

SUMMARY
The A2a adenosine receptor, a member of the G protein-coupled receptor family, is important in the regulation of dopaminergic pathways of the brain and in platelet and cardiovascular functions. In this study, the role of extracellular loops in ligand binding to the human A2a receptor was explored through site-directed mutagenesis. Four glutamate/aspartate residues (Glu151, Glu161, Glu169, and Asp170) in the second extracellular loop (E2) and a cysteine residue (Cys262) in the third extracellular loop (E3) were individually replaced with alanine and other amino acids. A proline residue (Pro173) in E2 was mutated to arginine, the homologous amino acid in A3 receptors. The binding properties of the resultant mutant receptors were determined in transfected COS-7 cells. The mutant receptors were tagged at their amino terminus with a hemagglutinin epitope, thus allowing their detection in the plasma membrane with immunological techniques. High affinity specific binding of [3H]2-[4-[(2-carboxyethyl)phenyl]ethyl-amino]-5′-N-ethylcarboxamidoadenosine (15 nm) and [3H]8-[4-[[[[(2-aminoethyl)-amino]carbonyl]methyl]oxy]phenyl]-1,3- dipropylxanthine (4 nm), an A2a agonist and antagonist, respectively, was not observed with four of the mutant receptors, E151A, E151Q, E151D, and E169A, although they were well expressed at the cell surface. The E151A and E169A mutant receptors showed nearly full stimulation of adenylyl cyclase at ~103-fold higher concentrations of 2-[4-[(2-carboxyethyl)phenyl]ethyl-amino]-5′-N-ethylcarboxamidoadenosine. The E161A mutant receptor showed an increase in affinity for the nonxanthine adenosine antagonist 9-chloro-2-(furyl)[1,2,4]triazolo[1,5-c]quinazolin-5-amine (6-fold) but not for other ligands. An E169Q mutant gained affinity (5–22-fold) for adenosine derivatives (agonists) substituted at N6 but not at C2 or C5′ positions. Mutant receptors D170K and P173R were similar to wild-type receptors in binding of both agonist and antagonist radioligands. A C262G mutant also resembled the wild-type receptor in radioligand binding, indicating that a potential disulfide bridge with another cysteine residue in proximity is not required for the structural integrity of the receptor. Our data suggest that certain amino acids in the second extracellular loop may be directly or indirectly involved in ligand binding.
Endogenous adenosine is released locally within various organs such as the heart, brain, and liver, particularly in response to physiological stress (1). The regulation of blood pressure by centrally (2) and peripherally mediated (vascular) mechanisms (3) involves A2a receptors. Selective A2a agonists such as CGS 21630 (4), which does not readily cross the blood-brain barrier, have been evaluated as antihypertensive agents. In the brain, A2areceptors occur primarily in the olfactory tubercle and striatum, where they are colocalized with postsynaptic, striatopallidal D2 dopamine receptors (5). Adenosine acts in a manner opposite to dopamine and therefore depresses locomotor activity (6). Thus, therapeutic approaches for central nervous system disorders have been proposed based on activating A2a receptors, targeting diseases in which the dopaminergic system is hyperactive, e.g., schizophrenia (7), and Huntington’s disease (5). Parkinson’s disease, in which the dopaminergic system is hyporesponsive, may be amenable to treatment by selective antagonism of A2a receptors (5). A2a receptors have been cloned from thyroid, brain, and mast cell cDNA libraries (8). The A2a adenosine receptor activates adenylyl cyclase (9 and references therein) via coupling to Gs.
Most of the mutagenesis and modeling studies of the binding of small molecules, such as biogenic amines (10, 11), to GPCRs in general and to adenosine receptors in particular (12–15) have implicated the TMs in ligand recognition. Several residues within TM5–7 of human A2a receptors that are facing the ligand binding cleft have been shown to be involved in ligand recognition (12). However, for peptide ligand binding to GPCRs, residues of the extracellular loops, both charged (16,17) and uncharged (18), often play a crucial role. For adenosine receptors, there is evidence that the binding of xanthine antagonists, although small molecules, involves at least one of the outer loops. Using chimeric A1/A3 receptors, Olah et al. (19) demonstrated the involvement of the carboxyl-terminal half of the second extracellular loop (E2) in antagonist binding at bovine A1 receptors.
In this study, the role of two of the extracellular loops in ligand binding was explored through site-directed mutagenesis of the human A2a receptor. It was noted that there is a predominance of negatively charged residues in the second extracellular loop and that these residues are somewhat conserved within the adenosine receptor family. Thus, these glutamic acid and aspartic acid residues as well as several uncharged residues (cysteine and proline) in the extracellular loops were targeted in this study. We demonstrate that mutation of specific amino acids in the second extracellular loop, especially of Glu151, has strong effects on ligand binding.
