

Abstract
In the present study we synthesize 18F-labeled insulin of high specific radioactivity. A new prosthetic group methodology, in which [18F]fluoride displaces a bromide group of 4-(bromomethyl)-benzoylamine intermediates, was used. The 4-(fluoromethyl)benzoyl product was chemically stable. 18F-Labeled insulin retains the essential biological properties of native insulin, as measured in vitro by binding to insulin receptors on human cells and stimulation of glucose metabolism in rat adipocytes. The overall process can be carried out speedily to yield a product of sufficient purity to permit in vivo studies. The method appears to be applicable to a wide variety of peptides.
Positron emission tomography (PET) is a powerful, noninvasive procedure for the quantitative regional mapping of biochemical processes in vivo (Kilbourn & Zalutsky, 1985). Positron-emitting radionuclides commonly used to label molecules for the development of PET-scanning agents include 11C, 13N, 15O, and 18F. The relatively long half-life of 18F (110 min) has made this nuclide particularly attractive. To date, hundreds of compounds have been labeled by these radioisotopes, including sugars, steroids, amino acids, and other metabolites and drugs. The broad list of medical applications of PET-scanning agents includes studies of cerebral blood flow, transport and metabolism of carbohydrates, protein synthesis, neurotransmitter biosynthesis and storage, aminergic receptor quantitation, and tumor visualization (Phelps & Mazziotta, 1985).
Despite the proven value of PET, [11C]Met-enkephalin appears to be the only reported example of a hormone-like peptide labeled site-specifically with a positron emitter (Nargen et al., 1986). A scheme for protein labeling with a positron emitting radionuclide has appeared recently (Kilbourn et al., 1987), utilizing fluoride substitution of an aromatic nitro group (18F product and precursor were not separated). Earlier, urokinase had been labeled with [18F]fluoroacetic acid through carbodiimide coupling (Müller-Platz et al., 1982). Neither of these methods, however, has been used in a site-specific manner or with peptide hormones. The complications inherent in the labeling of large molecules with short-lived radionuclides reflect the necessity of carrying out the preparation and purification of the compound to homogeneity under extreme time constraints.
Methodologies for introduction of 18F into compounds for PET scanning must address three main points. (1) Because of limitations in the amount of isotope available for synthesis, the radiochemical reaction must be extremely efficient and favor the formation of the desired product under conditions of a very low concentration of fluoride (10−4 M or less). Further, the large excess of organic substrate normally present subsequently must be removed quantitatively from the reaction mixture. (2) Because of the short half-life of the nuclide, the chemistry, including purification of the product to a degree suitable for administration, must be completed in a very short time. (3) The 18F-labeled product must retain biological activity.
Here we describe a prosthetic group methodology for the rapid introduction of 18F into insulin, a protein that has receptors on cells throughout the body including the central nervous system. This methodology, based on a rapid nucleophilic displacement of bromide by [18F] fluoride from a benzyl bromide precursor (Jacobson et al., 1988), should be applicable to a wide range of proteins for the potential development of PET-scanning agents.
Materials and Methods
For each compound in the series 4–6 and 8–10 the designation a or b refers to homologues having two or four methylene groups, respectively.
A1,B29-Di-Boc-insulin (2)
A solution of (Boc)2O (18.2 mg, 83 μmol), 19 μL of triethylamine, and 20 mg of N-hydroxysuccinimide in 50 μL of DMSO was mixed for 20 min. This solution of activated Boc formed in situ was added in three portions at intervals of 3 min to a rapidly stirred solution of 200 mg (33.3 μmol) of insulin (porcine, Sigma) in 4 mL of DMSO containing 5% triethylamine. After 20 min, amino-ethanol (5 μL) was added to terminate the reaction. The di-Boc-insulin was purified further to more than 95% homogeneity by RP-HPLC1 on a semipreparative column (Vydac, protein, 10 mm × 250 mm) using a gradient of 25% solvent B to 80% solvent B in 40 min (solvent A, 0.1% TFA in water; solvent B, 0.1% TFA in acetonitrile).
A1,B29-Di-Boc-B1-[(N-hydroxysuccinimidyl)suberoyl]-insulin (3)
A mixture of 10 mg (1.7 μmol) of A1,B29-di-Boc-insulin (2) and 6.2 mg (17 μmol) of disuccinimidyl suberate (DSS) in 0.10 mL of DMSO containing 5% (by volume) triethylamine was stirred for 3 min at ambient temperature. After the reaction, protein was precipitated by the addition of 1 mL of ether containing 5% acetonitrile and isolated by microcentrifugation. To remove disuccinimidyl suberate completely, successive resuspension of the precipitate in a 1:1 [or 50% (by volume)] ether-acetonitrile mixture and reisolation by centrifugation was carried out three times. The precipitate then was dissolved in 150 μL of DMSO and purified on a semipreparative C-4 column (Vydac, 10 mm × 250 mm) using isocratic conditions (34% aqueous acetonitrile). The peak that migrated from 24 to 33 min was collected and lyophilized. Rechromatography of this fraction on an analytical C-4 column (Vydac, protein, 4.1 mm X 250 mm) using gradient elution (solvent A, 0.1% TFA in water; solvent B, 0.1% TFA in acetonitrile, using a gradient of 25% solvent B to 80% solvent B in 40 min) confirmed the presence of a single pure product.
