ph, 2–Ph). HRMS (FAB+) calcd. 400.1314, found 400.1306. The HPLC retention time was 11.14 min.
2.2. Pharmacology
2.2.1. Antagonist activity at recombinant P2X receptors
Xenopus oocytes were harvested and prepared as previously described (King et al., 1997). Defolliculated oocytes were injected cytosolically with 40 nl of a solution of cRNA of rat P2X4 receptors (1 μg/ml) or rat P2X2 receptors (0.002 μg/ml) incubated for 24 h at 18°C in Barth’s solution and kept for up to 12 days at 4°C until used in electrophysiological experiments.
ATP-activated membrane currents (Vh= −50 mV) were recorded from cRNA-injected oocytes using the twin-electrode voltage-clamp technique (Axoclamp 2B amplifier). Voltage recording and current-recording microelectrodes (1–5 MΩ tip resistance) were filled with 3.0 M KCl. Oocytes were held in an electrophysiological chamber and superfused with Ringer’s solution (5 ml/min, at 18°C) containing (mM) NaCl, 110; KCl, 2.5; HEPES, 5; BaCl2, 1.8, adjusted to pH 7.5.
ATP was superfused over oocytes for 120 s then washed out for a period of 20 min. The agonist concentration (10 μM for P2X2 or 30 μM for P2X4) was approximately equal to the EC70 value at each subtype. For inhibition curves, data were normalized to the current evoked by ATP at pH 7.5. Test substances were added for 5 min prior to ATP exposure; all compounds were tested for reversibility of their effects. The concentration required to inhibit the ATP-response by 50% (IC50) was taken from Hill plots constructed using the formula log (I/Imax − I), where I was the current evoked by ATP in the presence of an antagonist. Data are presented as mean±S.E.M. (n=4) for data from different batches of oocytes.
3. Results
Nicardipine, a representative DHP, was tested in functional assays of recombinant rat P2X2 and P2X4 receptors expressed in Xenopus oocytes (Fig. 1). Its potency (IC50) in inhibiting ATP-elicited membrane currents was 24±5 μM at P2X2 receptors and ~220 μM at P2X4receptors. At Group I (P2X1 and P2X3) receptors the potency was not determined, however the closely related DHP nifedipine was inactive at rat smooth muscle P2X1-like receptors (Blakeley et al., 1981) and at inhibitory P2Y receptors in pig ileum (Soto et al., 1999). Nicardipine was inactive at 100 μM as an antagonist of the effects of 2-MeSATP at turkey erythrocyte P2Y1 receptors (J. Boyer, T.K. Harden, unpublished).
Fig. 1.

Effects of the DHP nicardipine on current induced at recombinant rat P2X2 (■) and P2X4 (●) receptors, expressed in Xenopus oocytes (n=4). The twin-electrode voltage-clamping technique was used (Vh=−50 mV). The medium consisted of Ba2+ Ringer’s buffer at pH 7.50. The structure of nicardipine is shown.
In addition to the optimization of this lead for the design of 1,4-DHP 3,5-diesters, such as nicardipine as P2X2 receptor antagonists, we have modified the DHP with a predicted P2 receptor recognition moiety in the form of a negatively charged phosphonate group. Thus, we have introduced a phosphonate group in place of the 5-ester group of the DHP template.
Fig. 2 outlines the synthesis of three such 4-phenyl-5-phosphonato-1,4-DHP derivatives, 5a–c: MRS 2154 (2,6-dimethyl), MRS 2155 (6-methyl-2-phenyl), and MRS 2156 (2-methyl-6-phenyl). All were synthesized through three-component (1–3) condensation reactions (Morita et al., 1987). The phosphonate methyl ester groups of the product, 4, were deprotected using trimethylsilyl bromide to give the free phosphonates, 5a–c. A phenyl group was necessary at the 4-position, since the product proved to be unstable during the deprotection reaction when the same synthetic route was applied to a 4-methyl analogue.