Experimental Procedures
Materials
Human A2a adenosine receptor cDNA (pSVLA2a) was provided by Dr. Marlene A. Jacobson (Merck Research Labs, West Point, PA). Taq polymerase for the PCR was purchased from Perkin Elmer Cetus (Emeryville, CA). All enzymes used in this study were obtained from New England Biolabs (Beverly, MA). The agonists CGS 21680, NECA, R–N6-phenylisopropyladenosine, 2-chloroad-enosine, and DPMA and the antagonists XAC and CGS 15943 were purchased from RBI (Natick, MA). [3H]CGS 21680 (38.3 Ci/mmol), [3H]NECA (25.7 Ci/mmol), and [3H]XAC (118 Ci/mmol) were obtained from DuPont-NEN (Boston, MA), and [3H]adenine (15 Ci/mmol) was supplied by American Research Chemicals (St. Louis, MO). IB-MECA was prepared as described (20). Chemical structures of ligands used in this study are given in Table 1. FBS and o-phenylenediamine dihydrochloride were purchased from Sigma Chemical Co. (St. Louis, MO). The Sequenase kit, ATP, and cAMP were acquired from United States Biochemical (Cleveland, OH). All oligonucleotides used were synthesized by Bioserve Biotechnologies (Laurel, MD). A monoclonal antibody (12CA5) against an HA epitope was purchased from Boehringer Mannheim Biochemicals (Indianapolis, IN), and goat anti-mouse IgG (γ chain specific) antibody conjugated with horseradish peroxidase was purchased from Sigma. DEAE-Dextran was obtained from Pharmacia-LKB (Piscataway, NJ). Rolipram was a gift from Schering AG (Berlin, Germany).
TABLE 1. Ligand binding properties of wild-type, E161A, and E169Q human A2a, receptors.
Agonist and antagonist binding affinities (Ki, values) were determined in [3H]CGS 21680 (15 nm) competition binding studies using membrane homogenates prepared from transiently transfected COS-7 cells, as described in Experimental Procedures. Ki, values were calculated from IC50 value with the Kaleidagraph program. Approximately 15 μg of membrane protein/tube were used. The following Kd (nm) and Bmax values for [3H]CGS 21680 (pmol/mg protein, in parentheses) were determined: wild-type, 22.3 ± 4.6 (15.5 ± 0.1); E161A, 41.7 ± 9.2 (17.0 ± 2.7); and E169Q, 57.0 ± 1.8 (9.26 ± 0.37). Values are mean ± standard error of two or three independent experiments, each performed in duplicate.
| ligand | Position of substitution (agonists) |
Ki
|
||
|---|---|---|---|---|
| Wild-type | E161A | E160Q | ||
| nm | ||||
| Agonist 2-CADO 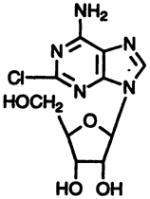
|
2 | 144 ± 33 | 221 ± 71 | 946 ± 20 |
DPMA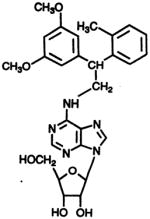
|
N6 | 36.3 ± 1.2 | 61.2 ± 0.2 | 1.62 ± 0.94 |
R-N6-Phenyiisopropyladenosine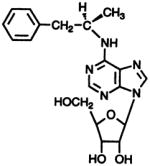
|
N6 | 158 ± 19 | 264 ± 21 | 33.9 ± 2.8 |
| Agonist IB-MECA 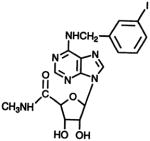
|
N6,5′ | 456 ± 56 | 433 ±13 | 53.4 ± 2.6 |
NECA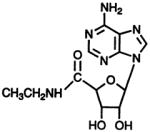
|
5′ | 11.4 ± 4.0 | 23.9 ± 0.2 | 253 ± 40 |
| Antagonist CGS 15943 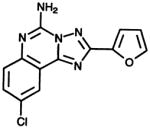
|
1.71 ± 0.28 | 0.287 ± 0.16 | 2.26 ± 1.4 | |
| XAC |
6.98 ± 1.6 | 7.19 ± 1.70 | 11.5 ± 2.8 | |
Plasmid construction and site-directed mutagenesis
The coding region of pSVLA2a was subcloned into the pCD-ps cDNA expression vector (21), yielding pcDA2a. All mutations were introduced into pcDA2a with standard PCR mutagenesis techniques (22). The accuracy of all PCR-derived sequences was confirmed by dideoxy sequencing of the mutant plaamids (23).
Epitope tagging
A 9-amino acid sequence derived from the influenza virus HA protein (TAC CCC TAC GAC GTC CCC GAC TAC GCC; peptide sequence: YPYDVPDYA) was inserted after the second methionine (residue 4) at the extracellular amino terminus of the A2a adenosine receptor gene as described previously (12). The first methionine (residue 1) was deleted as described by Kim et al. (12).
Transient expression of mutant receptors in COS-7 cells
COS-7 cells (2 × 106) were seeded into 100-mm culture dishes containing 10 ml Dulbecco’s modified Eagle’s medium supplemented with 10% FBS. Cells were transfected with plasmid DNA (4 μg DNA/dish) with the DEAE-dextran method (24) ~24 hr later and grown for an additional 72 hr at 37°.
Membrane preparation and radioligand binding assay
Cells were scraped into ice-cold lysis buffer (4 ml of 50 mM Tris, pH 6.8 at room temperature, containing 10 mM MgCl2). Harvested cells were homogenized with a Polytron homogenizer and then spun at 27,000 × g for 15 min. Cell membranes (pellet) were resuspended in the same buffer.