The chemical reactivity of 3 was confirmed by its rapid reaction with primary amines. For example, exposure to an excess of aminoethanol for 5 min produced a new compound as demonstrated by a shift in the HPLC retention time from 24.5 to 22.5 min. A quantitative conversion was indicated by the peak areas of the HPLC profiles.
B1-[[[4-[[4-(Fluoromethyl)benzoyl]amino]butyl]amino]-suberoyl]insulin [Fluoroinsulin (5b)]
Compound 9b (537 μg, 1.66 μmol) was treated with 50 μL of TFA for 10 min. After removal of the TFA under a stream of nitrogen, 5 mg (0.83 μmol) of activated insulin, 3, in 50 mL of DMSO containing 5% triethylamine was added to the residue. After 1 h at 55 °C, an excess of ethylenediamine was added. This step effected the aminolysis of the excess activated insulin, producing a compound that, because of the resulting shift of retention time from 22.8 to 20.1 min, could be separated readily from the desired product by HPLC. The yield of 4b was 60% as judged by HPLC peak areas. After lyophilization of the HPLC fractions containing 4b, the residue was treated for 10 min with 50 μL of TFA to remove the Boc protecting groups. Evaporation of TFA under a stream of nitrogen gave pure 5b, identified by N-terminal amino acid sequencing and by total protein hydrolysis following dansylation. The product was tested for hormone activity by bioassay with rat adipocytes, binding to insulin receptors, and binding to anti-insulin antibodies.
Mono-N-(tert-butyloxycarbonyl)ethylenediamine (6a)
Two grams (33.3 mmol) of ethylenediamine (Aldrich Chemical Co., Milwaukee, WI) was dissolved in 10 mL of acetone. To this solution was added with stirring 726 mg (3.33 mmol) of di-tert-butyloxy dicarbonate (Pierce Co., Rockford, IL). After 5 min, the excess of ethylenediamine was removed by evaporation under a stream of nitrogen, and the residue was purified by TLC on silica with acetone. A band migrating at Rf = 0.25 was isolated as an oil upon elution with acetonitrile and was identified as the product by MS. The product gave a positive ninhydrin test and, after treatment with TFA, comigrated with ethylenediamine.
N-[4-(Bromomethyl)benzoyl]-N′-(tert-butyloxycarbonyl)-ethylenediamine (8a)
Three hundred milligrams (1.4 mmol) of 4-α-bromotoluic acid (Aldrich) was dissolved in 5 mL of acetonitrile, and 223 mg (1.4 mmol) of 7a was added in portions with stirring. After dissolution was complete, 1.6 mL of 1 M dicyclohexylcarbodiimide in methylene chloride was added in portions, and stirring was continued for 1 h. After removal of the precipitate that had formed, evaporation of the acetonitrile gave a solid product. Purification by TLC on silica with ethyl acetate-petroleum ether (3:2) and elution with acetonitrile gave 200 mg (38%) of 8a, mp 147 °C. MS (CI-NH3) showed peaks at 358 and 258 m/z, corresponding to loss of the tert-butyloxycarbonyl group. The structure was confirmed by NMR, and purity was verified by HPLC (Altex C-18 4.4 × 250 mm column, flow rate 0.9 mL/min, retention time 25.4 min; solvent A, 0.1% TEA in water; solvent B, 0.1% TFA in acetonitrile, using a gradient of 25% solvent B to 80% solvent B over 45 min).
N- [4-(Bromomethyl)benzoyl]-N′-(tert-butyloxycarbonyl)-butane-1,4-diamine (8b), mp 139 °C, was prepared from N-succinimidyl 4-(bromomethyl)benzoate (7) (Jacobson et al., 1988) and mono-N-(tert-butyloxycarbonyl)butane-1,4-diamine (6b) in an overall yield of 70%. HPLC purification was performed on a C-4 Vydac column under the same conditions as for 8a, retention time of 20.3 min. The product was identified by MS (peaks at 386 and 286 m/z) and 1H NMR in CDCl3, with characteristic resonances at δ 7.41 and 7.36 (each d, 2 H, Ar) and 4.58 (s, 2 H, CH2Br).