Fig. 2.

Synthesis of 1,4-DHP 5-phosphonate derivatives, 5a–c.
In the biological assay, defolliculated oocytes were used to express recombinant P2X2 receptors and membrane currents were recorded under twin-electrode voltage-clamp at −50 mV. All three substances (MRS2154/5/6) were dissolved in DMSO and tested over a range of 0.3 to 100 μM against ATP-responses (3 or 10 μM) at P2X2 receptors. The phosphonates, applied in increasing, cumulative concentrations, were superfused on the oocytes expressing P2X2 receptors for 5 min prior to applying ATP as agonist. The pH of the bathing medium was not affected.
The 4-phenyl-5-phosphonato-1,4-DHP derivatives were not pure antagonists of the effects of ATP at P2X2 receptors, rather were either inactive (MRS 2156) or potentiated the effects of ATP in a concentration-dependent manner (MRS 2154 in the 0.3 to 10 μM range and MRS 2155 at >1 μM). The maximal agonist effect in the presence of MRS 2154 was roughly twice the response to ATP alone (Fig. 3). Antagonism of the effects of ATP at the P2X2 receptor superimposed on the potentiation was also observed at >10 μM (MRS 2154) or 0.3–1 μM (MRS 2155) as shown in Fig. 3. Inhibition by MRS 2154 reached the amplitude of control responses, i.e. full reversal of the potentiating effect at 100 μM. MRS 2155 was tested over a range of 0.3 to 30 μM against P2X2 receptor responses to ATP (10 μM). There was evidence of inhibition at 0.3 μM, but this was followed by a concentration-dependent potentiation of ATP-elicited responses. MRS 2156 in a concentration range of 1–100 μM did not alter the response to ATP, either as antagonist or potentiator (data not shown). The potentiating effects of MRS 2154 and MRS 2155 were readily reversed upon washout with fresh medium.
Fig. 3.
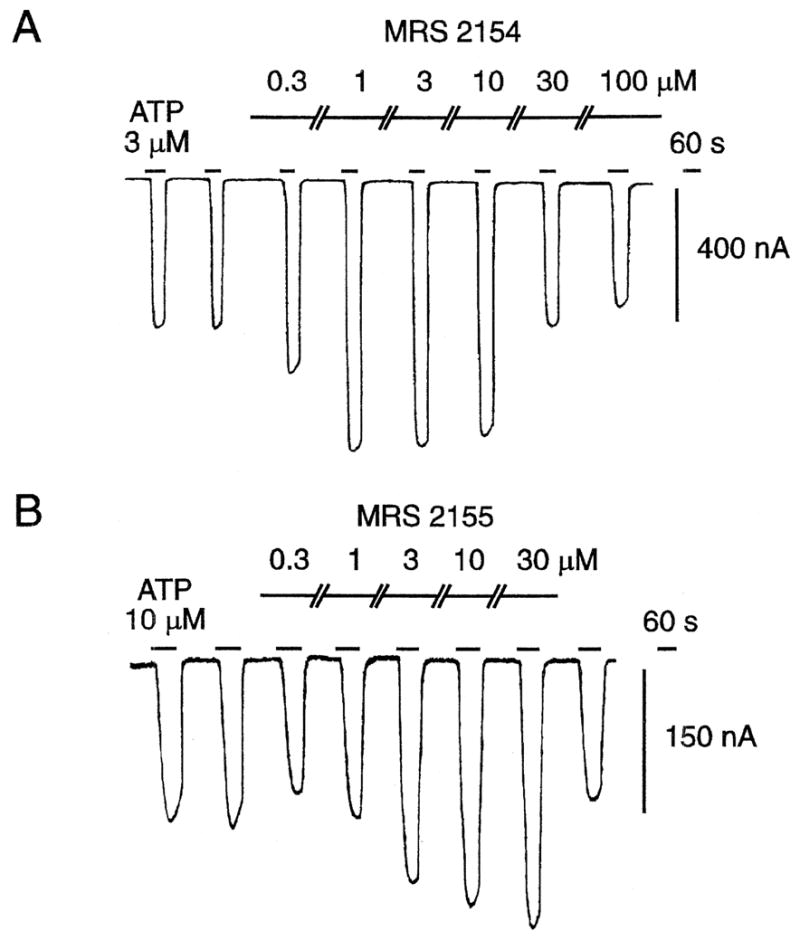
Effects of DHP phosphonate derivatives MRS 2154 (A) and MRS 2155 (B) on current induced by activation of recombinant rat P2X2 receptors expressed in Xenopus oocytes. The twin electrode–voltage clamping-technique was used; Vh=−50 mV. The medium consisted of Ba2+ Ringer’s buffer at pH 7.50. MRS 2156 (≤100 μM) had no effect on ATP-induced ion flux (data not shown).