For saturation and competition binding experiments with [3H]CGS 21680 or [3H]NECA at human A2a receptors expressed in COS-7 cell membranes, each tube contained 100 μl of membrane suspension (containing 2 units/ml adenosine deaminase; Boehringer Mannheim Biochemicals), 50 μl of radioligand, and either 50 μl of buffer/competitor (50 mM Tris, pH 6.8, 10 mM MgCl2) or 50 μl of either 60 nM ([3H]CGS 21680 binding) or 160 nM ([3H]NECA binding) CADO in buffer (to define nonspecific binding). The mixtures were incubated at 25° for 120 min, filtered, and washed three times with ~5 ml ice-cold buffer/wash with use of a Brandel cell harvester. Saturation curves were established at ≥10 different concentrations. Data analysis was performed with the KaleidaGraph program (Abel-beck Software, version 3.01).
cAMP determination
cAMP levels were determined by measuring the conversion of [3H]ATP to [3H]cAMP. One day after transfection, cells were transferred from 100-mm dishes into six-well dishes (~3 × 105 cells/well) and incubated with culture media containing 2 μCi/ml [3H]adenine. After 24 hr, the cultures were washed and incubated with 1 ml/well Hanks’ balanced salt solution containing 0.1 mM rolipram for 15 min at 37°. The cells were incubated with different concentrations of the agonist CGS 21680 (in culture media) for 30 min at 37°. The reaction was terminated by aspiration of the media and addition of 1 ml of ice-cold 5% trichloroacetic acid containing 1 mM ATP and 1 mM cAMP. After 30-min incubation at 4°, cell lysates were eluted through sequential chromatography on Dowex and alumina columns (25). cAMP formation is expressed as fold-stimulation of conversion of [3H]ATP into [3H]cAMP (26).
ELISA
For indirect cellular ELISA measurements, cells were transferred to 96-well dishes (4–5 × 104 cells/well) at 1 day after transfection. At ~48 hr after splitting, cells were fixed in 4% form-aldehyde in phosphate-buffered saline for 30 min at room temperature. After being washed with phosphate-buffered saline three times and blocked with Dulbecco’s modified Eagle’s medium (containing 10% FBS), cells were incubated with HA-specific monoclonal antibody (12CA5; 20/μg/ml) for 3 hr at 37°. Plates were washed and incubated with a 1:2000 dilution of a peroxidase-conjugated goat anti-mouse IgG antibody (Sigma) for 1 hr at 37°. H2O2 and o-phenylenediamine (each 2.5 mM in 0.1 M phosphate-citrate buffer, pH 5.0) served as substrate and chromogen, respectively. The enzymatic reaction was stopped after 30 min at room temperature with 1 M H2SO4 solution containing 0.05 M Na2SO3, and the color development was measured bichromatically with the BioKinetics reader (EL 312, Bio Tek Instruments, Winooski, VT) at 490 nm while using absorbance at 630 nm as the base-line.
Results
A sequence alignment of the human A2a receptor and other adenosine receptor clones was carried out manually, and the second and third extracellular domains (E2 and E3) are shown in Fig. 1. In these loop regions, there is a high degree of sequence homology between bovine and human A1 receptors and between rat and human A2a receptors but a low homology among subtypes of adenosine receptors; thus, the alignment is necessarily imprecise. It was noted that there is a predominance of acidic side chains in the E2, and accordingly such residues are aligned whenever possible in Fig. 1.
Fig. 1.

Location of mutations carried out in this study illustrated through an alignment of the second (E2) and third (E3) extracellular loops of selected adenosine receptor subtypes. Due to the low sequence homology, this alignment is necessarily imprecise. Periods, gaps inserted in the sequence for alignment purposes. Bold type, A2a receptor residues mutated in the present study. Underline, an 11-amino acid region of the bovine A1 receptor that restored the ability to bind xanthines when included in a chimeric bovine A1/rat A3 construct (19). Accession numbers are bA1 (bovine), P28190; hA1 (human), P30542; rA2a (rat), P30543; hA2a (human), P29274; hA2b (human), P29275; and hA3 (human), P33765. Numbers above sequences, amino acid positions in the human A2a receptor.
Mutation sites and expression of mutant A2a-adenosine receptors
The residues of the human A2a receptor, selected as targets for site-directed mutagenesis, are shown in bold type (Fig. 1). Each of these amino acid residues was individually replaced with alanine and/or other amino acids (see below). These mutation sites include glutamate residues (Glu151, Glu161, and Glu169), an aspartic acid residue (Asp170), a proline residue (Pro173), and a cysteine residue (Cys262). Residue Asp170 was mutated to lysine, the homologous residue in the A1 receptor. Residue Pro173 was mutated to arginine, the homologous residue in the sheep and human A3 receptors, based on a prediction that this may be a site responsible for enhanced affinity of acidic ligands (27). In E3, A1 and A2a receptors contain cysteine residues separated by two amino acid residues and thus are positioned to stabilize a β-turn by forming a disulfide bridge. The likelihood of such a β-turn at this position has been discussed (28).
Each mutant (except C262G) contained an epitope-tag sequence at the amino terminus for immunological detection. The pharmacological properties of the mutant receptors were compared with the wild-type receptor similarly modified.
Ligand binding properties of mutant A2a-adenosine receptors
Radioligand binding studies with a fixed concentration of either the agonist [3H]CGS 21680 (15 nM) or the antagonist [3H]XAC (4 nM) were carried out on the wild-type and mutant receptors. Specific binding of either ligand was greatly diminished (i.e., <1% of the specific binding of [3H]CGS 21680 observed with the wild-type receptor and <8% of the [3H]XAC binding) in the following mutants: E151A, E151Q, E151D, and E169A At position 169, a glutamine residue, which, like glutamic acid, has hydrogen bond donating and accepting properties but is uncharged, may substitute (see below). The Kd (nM) and Bmax values for [3H]CGS 21680 binding at the E169Q mutant receptor were found to be 57.0 ± 1.8 nM and 9.26 ± 0.37 pmol/mg protein, respectively (three experiments). The comparable values for the wild-type receptor expressed in COS-7 cells were 22.3 ± 4.6 nM and 15.5 ±0.1 pmol/mg protein (four experiments). In contrast to substitution of Glu169, at position 151, the requirement for glutamic acid to achieve high affinity ligand binding is absolute.