N-[4-(Fluoromethyl)benzoyl]-N′-(tert-butyloxycarbonyl)-ethylenediamine (9a)
To a solution of 10 mg (2.8 mmol) of 8a in 300 mL of dry acetonitrile was added, with stirring, 28 mL (28 mmol) of a 1 M solution of tetramethylammonium fluoride in tetrahydrofuran. After 10 min at 50 °C, the product (Rf = 0.46) was isolated by preparative TLC on silica with ethyl acetate–petroleum ether (3:2). HPLC showed the presence of a single pure product, retention time of 20.6 min, using the gradient described above for analysis of 8a. MS (CI-NH3) showed a molecular ion at 297 and a fragment at 197, corresponding to the loss of the tert-butyloxycarbonyl group.
N-[4-(Fluoromethyt)benzoyl]-N′-(tert-butyloxycarbonyl)-butane-1,4-diamine (9b) was synthesized in a similar fashion and purified by HPLC (conditions as for 8b, retention time of 18.9 min). MS and NMR (characteristic FCH2 doublet in DMSO-d6 at δ 5.45, J = 47.4 Hz) were consistent with the assigned structure.
Radiochemical Syntheses
(a) [18F]-B1-[[[4-[[4-(fluoromethyl)benzoyl]amino]butyl]amino]suberoyl]insulin ([18F]-5b)
18F-Labeled compound 10b (from the previous step but described later) was allowed to react with 1.5 mg of activated insulin, 3, in 50 μL of DMSO containing 1 μL of triethylamine for 10 min at 50 °C. Following the reaction, the remaining unreacted active ester was removed by reaction with aminomethylated polystyrene (1% cross-linked, 50 mesh, PCJ Co., Nes Ziona, Israel) for 10 min at 50 °C, followed by reaction with 5 μL of ethylenediamine in DMSO (50 μg/μL) for 5 min at ambient temperature. The total mixture was injected onto a C-4 protein analytical HPLC column (Vydac, 214TP, 4.6 mm × 250 mm) and eluted with 32% aqueous acetonitrile containing 0.1% TFA at a rate of 0.9 mL/min. The eluent was monitored both for radioactivity and UV absorbance (208 nm), and the radioactive fraction was collected (retention time of 31.6 min). The product, [18F]-4b, was removed from the HPLC eluent by adsorption on a C-8 cartridge (1 mL of Bond-Elute) and then eluted with 0.5 mL of 80% aqueous acetonitrile containing 0.5% TFA. The solvent was evaporated at 50 °C under a stream of argon, and the Boc protecting groups were removed by treatment with 0.2 mL of TFA for 5 min at ambient temperature. The TFA was evaporated under a stream of argon and the final product was formulated in 10 mL of pH 7.2 isotonic phosphate buffer and sterilized by passage through a 0.22-μm filter (Millex 6V) prewashed with buffer containing 1% bovine serum albumin (BSA).
(b) [18F]-N-[4-(Fluoromethyl)benzoyl]-N′-(tert-butyloxycarbonyl)butane-1,4-diamine ([18F]-9b)
[18F]Fluoride was produced by proton bombardment of 95% 18O-enriched water at the NIH cyclotron facility. The aqueous 18F was added to the reaction vessel which contained 1.8 μmol of tetramethylammonium hydroxide. The 18OH2 was evaporated at 100 °C with a stream of argon, and residual traces of water were removed by three successive evaporations with 300 μL of anhydrous acetonitrile. After the final evaporation, 400 μL of acetonitrile containing 3 mg (8 μmol) of 8b was added. The resulting mixture was heated at 100 °C for 15 min. After evaporation of the acetonitrile, the residue was applied to a silica column (3 mL of Bond-Elute) with 2 × 0.3 mL of chloroform. The column was eluted with ethyl acetate-hexane (1:1), and the fraction (6 mL) containing the 18F radioactivity was collected. The solvent was removed by evaporation at 50 °C under a stream of argon. The residue in 0.3 mL of acetonitrile–H2O (1:1) was injected onto an HPLC semipreparative column (Altex ODS, 10 mm × 250 mm) and eluted isocratically with 32% aqueous acetonitrile at a rate of 4 mL/min. The eluent was monitored both for radioactivity and UV absorbance (208 nm), and the fraction containing radioactivity was collected (retention time of 30.8 min). The HPLC fraction was evaporated partially under a stream of argon at 50 °C and then loaded onto a C-18 cartridge (3 mL of Bond-Elute). All of the radioactivity was retained, and after air drying the cartridge, it was eluted with 2 mL of acetonitrile to yield 9b. After evaporation of the acetonitrile under a stream of argon, the Boc group of 9b was removed by treatment with TFA (0.2 mL) for 10 min. The acid was evaporated to afford pure [18F]-1-[[4-(fluoromethyl)benzoyl]amino]butane-4-amine (10b) as a trifluoroacetate salt.