4. Discussion
Previously, the 1,4-DHP nifedipine was found to be inactive in blocking the effects of ATP at P2X1-like receptors in the rat vas deferens (Blakeley et al., 1981). Thus far, the new generation of P2X receptor antagonists tends to show good activity at the P2X1 and P2X3 subunits (see Section 1) but reduced activity at the P2X2 and P2X4 subunits. To this extent, substances which preferentially select P2X2 and P2X4 receptors are very desirable. Present results suggest that the 4-(3-nitrophenyl)-1,4-DHP nicardipine is a weak antagonist of the rat P2X2 receptor, with a ninefold selectivity versus the P2X4 receptor. There is presently no evidence that P2X2 receptor inhibition occurs at clinically relevant doses of DHPs, when used as potent blockers of L-type calcium channels.
Thus, DHPs represent a suitable lead for enhancement of affinity and possibly receptor subtype selectivity through chemical synthesis. We are currently screening libraries of 1,4-DHPs and related molecules, with the aim of increasing affinity at P2 receptors and eliminating binding to L-type calcium channels.
An attempt was made to enhance the antagonist properties of DHPs, by a departure from the classical 1,4-DHP structure, i.e. through the incorporation of a 5-phosphonate group. A phosphonate group might act similarly to the phosphate groups of nucleotide ligands, which form putative electrostatic bonds with positively-charged groups on the P2 receptors (North and Barnard, 1997; Moro et al., 1998). The incorporation of a 5-phosphonate in the 4-phenyl-1,4-DHPs MRS 2154 and MRS 2155 (differing only in the substitution at the 2-position with methyl or phenyl) resulted not in pure antagonists, but in potentiators of the action of ATP at P2X2 receptors. The potentiation along with a superimposed antagonism at either high (MRS 2154) or low concentrations (MRS 2155) was demonstrated in an electrophysiological assay at the recombinant rat P2X2 receptor. Thus, while a conventional DHP structure, nicardipine, was found to antagonize rat P2X2 receptors, the effects of novel, anionic 5-phosphonate analogues at the receptor were more complex.
The potency of ligands at various P2X receptor subtypes have been compared (Bianchi et al., 1999), but selective agonists and antagonists for these subtypes are not well developed. Potentiation of the effects of ATP at P2X1 receptors by a pyridoxine cyclic phosphate and other antagonists (Jacobson et al., 1998) has been described, but this is the first example of potentiation of the agonist effects at P2X2 receptors. It will be useful to study the novel potentiators described in the present study at other receptor subtypes and to systematically modify their molecular structures.