An immunological method (12) was used to determine whether the pharmacologically inactive mutant receptors were retained in an intracellular compartment or properly delivered to the cell surface. A nonapeptide tag derived from the HA epitope was attached near the amino terminus of the receptor. Previously, this modification was found not to interfere with binding or adenylyl cyclase activation by the A2a receptor (12). To quantify the amount of HA-tagged receptor protein present on the cell surface, an indirect cellular ELISA was used (see Experimental Procedures). Essentially, nonpermeabilized cells expressing an HA-tagged receptor were incubated with a monoclonal antibody (12CA5) directed against this epitope, followed by the addition of a secondary peroxidase-conjugated antibody and the photometric determination of peroxidase activity. Previous studies showed that the absorbance readings obtained with the ELISA system were directly proportional to the amount of receptor protein present on the cell surface (12). This ELISA procedure did not interfere with the integrity of the plasma membrane barrier, as determined in control experiments with carboxyl-terminal tagged receptors (12). Thus, the ELISA measurements are specific for receptor molecules properly inserted in the cell membrane.
All of the mutant receptors that were undetectable with the use of radioligands were found to be well expressed on the cell surface with the ELISA assay. The following expression levels (as percentage of expression of wild-type receptors) were determined: E151A, 90.8 ± 4.1%; E151D, 95.1 ± 5.3%; E151Q, 88.9 ± 2.8%; and E169A, 93.2 ± 4.0%.
To eliminate the possibility that the lack of high affinity binding of [3H]CGS 21680 to the various mutant receptors is not simply due to steric interference by the bulky chain located at the 2-position of the ligand, binding studies were also carried out with a high concentration of [3H]NECA (100 nM). Nonspecific binding was determined with a final concentration of 40 μM CADO. As reported above for [3H]CGS 21680 binding, <10% of the specific binding of [3H]NECA observed with the wild-type receptor was found with the following mutants: E151A, E151Q, E151D, and E169A. Under these conditions, wild-type receptors displayed specific binding consisting of ~90% of total binding.
In addition to the E169Q mutant receptor, the E161A mutant receptor retained the ability to bind ligands with high affinity (Table 1), indicating that this site is not involved in ligand recognition. The Kd (nM) and Bmax values for [3H]CGS 21680 binding at the E161A mutant receptor were found to be 41.7 ± 9.2 nM and 17.0 ± 2.7 pmol/mg protein, respectively (three experiments). Ki values for the binding of a variety of agonists and antagonists versus [3H]CGS 21680 (Table 1) were found to be comparable to those found with the wild-type receptor; however, some differences were noted. For example, the mutant receptor E161A demonstrated selectively enhanced affinity (6-fold) for the antagonist CGS 15943 (Fig. 2A). For all other ligands, the affinities at E161A mutant receptors were very similar to the corresponding wild-type receptor values. At the E169Q mutant receptor, antagonists bound with affinities within 2-fold of wild-type receptors, but agonists, depending on the site of modification, were considerably more or less potent than at wild-type receptors (Table 1 and Fig. 2B). The affinity of the following N6-substituted agonists was enhanced in the E169Q mutant receptors: IB-MECA (8.5-fold), DPMA (22-fold), and R-N6-phenylisopropyladenosine (4.7-fold). The agonists NECA and CADO, which were substituted exclusively at positions other than N6, displayed 22-fold and 6.6-fold diminished affinity, respectively, for the E169Q mutant receptors.
Fig. 2.

Displacement of specific binding of the agonist radioligand [3H]CGS 21680 from epitope-tagged A2a wild-type (WT) and E161 A (E161A) and E169Q (E169Q) mutant receptors expressed in COS-7 cells. Competitors used were CGS15943 (A) and IB-MECA (B). Competition binding studies were carried out with membrane homogenates prepared from transfected COS-7 cells, as described in Experimental Procedures. Data presented are the results of a representative experiment carried out in duplicate.
Mutant receptors D170K, P173R, and C262G were similar to wild-type receptors in binding of both agonist and antagonist radioligands (determined with 15 nM [3H]CGS 21680 and 4 nM [3H]XAC), indicating that these residues are not a requirement for ligand recognition.
Functional assay
To determine whether the two alanine mutant receptors (E151A and E169A) that lacked high affinity radioligand binding were still functional at high agonist concentrations, their ability to mediate increases in intracellular cAMP levels was studied (Fig. 3). In the presence of the PDE inhibitor rolipram (0.1 mM), the wild-type receptor expressed in COS-7 cells stimulated adenylyl cyclase with an EC50 value of 5.73 ± 1.24 nM (Fig. 3). The E151A and E169A mutant receptors stimulated adenylyl cyclase only at ≥10 μM CGS 21680. In neither case was a full degree of stimulation reached at 30 μM CGS 21680; thus, it was not possible to determine EC50 values for the mutant receptors. Nevertheless, it is clear that the concentration-response curves for E151A and E169A mutant receptors are shifted to the right by ~3 orders of magnitude and that substantial agonist efficacy is present at the high concentrations. Under the same conditions, CGS 21680 (≤30 μM) failed to stimulate adenylyl cyclase in the untransfected COS-7 cells.
Fig. 3.