Results and Discussion
Insulin Modification
Since it is critical that the modified analogue of insulin retain high affinity for the receptor similar to that of native insulin, we chose the N-terminal amino group of the insulin B-chain (B1 phenylalanine) as the site for modification. Previous investigators have shown that modification of insulin at the amino group of either A1 glycine or B29 lysine causes reduction of both receptor binding and the ability to stimulate glucose uptake (Pullen et al., 1976; Yeung et al., 1980). In contrast, the B1 residue seems to be less essential for receptor binding and for biological activity. Thus, acylation of this N-terminal amine with neutral groups has little effect on receptor binding and biological activity. However, extension of the peptide chain at the B1 terminus by the addition of charged arginine or lysine residues results in a significant parallel loss of both binding and biological potency (Yeung et al., 1979).
Selective acylation of the amino groups of insulin has been described [review by Schüttler et al. (1984)]. The N-terminus of the B-chain was the site of selective incorporation of 125I (Assoian & Tager, 1981; Bahrami et al., 1980). To modify insulin selectively at the B1 phenylalanine residue (Figure 1), we first protected both A1 and B29 amino residues with tert-butyloxycarbonyl (Boc) groups (Krail et al., 1975; Hofmann et al., 1977, 1984). Since this reaction was extremely sensitive to reaction conditions, we studied several parameters to optimize yields in this critical step. Using RP-HPLC to monitor the reaction, we found the best conditions to be the use of 2.5 equiv of (Boc)2O, with N-hydroxysuccinimide as an additive, in DMSO, with a reaction time of 20 min. Following quenching of the excess (Boc)2O by excess aminoethanol, A1,B29-di-Boc-insulin (2) was purified to >99% homogeneity by RP-HPLC. N-Terminal sequencing confirmed the structural assignment by showing that the amine content consisted of >99% phenylalanine. Total hydrolysis after dansylation gave only dansylphenylalanine. These conditions consistently gave high yields of 2 (68%) and were superior to those using other solvents or acylating agents. With longer reaction times the desired product was contaminated with another insulin derivative (probably A1,B1-di-Boc) which was difficult to separate. Two other products present in the reaction mixture were identified as A1-mono-Boc- and A1,B29,B1-tri-Boc-insulin by N-terminal sequencing and by hydrolysis after dansylation.
FIGURE 1.

Synthesis of 18F-labeled insulin, 5, using an intermediate containing the FMB prosthetic group. (Left column) Synthesis of an amino intermediate containing the (fluoromethyl)benzoyl (FMB) prosthetic group, 10. (Right column) Modification of native porcine insulin, 1. See Results for description. For series a, n = 2; for series b, n = 4.
Under these conditions, the preference for sites of acylation of insulin was in the order of A1 > B29 > B1. In contrast, in dimethylformamide the preference was B29 > A1 > B1. The preference of B29 over A1 during modification also was found in earlier studies using the (azidophenylacetyl)-N-hydroxysuccinimide active ester for modification of insulin (Yip et al., 1980).
Prosthetic Group Attachment
In the next step, a prosthetic group for the introduction of 18F was coupled to the protected insulin derivative at the remaining free amine (B1). Recently we have shown that α-bromotoluic acid can be coupled to amino groups as well as to dipeptides to give the corresponding (bromomethyl)benzoyl (BMB) derivatives (Jacobson et al., 1988). We also demonstrated a facile and quantitative displacement of bromide by fluoride in dry acetonitrile or dimethylformamide. The reaction was rapid, even at very low concentrations of reagents.
In theory there are two routes available for the radiolabeling of proteins using a prosthetic group, differing in the step at which the radioisotope is introduced. Thus, the radiolabel can be incorporated into an intermediate (e.g., 10, Figure 1) to be coupled subsequently to the protein (e.g., the activated insulin derivative 3), or, alternatively, a conjugate of the protein and prosthetic group can be radiolabeled as a final step (Figure 2). In this study only the former route, which obviates the exposure of the protein to nucleophilic fluoride, was successful.
FIGURE 2.

Attempted synthesis of 18F-labeled insulin by fluoride exchange on the (bromomethyl)benzoyl (BMB) labeled B-chain with or without a glycine spacer. See Results for description (method A).