The P2X2 receptor, which has now been cloned from human (Lynch et al., 1999) and guinea pig (Parker et al., 1998), is present in PC12 cells, in which it stimulates MAP kinases (Swanson et al., 1998), and in the central nervous system (principally in the cerebellum, hypothalamus, brain stem, dorsal horn, and cochlea) (Kanjhan et al., 1999). The action of ATP as a neurotransmitter in the hippocampus has been described (Pankratov et al., 1998). P2X2-like receptors occur on rat cerebellar neurons (Borderies et al., 1997) and possibly rat hippocampal neurons where ATP acts as a neurotransmitter (Pankratov et al., 1998). The use of DHP-based antagonists and potentiators of P2X2 receptor function may help to clarify the role of ATP as a neurotransmitter. Antagonists that are selective for P2X2/P2X3 heteromeric receptors may be useful in pain control (Burgard et al., 1999).
Acknowledgments
We thank Dr. Lewis Pannell and Wesley White for determination of HRMS and NMR.
Abbreviations
- ATP
adenosine 5′-triphosphate
- DHP
1,4-dihydropyridine
- DMSO
dimethylsulfoxide
- EC70
70% effective concentration
- EFS
electrical field stimulation
- EtOH
ethanol
- H-NMR
proton nuclear magnetic resonance
- IC50
50% inhibitory concentration
- KB
Schild constant
- 2-MeSATP
2-methylthioadenosine 5′-triphosphate
- MRS 2154
2,6-dimethyl-3-(ethoxycarbonyl)-4-phenyl-1,4-(±)-dihydropyridine-5-phosphonate
- MRS 2155
2,4-diphenyl-3-(ethoxycarbonyl)-6-methyl-1,4-(±)-dihydropyridine-5-phosphonate
- MRS 2156
4,6-diphenyl-3-(ethoxycarbonyl)-2-methyl-1,4-(±)-dihydropyridine-5-phosphonate
- PPA-DS
pyridoxal 5′-phosphate-6-phenylazo-2,4-disulfonate
- SAR
structure– activity relationship
- TMS-Br
trimethylsilyl bromide
References
- Bianchi BR, Lynch KJ, Touma E, Niforatos W, Burgard EC, Alexander KM, Park HS, Yu H, Metzger R, Kowaluk E, Jarvis M, Van Biesen T. Pharmacological characterization of recombinant human and rat P2X receptor subtypes. Eur J Pharmacol. 1999;376:127–138. doi: 10.1016/s0014-2999(99)00350-7. [DOI] [PubMed] [Google Scholar]
- Blakeley AG, Brown DA, Cunnane TC, French AM, McGrath JC, Scott NC. Effects of nifedipine on electrical and mechanical responses of rat and guinea pig vas deferens. Nature. 1981;294:759–761. doi: 10.1038/294759a0. [DOI] [PubMed] [Google Scholar]
- Borderies JR, Goñalons E, Angel F, Vergara P, Jiménez M. Effect of different calcium channel blockers on inhibitory junction potentials and slow waves in porcine ileum. Life Sci. 1997;60:883–892. doi: 10.1016/s0024-3205(96)00670-4. [DOI] [PubMed] [Google Scholar]
- Boyer JL, Mohanram A, Camaioni E, Jacobson KA, Harden TK. Competitive and selective antagonism of P2Y1 receptors by N6-methyl 2′-deoxyadenosine 3′,5′-bisphosphate. Br J Pharmacol. 1998;124:1–3. doi: 10.1038/sj.bjp.0701837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgard EC, Niforatos W, Van Biesen T, Lynch KJ, Touma E, Metzger R, Kowaluk E, Jarvis M. P2X receptor-mediated ionic currents in dorsal root ganglion neurons. J Neurophysiol. 1999;82:1590–1598. doi: 10.1152/jn.1999.82.3.1590. [DOI] [PubMed] [Google Scholar]