Stimulation of adenylyl cyclase in COS-7 cells transiently expressing epitope-tagged A2a wild-type or mutant A2a-adenosine receptors in the presence of 2 units/ml adenosine deaminase and 0.1 mM rolipram. The receptors studied were wild-type (WT), E151A (E151A), and E169A (E169A). Transfected COS-7 cells were incubated for 30 min at 37° (for details, see Experimental Procedures) with increasing concentrations of CGS 21680. In control experiments, untransfected COS-7 cell membranes did not show stimulation at ≤30 μM CGS 21680. Data are presented as fold increase in cAMP above basal levels (400–600 cpm/well) in the absence of CGS 21680. Each curve represents the mean fold-stimulation of two independent experiments, each carried out in duplicate.
Effects of divalent cations
Based on the hypothesis that the E2 loop of the A2a receptor might participate in ion complexation, we explored the effects of divalent metal cations on binding. This preliminary study was based on the observation that calcium and magnesium ion binding sites in other proteins such as calmodulin (29) often contain multiple glutamic acid and aspartic acid residues. In addition, a direct binding of divalent cations to the A2a receptor was proposed (30).
Using wild-type A2a receptors expressed in COS cells, we confirmed the findings of Johansson et al. (30) that at high concentrations, Ca2+ and Mg2+ cause an increase in the level of binding of the agonist radioligand (Fig. 4). The dependence of levels of [3H]CGS 21680 binding in the E161A and E169Q mutant receptors on the magnesium ion concentration was similar to that seen with the wild-type receptor, i.e., a gradual rise in binding on increasing the levels of the divalent cation from 10 mM to 1 M (Fig. 4). If the high affinity binding of [3H]CGS 21680 were dependent on the presence of a receptor-bound divalent cation, then the radioligand binding in the mutant receptors, which might have a lower affinity for the metal ions, might be expected to be driven by raising the Mg2+ concentration. Yet, in the E151D mutant receptor, even very high levels of Ca2+ and Mg2+ failed to restore specific [3H]CGS 21680 binding at a concentration of 15 nM. Either CaCl2 or MgCl2 added to the incubation medium at a concentration of 50 mM, 100 mM, or 1 M failed to boost the level of binding observed with the E151D mutant beyond 2% of the level of binding observed with the wild-type receptor. Thus, we were unable to experimentally support the hypothesis that the E2 loop participates in binding of Ca2+ and/or Mg2+.
Fig. 4.

The effect of magnesium ions on the specific binding of the radioligand [3H]CGS 21680 (15 nM) at epitope-tagged A2a wild-type (WT), E161A (E161A) and E169Q (E169Q) mutant receptors expressed in COS-7 cells. The amount of binding is expressed as a percentage of the binding in the presence of 10 mM Mg2+, the standard concentration used in all other binding experiments. The specific binding of [3H]CGS 21680 (15 nM) at E151D mutant receptors expressed in COS-7 cells was found to be undetectable (<2% of wild-type) in the presence of 10, 50, 100, or 1000 mM Mg2+ or in the presence of added Ca2+ at the same concentrations.
Discussion
A structural study of human A2a receptors has shown that residues in TM5, TM6, and TM7 are important for ligand binding (12). Recently, the importance of TM3 was also established through mutagenesis of human A2a receptors.1 In particular, two histidine residues in TM6 and TM7, which are conserved among A1 and A2 receptors, have been implicated in ligand binding (12, 32). Mutagenesis studies of bovine A1 receptors, involving single amino acid replacements and the use of chimeric A1/A3 receptors, have also identified regions that are involved in ligand recognition. Specifically, a hexapeptide segment in TM5 (13) and several residues (Ile274 and Ser277) in TM7 (14, 15) are important for binding of adenosine derivatives.
Neither of the molecular models proposed for A2a adenosine receptors (12, 33) takes into account possible contributions of the extracellular loops in ligand recognition. Nevertheless, results by Olah et al. (28) prompted us to reevaluate the functional importance of residues in these regions, as has been done for other GPCRs. For example, in peptide GPCRs the extracellular loops are often involved in ligand binding (16–18, 34). In rhodopsin, mutational analysis of the intradiscal loops has indicated that certain residues are essential for retinal binding (35). In addition, in muscarinic acetylcholine receptors, a tryptophan residue in the first extracellular loop was required for high affinity ligand binding (36).
In this study, anionic residues located in the E2 loop were targeted for mutagenesis because the occurrence of glutamate and aspartate residues in E2 of A2a receptors and other GPCRs is greater than the statistical average. Many GPCRs contain two or more negative charges in the E2 loop (16). For example, other receptors that contain a glutamate residue at positions close to Glu169 of the A2a receptor, with respect to the sequence of TM5, are dopamine D2, red opsin, neurokinin3, human thromboxane A2, and somatostatin2 receptors (34). Interestingly, glutamate and aspartate residues have often been shown to be involved in carbohydrate recognition by various enzymes and other proteins (37, 38).