A fluorine-labeled intermediate carrying an amino group for subsequent reaction with the activated insulin was prepared, as shown in the left column of Figure 1. First, N-alkyldiamines, such as ethylenediamine and 1,4-diaminobutane, were partially protected as the mono-Boc derivatives, 6. For each compound, the designation a or b refers to homologues having two or four methylene groups, respectively. These protected diamines then were coupled to (bromomethyl)benzoyl (BMB) N-hydroxysuccinimide ester (7). The resulting fully blocked BMB-diamine derivatives, 8, prepared as fluorination substrates, as stable either in solution or as crystalline powders.
The protected BMB derivatives, 8, reacted with tetra-alkylammonium fluoride in acetonitrile at 70 °C within 10 min. Use of the BMB-diaminobutane derivative 8b gave better yields for the fluoride exchange reaction under mild conditions than did the two-carbon homologue 8a. A major side product (approximately 20%) of the fluorination reaction was shown by MS and NMR to be the corresponding (hydroxymethyl)benzoyl derivative. For peptides and other substrates having limited solubility in acetonitrile, the fluorination reaction may be carried out in other nonreactive, polar, aprotic solvents such as tetrahydrofuran or dimethylformamide. However, DMSO is not a suitable solvent for the fluorination reaction, since the benzyl bromide group reacts with this solvent. Following purification of the FMB intermediate, 9, by RP-HPLC, the Boc protecting group was removed (TFA, 5 min) to provide a free amino derivative of FMB, 10, the substrate for coupling with an active ester of insulin, 3. The fluoromethyl group of 10a or 10b is not subject to nucleophilic attack by protein-bound nucleophiles. This was demonstrated by lack of reaction with a large excess of ethylenediamine in either acetonitrile or DMSO at 70 °C for 15 min.
A protected insulin derivative containing an N-succinimidyl alkyldiacyl group coupled at the B1-terminal amine, 3, was prepared. This reactive peptide derivative is similar to the activated o-nitrophenyl ester derivative reported by Schüttler and Brandenburg (1982) and was found to be a suitable reactive intermediate. Such N-hydroxysuccinimide active esters are very good acylating reagents, having selectivity for amines, but yet are relatively stable and can be stored dry for long periods. A1,B29-di-Boc-insulin (2) was treated for 2 min with a large excess of disuccinimidyl suberate (DSS) in DMSO in the presence of triethylamine to give a single major product, as judged by RP-HPLC. After isolation (see Materials and Methods), the product, A1,B29-di-boc-B1-(succinimidylsuberoyl)insulin (3) was purified rigorously by RP-HPLC on a semipreparative C-4 column to >99% homogeneity. The chemical reactivity of this derivative was demonstrated by its immediate and quantitative reaction with primary amines such as aminoethanol and ethylenediamine. As a lyophilized solid or in anhydrous DMSO solution, the product was stable for several weeks at 0 °C.
This route offered a convenient procedure for product purification after the coupling reaction between the insulin and the prosthetic group. Following coupling, the large excess of N-hydroxysuccinimide ester of the insulin (11) present in the reaction was removed readily by a rapid reaction with an amine-functionalized polymeric support, such as poly(benzylamine) resin.
The two FMB-amine intermediates, 10a and 10b, were compared for efficiency of acylation by A1,B29-di-Boc-B1-(succinimidylsuberoyl) insulin (3). The reaction of 5 nmol of the activated insulin, 7 nmol of FMB-diaminobutane (10b), and an excess of triethylamine in DMSO was complete within 10 min at 70 °C. In contrast, several hours were required for the analogous reaction with 10a. Because of this greater nucleophilicity, the FMB-amine 10b was chosen for subsequent studies leading to 18F-labeled insulin.
To complete the synthesis, A1,B29-di-Boc-B1-[[[(FMB-amino)butyl]amino]suberoyl]insulin (4b) was prepared, purified by RP-HPLC, and lyophilized. The Boc protecting groups were removed from this insulin derivative by brief treatment with TFA. RP-HPLC analysis showed that a single pure product was obtained. Verification that this material is the desired B1-[[[(FMB-amino)butyl]amino]suberoyl]insulin[fluoroinsulin (5b)] was obtained by N-terminal sequencing, which indicated that the free α-amino content consisted of >99% glycine. By total hydrolysis after dansylation, only dansylglycine and ε-dansyllysine were obtained.
Fluoroinsulin (5b) competed for the binding of 125I-labeled insulin to insulin receptors on human lymphoblastoid (IM-9) cells (Lesniak et al., 1987) in a manner indistinguishable from that of insulin (Figure 3A). Fluoroinsulin also stimulated glucose metabolism in adipocytes of young rats (Gliemann et al., 1984) in a manner roughly similar to that of unlabeled insulin; because standardization was inexact we cannot provide precise limits for the bioactivity of the fluoroinsulin. In all of the studies, the fluoroinsulin was effectively free of underivatized hormone, indicating that fluoroinsulin itself has an affinity for insulin receptors quite like that of insulin and an intrinsic activity at least roughly similar to that of insulin.