- Camaiono E, Boyer JL, Mohanram A, Harden TK, Jacobson KA. Deoxyadenosine-bisphosphate derivatives as potent antagonists at P2Y1 receptors. J Med Chem. 1998;41:183–190. doi: 10.1021/jm970433l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damer S, Niebel B, Czeche S, Nickel P, Ardanuy U, Schmalzing G, Rettinger J, Mutschler E, Lambrecht G. NF279, a novel potent and selective antagonist of P2X receptor-mediated responses. Eur J Pharmacol. 1998;350:R5–6. doi: 10.1016/s0014-2999(98)00316-1. [DOI] [PubMed] [Google Scholar]
- Fischer B. Therapeutic applications of ATP-(P2) receptors agonists and antagonists. Exp Opin Ther Patents. 1999;9:385–399. [Google Scholar]
- Humphreys BD, Virginio C, Surprenant A, Rice J, Dubyak GR. Isoquinolines as antagonists of the P2X7 nucleotide receptor: high selectivity for the human versus rat receptor homologues. Mol Pharmacol. 1998;54:22–32. doi: 10.1124/mol.54.1.22. [DOI] [PubMed] [Google Scholar]
- Ingall AH, Dixon J, Bailey A, Coombs ME, Cox D, McInally JI, Hunt SF, Kindon ND, Teobald BJ, Willis PA, Humphries RG, Leff P, Clegg JA, Smith JA, Tomlinson W. Antagonists of the platelet P2T receptor: a novel approach to antithrombotic therapy. J Med Chem. 1999;42:213–220. doi: 10.1021/jm981072s. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Kim YC, Camaioni E, van Rhee AM. Structure activity relationships of P2 receptor agonists and antagonists. In: Turner JT, Weisman G, Fedan J, editors. The P2 Nucleotide Receptors, The Receptors. Chapter 4 Humana Press; Clifton, NJ: 1997. pp. 81–107. [Google Scholar]
- Jacobson KA, Kim YC, Wildman SS, Mohanram A, Harden TK, Boyer JL, King BF, Burnstock G. A pyridoxine cyclic-phosphate and its 6-arylazo-derivative selectively potentiate and antagonize activation of P2X1 receptors. J Med Chem. 1998;41:2201– 2206. doi: 10.1021/jm980183o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JL, Van Rhee AM, Chang L, Patchornik A, Evans P, Melman N, Jacobson KA. Structure activity relationships of 4-phenylethynyl-6-phenyl-1,4-dihydropyridines as highly selective A3 adenosine receptor antagonists. J Med Chem. 1997;40:2596–2608. doi: 10.1021/jm970091j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanjhan R, Housley GD, Burton LD, Christie DL, Kippenberger A, Thorne PR, Luo L, Ryan AF. Distribution of the P2X2 receptor subunit of the ATP-gated ion channels in the rat central nervous system. J Comp Neurol. 1999;407:11–32. [PubMed] [Google Scholar]
- Kim YC, Brown SG, Harden TK, Boyer JL, Dubyak G, King BF, Burnstock G, Jacobson KA. Structure activity relationships of pyridoxal phosphate derivatives as potent and selective antagonists of P2X1 receptors. J Med Chem. 2000 doi: 10.1021/jm9904203. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King BF, Wildman SS, Ziganshina LE, Pintor J, Burnstock G. Effects of extracellular pH on agonism and antagonism at a recombinant P2X2 receptor. Br J Pharmacol. 1997;121:1445–1453. doi: 10.1038/sj.bjp.0701286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King BF, Liu M, Pintor J, Gualix J, Miras-Portugal MT, Burnstock G. Diinosine pentaphosphate (IP5I) is a potent antagonist at recombinant rat P2X1 receptors. Br J Pharmacol. 1999;128:981–988. doi: 10.1038/sj.bjp.0702876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht G, Ardanuy U, Baumert HG, Bo X, Hoyle CHV, Nickel P, Pfaff O, Ralevic V, Windschief U, Zigashin AU, Ziyal R, Mutscheler E, Burnstock G. Design and pharmacological characterization of selective P2-purinoceptor antagonists. Perspectives in Receptor Research. 1996:337–350. [Google Scholar]