Several residues in the E2 loop of the human A2a receptor were found to be essential for both agonist and antagonist binding. Of the three glutamic acid residues mutated, Glu151 was found to be essential for ligand recognition, whereas Glu161 was not. The third glutamic acid residue (Glu169) was important for recognition, but some modification was tolerated. All three point mutations at Glu151 (alanine, aspartic acid, and glutamine) prepared in this study were unable to bind [3H]CGS 21680 at 15 nM. The fact that mutant receptor E151D (differing from the wild-type receptor by only one methylene unit) was unable to bind radioligand is indicative of the strict structural requirements at this position. Curiously, in rat A2a receptors an aspartic acid residue occurs in the same region of the loop (Fig. 1). Mutagenesis at Glu169 was tolerated only for a side chain of comparable size, polarity, and hydrogen bonding capability (glutamine), but preservation of the charge was not required. Because the amino termini of mutant receptors E151A, E151D, E151Q, and E169A were expressed on the outer surface of the plasma membrane, the lack of high affinity radioligand binding is not a result of sequestering of the mutant receptors intracellularly. It is likely related to ligand recognition, assuming proper folding of the receptor. Moreover, for the two alanine mutant receptors studied in a functional assay, the stimulation of adenylyl cyclase achieved at high concentrations of CGS 21680 indicates proper folding. These results suggest that both residues Glu151 and Glu169 appear to be involved, either directly or indirectly, in the molecular recognition of both adenosine agonists and antagonists. The affinity for the N6-substituted adenosine agonist DPMA was enhanced by 34-fold at the E169Q mutant receptor versus the wild-type receptor, and the affinity of other N6-substituted adenosine analogues was also enhanced but to a lesser degree. The disubstituted IB-MECA resembled other N6-substituted adenosine analogues in this respect (Fig. 2B), indicating that 5′-substitution does not preclude the affinity enhancement in the E169Q mutant receptor. Residue Glu161 proved to be nonessential for binding of either agonist or antagonist radioligand. However, the E161A mutant receptor displayed a selectively enhanced affinity for the nonxanthine antagonist CGS 15943 (Fig. 2A).
It remains unclear whether the E2 loop of the A2a receptor is in direct contact with ligands. A direct contact is conceivable, especially in light of the putative disulfide bridge formed between Cys166 (E2 loop) and a cysteine residue near the amino terminus of TM3 (28), which would covalently attach the E2 loop in physical proximity to the ligand binding site. A folded loop projecting into the binding cavity of the receptor would be conformationally possible due to flexibility of this region of the protein. Hydrophobic residues in the loop might favor association with transmembrane domains. A propensity for β-turns was predicted for the E2 loop of canine A2a receptors through the use of two algorithms (28). Conformational modeling studies are under way in our laboratory. An alternate hypothesis is that the E2 loop is not in direct contact with the ligand but can modulate the assembly of the transmembrane domains (39), which are primarily involved in ligand binding. The major sequence differences between species might argue against a specific contact between the glutamic acid residues and the A2a receptor-bound ligand. Nevertheless, this loop region has a strong influence on ligand binding and might relate to differences among species and among subtypes of adenosine receptors.
In certain ion channels proteins (40), the extracellular loops are known to bend inward into the pore and to participate in the complexation of small molecular species. Thus, there is structural precedent for the proposal that the E2 loop in human A2a adenosine receptors, and possibly in GPCRs in general, may be in direct contact with the ligand by projecting into the cavity. Consistent with this proposal are the findings of Olah et al. (19), which state that the carboxyl-terminal 11 amino acids of this loop in bovine A1 receptors participate in antagonist recognition.
A conceivable role for multiple glutamate residues in the E2 is that of metal complexation, although in this study we were unable to restore binding properties to the mutant receptor in the presence of high concentrations of divalent cations. It has already been noted that the binding of [3H]CGS 21680 is unusually dependent on both monovalent and divalent cations (30) in a manner that is only partly explained by a change in coupling to G proteins. Thus, Johansson et al. (30) concluded that the effects of magnesium ions are probably exerted by binding to the receptor itself. In this study, the glutamate mutants that had [3H]CGS 21680 binding properties nearly identical to those of wild-type receptors (E161A and E169Q) also displayed the same dependence of the level of binding on divalent cations as did wild-type receptors (Fig. 4). Thus, neither residue Glu161 nor the carboxylate group of Glu169 is essential for ion complexation. A mutant receptor that totally lacked high affinity ligand binding (E151D) was not “pharmacologically rescued” by very high concentrations of magnesium or calcium ions. Nevertheless, it is still possible that the residue Glu151 is essential for ion complexation with strict structural requirements. The role of divalent cations on A2a mutant and wild-type receptor binding is presently being further explored.
Acknowledgments
We thank Prof. Gary Stiles and Dr. Mark Olah (Department of Medicine and Pharmacology, Duke University, Durham, NC) and Dr. A. M. van Rhee (LBC, National Institute for Diabetes and Digestive and Kidney Diseases, Bethesda, MD) for helpful discussions and Dr. Marlene Jacobson (Merck, Dept. of Pharmacology, West Point, PA) for providing the human A2a plasmid. We thank Dr. Neli Melman (LBC, National Institute for Diabetes and Digestive and Kidney Diseases, Bethesda, MD) for assisting with binding experiments. We are grateful to Dr. Stephen D. Hurt of DuPont-NEN Products (Boston, MA) for synthesizing the [3H]XAC used in these experiments.