FIGURE 3.

Receptor binding assays. (A) IM-9 lymphoblasts (American Type Culture Collection, Rockville, MD), approximately 3 × 106 cells/mL, were incubated at pH 7.8,13 °C, for 90 min (Lesniak et al., 1987), with 125I-labeled insulin in the absence and presence of a range of concentrations of unlabeled insulins [porcine insulin (+) or fluoroinsulin (5b) (△)] or 18F-labeled insulin, 5b, after decay (●). The vertical axis indicates the specific inhibition of 125I-labeled insulin binding to IM-9 lymphoblasts as a percent of maximum inhibition. The solid line depicts the inhibition curve for porcine insulin. The concentration of each insulin was determined by absorbance at 208 nm and/or radioimmunoassay. (B) Lymphoblasts were incubated for 75 min under the same conditions as in (A), with either 125I-labeled insulin (~15 pg/500 μL) (●) or 18F-labeled insulin (~15 pg/500 μL) (△) in the absence and presence of unlabeled porcine insulin. Specific inhibition of binding of labeled hormone is plotted as a function of total hormone concentration. The radioactivity measurements using 18F were corrected for decay.
In the alternate route shown in Figure 2, we also attempted derivatization of di-Boc-insulin (2) with the BMB prosthetic group for direct fluorination of a modified peptide. However, reaction of 2 with the N-hydroxysuccinimide active ester of BMB (7) gave no detectable product, 11, after 24 h. The N-hydroxysuccinimide ester of BMB-glycine (12) reacted cleanly with 2 over 24 h, resulting in the selective attachment of BMB-glycine to the B1-terminal amine of protected insulin. The product, 13, was isolated and purified to greater than 99% homogeneity by RP-HPLC, but in the presence of tetra-butylammonium fluoride extensive decomposition occurred even at room temperature. None of the desired product, 14, of nucleophilic displacement of bromide could be identified. The many protein-derived side products suggest that the high nucleophilicity of fluoride in organic aprotic solvents may compromise procedures that require nucleophilic reactions of fluoride directly on protein derivatives, particularly those proteins containing disulfide linkages.
Radiochemical Synthesis
[18F]-B1-[[[(FMB-amino)butyl]amino]suberoyl]insulin ([18F]-5b) was synthesized by the sequence of reactions in Figure 1 using [18F]fluoride (10 Ci/μmol at end of bombardment) and with some technical modifications. The first step in the radiosynthetic sequence, the SN2 displacement of bromine in compound 8b, was carried out by using tetramethylammonium hydroxide both as a pseudocarrier of fluoride as well as a phase-transfer catalyst. Following the fluoride displacement step, radioactivity was present in only two organic products, in addition to unreacted fluoride adsorbed to the glass. They comigrated with the desired N-Boc-FMB derivative (9b) and with the deprotected FMB-amine (10b) and occurred in a ratio of 20:1, respectively. A small solid-phase extraction column was used to reduce the mass of the crude reaction mixture, thereby simplifying HPLC purification. The nonradioactive products were all well separated, and the product was obtained in greater than 95% chemical and radiochemical purity. In a typical experiment this corresponded to a total mass of 2 μg Without rigorously optimizing the reaction conditions, the radiochemical recovery in the HPLC of purified [18F]-9b isolated in a total synthesis time of 80 min was 52% after correcting for 18F decay.
The second step in this sequence involved the reaction of [18F]FMB-butanediamine (9b) with the activated form of insulin 3. After 10 min at 50 °C, most of the unreacted active ester, which typically is present in a 100-fold molar excess, was removed by reaction with (aminomethyl)polystyrene. After this treatment, any residual insulin active ester was converted to a more polar adduct by reaction with ethylenediamine. The crude reaction mixture was then purified by HPLC under isocratic conditions that allowed excellent separation between the radioactive product and the other side products. The 18F-labeled FMB-(di-Boc) insulin eluted at the same time as the unlabeled FMB-(di-Boc)insulin (4b) (Figure 4). The purity of the product was greater than 95%, as indicated by the radioactivity and UV (208 nm) profiles. The Boc groups were removed with treatment of TFA, and the product, B1-[18F] [[[(FMB-amino)butyl]amino]suberoyl]insulin([18F]fluoroinsulin, [18F]-5b), was used without further purification. The total time required for the synthesis, purification, and formulation of [18F]fluoroinsulin was approximately equivalent to 2 half-lives of 18F. The specific activity of the final product 4 h after bombardment was greater than 1 Ci/μmol.
FIGURE 4.