- Lewis CJ, Surprenant A, Evans RJ. 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate (TNP-ATP) — a nanomolar affinity antagonist at rat mesenteric artery P2X receptor ion channels. Br J Pharmacol. 1998;124:1463–1466. doi: 10.1038/sj.bjp.0702001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KJ, Touma E, Niforatos W, Kage KL, Burgard EC, Van Biesen T, Kowaluk EA, Jarvis MF. Molecular and functional characterization of human P2X(2) receptors. Mol Pharmacol. 1999;56:1171–1181. doi: 10.1124/mol.56.6.1171. [DOI] [PubMed] [Google Scholar]
- Mateo J, Garcia-Lecea M, Miras-Portugal MT, Castro E. Ca2+ signals mediated by P2X-type purinoceptors in cultured cerebellar Purkinje cells. J Neurosci. 1998;18:1704–1712. doi: 10.1523/JNEUROSCI.18-05-01704.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita I, Tada S, Kunimoto K, Tsuda M, Kise M, Kimura K. Synthesis and antihypertensive activities of 1,4-dihydropyridine-5-phosphonate derivatives. I Chem Pharm Bull (Tokyo) 1987;35:3898–3904. doi: 10.1248/cpb.35.3898. [DOI] [PubMed] [Google Scholar]
- Moro S, Guo D, Camaioni E, Boyer JL, Harden TK, Jacobson KA. Human P2Y1 receptor. Molecular modeling and site-directed mutagenesis as tools to identify agonist and antagonist recognition sites. J Med Chem. 1998;41:1456–1466. doi: 10.1021/jm970684u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandanan E, Jang SY, Moro S, Kim H, Siddiqi MA, Russ P, Marquez V, Busson R, Herdewijn P, Harden TK, Boyer JL, Jacobson KA. Synthesis, biological activity, and molecular modeling of ribose-modified adenosine bisphosphate analogues as P2Y1 receptor ligands. J Med Chem. 2000;43:829–842. doi: 10.1021/jm990249v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA, Barnard EA. Nucleotide receptors. Curr Opin Neurobiol. 1997;7:346–357. doi: 10.1016/s0959-4388(97)80062-1. [DOI] [PubMed] [Google Scholar]
- Pankratov Y, Castro E, Miras-Portugal MT, Krishtal O. A purinergic component of the excitatory postsynaptic current mediated by P2X receptors in the CA1 neurons of the rat hippocampus. Eur J Neurosci. 1998;10:3898–3902. doi: 10.1046/j.1460-9568.1998.00419.x. [DOI] [PubMed] [Google Scholar]
- Parker MS, Larroque ML, Campbell JM, Bobbin RP, Deininger PL. Novel variant of the P2X2 ATP receptor from the guinea pig organ of Corti. Hear Res. 1998;121:62–70. doi: 10.1016/s0378-5955(98)00065-3. [DOI] [PubMed] [Google Scholar]
- Soto F, Lambrecht G, Nickel P, Stühmer W, Busch AE. Antagonistic properties of the suramin analogue NF023 at heterologously expressed P2X receptors. Neuropharmacology. 1999;38:141–149. doi: 10.1016/s0028-3908(98)00158-0. [DOI] [PubMed] [Google Scholar]
- Swanson KD, Reigh C, Landreth GE. ATP-stimulated activation of the mitogen-activated protein kinases through ionotrophic P2X2 purinoreceptors in PC12 cells. Difference in purinoreceptor sensitivity in two PC12 cell lines. J Biol Chem. 1998;273:19965–19971. doi: 10.1074/jbc.273.32.19965. [DOI] [PubMed] [Google Scholar]
- Triggle DJ. Drugs acting on ion channels and membranes. In: Emmett JC, editor. Comprehensive Medicinal Chemistry. Vol. 3. Pergamon Press; London: 1985. pp. 1047–1099. [Google Scholar]
- Williams M, Bhagwat SS. P2 purinoceptors: a family of novel therapeutic targets. Ann Rep Med Chem. 1996;31:21–30. [Google Scholar]