ABBREVIATIONS
- CGS 21680
2-[4-[(2-carboxyethyl)phenyl]ethyl-amino]-5′-N-ethylcarboxamidoadenosine
- CADO
2-chloroadenosine
- CGS 15943
9-chlorc-2-(furyl)[1,2,4]triazolo[1,5-c]quinazolin-5-amine
- DPMA
N6-[2-(3,5-dimethoxyphenyl)-2-(2-methyphenyl)ethyl]adenosine
- ELISA
enzyme-linked immunosorbent assay
- FBS
fetal bovine serum
- GPCR
G protein-coupled receptor
- HA
hemagglutinin
- IB-MECA
N6-(3- iodobenzyl)adenosine-5′-N-methyluronamide
- NECA
5′-N-ethylcarboxamidoadenosine
- PCR
polymerase chain reaction
- PDE
phosphodiesterase
- TM
transmembrane helical domain
- XAC
8-[4-[[[[(2-aminoethyl)-amino]carbonyl]methyl]oxy]phenyl]-1,3- dipropylxanthine
Footnotes
Q. Jiang, A.M. van Rhee, J.H. Kim, S. Yehle, J. Wess, and K.A. Jacobson. Hydrophilic side chains in the third and seventh transmembrane helical domains of human A2a adenosine receptors are required for ligand recognition. Submitted for publication.
References
- 1.Jacobson KA, van Galen PJM, Williams M. Adenosine receptors: pharmacology, structure activity relationships, and therapeutic potential. J Med Chem. 1992;35:407–422. doi: 10.1021/jm00081a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barraco RA, Martens K, Parizon M, Normile HJ. Role of adenosine A(2a) receptors in the nucleus-accumbens. Prog Neuropsychopharmacol Biol Psychiatry. 1994;18:545–553. doi: 10.1016/0278-5846(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 3.Olsson RA, Pearson JD. Cardiovascular purinoceptors. Pharmacol Rev. 1990;3:761–845. doi: 10.1152/physrev.1990.70.3.761. [DOI] [PubMed] [Google Scholar]
- 4.Jarvis MF, Schulz R, Hutchison AJ, Do UH, Sills MA, Williams M. [3H]CGS 21680, a selective A2 adenosine receptor agonist directly labels A2 receptors in rat brain. J Pharmacol Exp Ther. 1989;251:888–893. [PubMed] [Google Scholar]
- 5.Schiflman SN, Vanderhaegen J-J. Adenosine A2 receptor regulation of striatal gene expression. In: Belardinelli L, Pelleg A, editors. Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology. Kluver; Norwell, MA: 1995. pp. 5–14. [Google Scholar]
- 6.Nikodijević O, Jacobson KA, Daly JW. Acute treatment of mice with high-doses of caffeine: an animal-model for choreiform movement. Drug Dev Res. 1993;30:121–128. [Google Scholar]
- 7.Martin GE, Rossi DJ, Jarvis MF. Adenosine agonists reduce conditioned avoidance responding in the rat. Pharm Biochem Behav. 1993;45:951–958. doi: 10.1016/0091-3057(93)90146-k. [DOI] [PubMed] [Google Scholar]
- 8.Jacobson M. Molecular biology of adenosine receptors. In: Belardinelli L, Pelleg A, editors. Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology. Kluver; Norwell, MA: 1995. pp. 5–14. [Google Scholar]
- 9.Hide I, Padgett WL, Jacobson KA, Daly JW. A2a Adenosine receptors from rat striatum and rat pheochromocytoma PC12 cells: characterization with radioligand binding and by activation of adenylate cyclase. Mol Pharmacol. 1992;41:352–359. [PMC free article] [PubMed] [Google Scholar]
- 10.Strader CD, Candelore MR, Hill WS, Sigal IS, Dixon RAF. Identification of two serine residues involved in agonist activation of the β-adrenergic receptor. J Bid Chem. 1989;264:13572–13578. [PubMed] [Google Scholar]
- 11.Wess J, Blin N, Mutschler E, Blüml K. Muscarinic acetylcholine receptors: structural basis of ligand binding and G protein coupling. Life Sci. 1995;56:915–912. doi: 10.1016/0024-3205(95)00028-5. [DOI] [PubMed] [Google Scholar]
- 12.Kim JH, Wess J, van Rhee AM, Schöneberg T, Jacobson KA. Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2a adenosine receptor. J Biol Chem. 1995;270:13987–13997. doi: 10.1074/jbc.270.23.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olah ME, Jacobson KA, Stiles GL. Identification of an adenosine receptor domain specifically involved in binding of 5′-subetituted adenosine agonists. J Biol Chem. 1994;269:18016–18020. [PMC free article] [PubMed] [Google Scholar]
- 14.Townsend-Nicholson A, Schofield PR. A threonine residue in the 7th transmembrane domain of the human A1-adenoaine receptor mediates specific agonist binding. J Biol Chem. 1994;269:2373–2376. [PubMed] [Google Scholar]
- 15.Tucker A, Robeva AS, Taylor HE, Holeton D, Bockner M, Lynch KR, Linden J. A1 adenosine receptors: 2 amino-acids are responsible for species differences in ligand recognition. J Biol Chem. 1994;269:27900–27906. [PubMed] [Google Scholar]
- 16.Walker P, Munoz M, Martinez R, Peitsch MC. Acidic residues in extracellular loops of the human Y1 neuropeptide Y receptor are essential for ligand binding. J Biol Chem. 1994;269:2863–2869. [PubMed] [Google Scholar]
- 17.Flanagan CA, I, Becker I, Davidson JS, Wakefield IK, Zhou W, Sealfon SC, Millar RP. Glutamate 301 of the mouse gonadotropin-releasing hormone receptor confers specificity for arginine 8 of mammalian gonadotropin-releasing hormone. J Biol Chem. 1994;268:22636–22641. [PubMed] [Google Scholar]