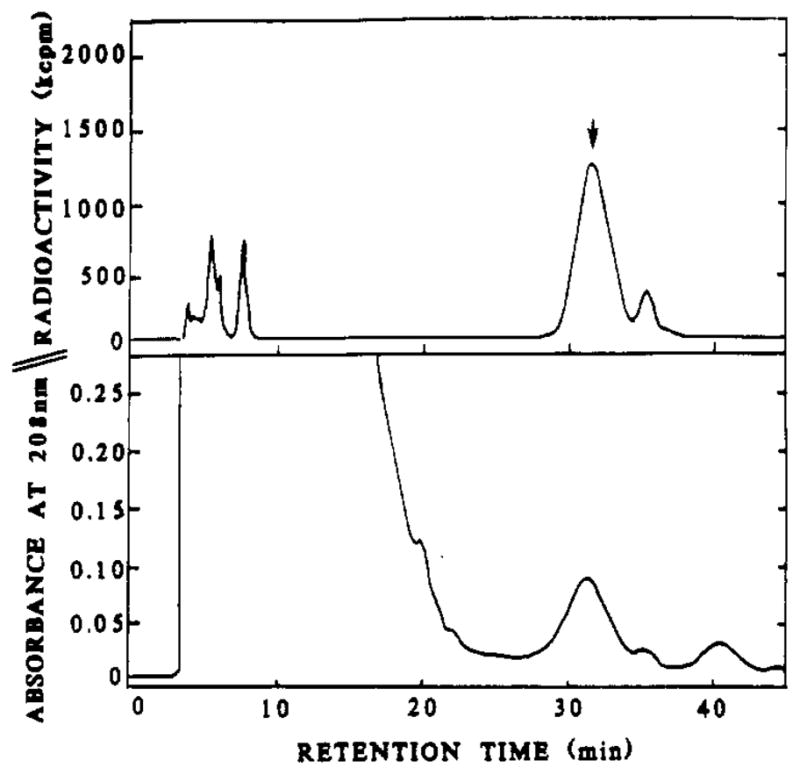
Purification of [18F]FMB-insulin ([18F]-4b) using RP-HPLC. The peak corresponding to [18F]-4b is denoted by the arrow. For conditions, see Materials and Methods.
The [18F]fluoroinsulin, studied over the concentration range 0.02–9 ng/mL, bound to insulin receptors on human lymphoblastoid (IM-9) cells with an affinity that was equal to or better than that observed for the binding of [125I]iodoinsulin (Figure 5). [18F]Fluoroinsulin that had undergone radioactive decay competed for [125I]iodoinsulin binding to insulin receptors in a manner indistinguishable from that of unlabeled insulin (Figure 3A). Further, unlabeled insulin competed with [18F]fluoroinsulin in a fashion similar to its competition for [125I]iodoinsulin (Figure 3B), providing further support for the close resemblance of the fluoro derivative to the iodo form as well as the native form of the molecule.
FIGURE 5.

Specific binding of 18F-labeled insulin to insulin receptors. In two separate experiments IM-9 cells were incubated for 75 min with 18F-labeled insulin (○) or 125I-labeled insulin (▲) over a range of concentrations from 0.01 to 9 ng/mL under conditions described in the legend for Figure 3. The concentrations of 18F-labeled insulin used were determined by radioimmunoassay and by UV absorbance at 208 nm of concentrated solution using RP-HPLC as described under Materials and Methods. The difference in bound to free labeled hormone (B/F) for the two insulin derivatives in part reflects differences in cell numbers.
Summary
We have synthesized and purified 18F-labeled insulin rapidly and have shown that this derivative binds in a saturable and specific fashion to a membrane receptor site. On the basis of these results, the radioactive FMB-insulin derivative holds promise as an imaging agent for PET studies of insulin receptors in vivo.
The methodology introduced here for coupling an 18F-prelabeled prosthetic group to a protein is presented in the context of a general method for peptide and protein labeling. Advantages of the FMB prosthetic group for radiofluorination include: (1) use of fluoride as source of 18F (no carrier added); (2) attachment methodology preserves sensitive groups on ligand; (3) rapid and efficient reaction; (4) no elimination reactions; (5) the FMB group is chemically stable to nucleophiles and heating; and (6) BMB starting materials can be stored at room temperature. A requirement is the presence of a nonessential functional group on the protein that can be activated for condensation with an amine.
The modification of insulin at the B1 site resulted in no change or a small enhancement (data not shown) of affinity at insulin receptors. This residue thus is a good site for the attachment of reporter groups in the preparation of molecular probes for the insulin receptor. We have presented a sequence of steps by which insulin may be derivatized selectively at this residue in high yield. The exceptionally high purity of insulin intermediates used in this sequence is achieved by using semipreparative RP-HPLC techniques.
Additional studies will be required to establish whether this derivative of insulin will have a sufficiently long half-life in vivo to image insulin receptors effectively and whether the utility will encompass insulin receptors in the central nervous system as well as in the periphery.
Acknowledgments
We thank Dr. Larry Wardzala, Diabetes Branch, NIDDK, for insulin bioassays, Dr. Nina Raben, Diabetes Branch, NIDDK, for radioimmunoassays, Dr. Brian M. Martin, Chemical Neurosciences Branch, NIMH, for end-group analyses, and Professor Klaus Hofmann, University of Pittsburgh, for an authentic sample of di-Boc-insulin and for helpful discussions.
Footnotes
Abbreviations: BMB, 4-(bromomethyl)benzoyl; FMB, 4-(fluoromethyl)benzoyl; RP-HPLC, reversed-phase high-pressure liquid chromatography; NHSu, N-hydroxysuccinimide; TLC, thin-layer chromatography.
These results were presented in part at the 196th National Meeting of the American Chemical Society, Los Angeles, Sept 29, 1988, Abstract MEDI 40.
References
- Assoian RK, Tager HS. J Biol Chem. 1982;257:4042–4049. [PubMed] [Google Scholar]
- Bahrami S, Zahn H, Brandenburg D, Machulla HJ, Dutschka K. Radiochem Radioanal Lett. 1980;45:221–226. [Google Scholar]
- Gliemann J, Rees WD, Foley JA. Biochim Biophys Acta. 1984;804:68–76. doi: 10.1016/0167-4889(84)90100-9. [DOI] [PubMed] [Google Scholar]
- Hofmann K, Finn FM, Friesen HJ, Diaconescu C, Zahn H. Proc Natl Acad Sci USA. 1977;74:2697–2700. doi: 10.1073/pnas.74.7.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K, Zhang WJ, Romovacek H, Finn FM, Bothner-by AA, Mishra PK. Biochemistry. 1984;23:2547–2553. doi: 10.1021/bi00307a002. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Furlano DC, Kirk KL. J Fluorine Chem. 1988;39:339–347. doi: 10.1016/S0022-1139(00)81606-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilbourn MR, Zalutsky MR. J Nucl Med. 1985;26:655–662. [PubMed] [Google Scholar]
- Kilbourn MR, Dence C, Welch MJ, Mathias CJ. J Nucl Med. 1987;28:462–470. [PubMed] [Google Scholar]
- Krail G, Brandenburg D, Zahn H, Geiger R. Hoppe-Seyler’s Z Physiol Chem. 1975;352:1595–1598. [PubMed] [Google Scholar]
- Lesniak MA, Hedo JA, Grunberger G, Marcus-Samuels B, Roth J, Gorden P. Methods Enzymol. 1987;150:701–723. doi: 10.1016/0076-6879(87)50116-1. [DOI] [PubMed] [Google Scholar]
- Müller-Platz CM, Kloster G, Legler G, Stöcklein G. J Labelled Compd Radiopharm. 1982;19:1645–1646. [Google Scholar]
- Nargen K, Ragnarsson V, Langstromm B. Appl Radiat Isot. 1986;37:533–539. [Google Scholar]
- Phelps ME, Mazziotta JC. Science. 1985;228:799–809. doi: 10.1126/science.2860723. [DOI] [PubMed] [Google Scholar]
- Pullen RA, Lindsay DG, Wood SP, Tickle IJ, Blundell TL, Wollmer A, Krail G, Brandenburg D, Zahn H, Gliemann J, Gammeltoft S. Nature (London) 1976;259:369–373. doi: 10.1038/259369a0. [DOI] [PubMed] [Google Scholar]
- Schüttler A, Brandenburg D. Hoppe-Seyler’s Z Physiol Chem. 1982;363:317. doi: 10.1515/bchm2.1982.363.1.317. [DOI] [PubMed] [Google Scholar]
- Schüttler A, Gattner HG, Brandenburg D. In: Methods in Diabetes Research, Vol I: Laboratory Methods, Part A. Larner J, Pohl S, editors. Wiley; New York: 1984. [Google Scholar]
- Yeung CWT, Moule ML, Yip CC. J Biol Chem. 1979;254:9453–9457. [PubMed] [Google Scholar]
- Yeung CWT, Moule ML, Yip CC. Biochemistry. 1980;19:2196–2203. doi: 10.1021/bi00551a031. [DOI] [PubMed] [Google Scholar]
- Yip CC, Yeung CWT, Moule ML. Biochemistry. 1980;19:70–76. doi: 10.1021/bi00542a011. [DOI] [PubMed] [Google Scholar]