- 18.Chini B, Mouillac B, Ala Y, Balestre M, Trumpp-Kallmeyer S, Hoflack J, Elands J, Hibert M, Manning M, Jard S, Barberis C. Tyr 115 is the key residue for determining agonist selectivity in the V1A vasopressin receptor. EMBO J. 1995;14:2176–2182. doi: 10.1002/j.1460-2075.1995.tb07211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olah ME, Jacobson KA, Stiles GL. Role of the 2nd extracellular loop of adenosine receptors in agonist and antagonist binding-analysis of chimeric A1/A3-adenosine receptors. J Biol Chem. 1994;269:24698–24698. [PMC free article] [PubMed] [Google Scholar]
- 20.Gallo-Rodriguez C, Ji XD, Melman N, Siegman BD, Sanders LH, Orlina J, Fischer B, Pu QL, Olah ME, van Galen PJM, Stiles GL, Jacobson KA. Structure-activity-relationships of N-6-benzylad-enosine-5′-uronamides as A3-selective adenosine agonists. J Med Chem. 1994;37:636–646. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okayama H, Berg PA. A cDNA cloning vector that permits expression of cDNA inserts in mammalian cells. Mol Cell Biol. 1983;3:280–289. doi: 10.1128/mcb.3.2.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Higuchi R. Using PCR to engineer DNA. In: Ehrlich HA, editor. PCR Technology. Stockton Press; New York: 1989. pp. 61–70. [Google Scholar]
- 23.Sanger R, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cullen BR. Use of eukaryotic expression technology in the functional analysis of cloned genes. Methods Enzymol. 1987;152:684–704. doi: 10.1016/0076-6879(87)52074-2. [DOI] [PubMed] [Google Scholar]
- 25.Enjalbert AJ, Bockaert J. Pharmacological characterization of the D2 dopamine receptor negatively coupled with adenylate cyclase in rat anterior pituitary. Mol Pharmacol. 1983;23:576–584. [PubMed] [Google Scholar]
- 26.Weiss S, Sebben M, Garcia-Sainz JA, Bockaert J. D2-Dopamine receptor-mediated inhibition of cyclic AMP formation in striatal neurons in primary culture. Mol Pharmacol. 1985;27:595–599. [PubMed] [Google Scholar]
- 27.Ji XD, Lubitz DKJE, Olah ME, Stiles GL, Jacobson KA. Species differences in ligand affinity at central A3-adenosine receptors. Drug Dev Res. 1994;33:51–59. doi: 10.1002/ddr.430330109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Galen PJM, Stiles GL, Michaels G, Jacobson KA. Adenosine A1 and A2 receptors: structure-function relationships. Med Res Rev. 1992;12:423–471. doi: 10.1002/med.2610120502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finn BE, Forsen S. The evolving model of calmodulin structure, function and activation. Structure. 1995;3:7–11. doi: 10.1016/s0969-2126(01)00130-7. [DOI] [PubMed] [Google Scholar]
- 30.Johansson B, Parkinson FE, Fredholm BB. Effects of mono- and divalent ions on the binding of the adenosine analogue CGS 21680 to adenosine A2 receptors in rat striatum. Biochem Pharmacol. 1992;44:2365–2370. doi: 10.1016/0006-2952(92)90681-8. [DOI] [PubMed] [Google Scholar]
- 31.Deleted in proof.
- 32.Olah ME, Ren HZ, Ostrowski J, Jacobson KA, Stiles GL. Cloning, expression, and characterization of the unique bovine A1 adenosine receptor studies on the ligand binding site by site-directed mutagenesis. J Biol Chem. 1992;267:10764–10770. [PMC free article] [PubMed] [Google Scholar]
- 33.Ijzerman AP, Van Galen PJM, Jacobson KA. Molecular modeling of adenosine receptors: the ligand-binding site on the rat adenosine A2a receptor. Eur J Pharmacol. 1994;268:95–104. doi: 10.1016/0922-4106(94)90124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fitzpatrick DV, Vandlen RL. Agonist selectivity determinants in somatostatin receptor subtypes I and II. J Biol Chem. 1994;269:24621–24526. [PubMed] [Google Scholar]
- 35.Anukanth A, Khorana HG. Structure and function in rhodopsin: requirements of a specific structure for the intradiscal domain. J Biol Chem. 1994;269:19738–19744. [PubMed] [Google Scholar]
- 36.Matsui H, Lazareno S, Birdsall NJM. Probing of the location of the allosteric site on m1 muscarinic receptors by site-directed mutagenesis. Mol Pharmacol. 1995;47:88–98. [PubMed] [Google Scholar]
- 37.Gohda K, Oka K, Tomita K, Hakoshima T. Crystal structure of RNase T1 complexed with the product nucleotide 3′-GMP. J Biol Chem. 1994;269:17531–17536. doi: 10.2210/pdb1rls/pdb. [DOI] [PubMed] [Google Scholar]
- 38.Mowbray SL, Cole LB. 1.7 A X-ray structure of the periplasmic ribose receptor fromEscherichia coli. J Mol Biol. 1992;225:155–175. doi: 10.1016/0022-2836(92)91033-l. [DOI] [PubMed] [Google Scholar]
- 39.Schöneberg T, Liu J, Wess J. Plasma membrane localization and functional rescue of truncated forms of a G protein-coupled receptor. J Biol Chem. 1995;270:18000–18006. doi: 10.1074/jbc.270.30.18000. [DOI] [PubMed] [Google Scholar]
- 40.MacKinnon R. Pore loops: an emerging theme in ion channel structure. Neuron. 1995;14:889–892. doi: 10.1016/0896-6273(95)90327-5. [DOI] [PubMed] [Google Scholar]