

Abstract
Binding isotope effects for L-aspartate reacting with the inactive K258A mutant of PLP-dependent aspartate aminotransferase to give a stable external aldimine intermediate are reported. They provide direct evidence for electronic ground state destabilization via hyperconjugation. The smaller equilibrium isotope effect with deazaPLP-reconstituted K258A indicates that the pyridine nitrogen plays an important role in labilizing the Cα-H bond.
Pyridoxal 5′-phosphate (PLP; vitamin B6) is required by many enzymes in amine and amino acid metabolism. Together, PLP enzymes catalyze a wide variety of reaction types, including transamination, decarboxylation, racemization, retro-aldol cleavage, β- and γ-elimination, and others. The efficacy of PLP in catalyzing this multiplicity of reactions originates in its ability to promote heterolytic cleavage of bonds to the nitrogen-bound carbon by stabilizing negative charge on it through resonance.
Traditionally, the catalytic power of PLP has been thought of in terms of stabilization of negative charge in the transition and product states. Here, we identify ground state weakening of the σ bond to be broken as an important component of catalysis in the reaction catalyzed by the prototypical PLP enzyme aspartate aminotransferase (AAT).
AAT, a PLP enzyme of fold type I, is one of the best understood PLP enzymes.1–3 The transamination reaction catalyzed by AAT begins with formation of the external aldimine intermediate from free enzyme and amino acid substrate (Figure 1), followed by removal of a proton from Cα of the amino acid in the external aldimine intermediate, with a rate constant of ~550 s−1 (Figure 1).4 Deprotonation forms a resonance stabilized, carbanionic “quinonoid” intermediate. With a pKa of ~30 for the Cα-H of free amino acids in water, catalysis certainly entails making this bond more labile.1,5
Figure 1.

Formation and stabilization of the carbanionic intermediate in transamination.
The obvious means by which AAT can lower the pKa of Cα-H is resonance stabilization of the carbanionic product state, via the electrophilic π system formed by the Schiff base and pyridine ring. This effect will be partially exerted at the transition state, speeding the deprotonation step.6
Another means is through reactant (ground) state destabilization. In AAT, this would involve weakening of the Cα-H bond through hyperconjugation in the external aldimine intermediate (Figure 2). It would reduce the energy difference between the reactant and product states as well as the reactant and transition states.
Figure 2.
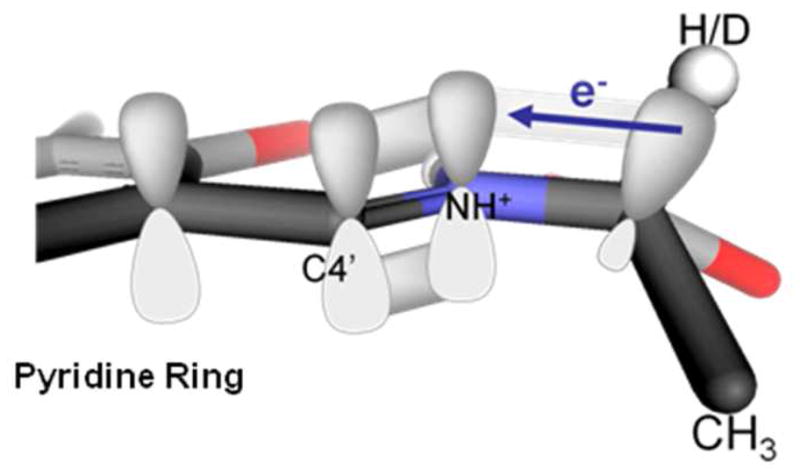
Alignment of the Cα-H σ bond with the p orbitals of the conjugated π system in PLP external aldimine intermediates, selectively weakening the Cα-H bond through hyperconjugation.
In 1966, Dunathan suggested that PLP enzymes in general have evolved to bind substrates so the appropriate bond to Cα is held parallel to the p orbitals of the π system. He noted the ground, transition, and product state effects of this orientation, and emphasized that such active site interactions provide a simple mechanism for controlling reaction specificity.7 Conformational constraint of substrates in PLP enzymes is documented in X-ray crystallographic studies.8,9 For example, the structure of the external aldimine formed from AAT and α-methyl-L-aspartate (PDB ID 1ART) shows that Arg292 and Arg386 make salt bridges to the carboxylates of the substrate, maintaining the Cα-methyl bond parallel to the p orbitals of the π system.
Hyperconjugation in the external aldimine intermediate would manifest itself as a weakening of the Cα-H bond in AAT, and correspondingly lower vibrational force constants for it. This change should result in a normal binding isotope effect (BIE), a discrimination by the enzyme against Cα deuterated substrate in formation of the external aldimine intermediate.
BIEs can occur through conformational restriction, bond angle distortion, and bond polarization, and they have been the topic of recent reviews.10–12 For example, a deuterium BIE of ~1.1 due to the weakening of the C4-H bond of NAD+ upon binding to lactate dehydrogenase was measured.13 A tritium BIE of ~1.1 was also reported for substrate binding to orotate phosphoribosyltransferase.14 Isotopic free energy profiles for another PLP enzyme, alanine racemase, identify a deuterium BIE of 1.26 ± 0.07 for external aldimine formation, suggesting hyperconjugation of the Cα-H bond with the π system of PLP.15
Direct measurement of BIEs with wild type AAT is not possible due to rapid deprotonation of the external aldimine intermediate. However, the K258A mutant of AAT forms external aldimine intermediates that are stable due to the absence of Lys258, the catalytic acid/base critical to the central 1,3-prototropic shift. The transamination activity of K258A is reduced 106–108-fold, but its activity can be rescued to high levels with exogenous amine catalysts. 16,17
The structure of K258A is very similar to wild type AAT with the exception of the site of mutation. This is illustrated in Figure 3A, which presents the structure of the K258A/deazaPLP/L-aspartate external aldimine intermediate determined here by X-ray crystallography to 1.5 Å resolution (PDB ID 4DBC). This is overlaid with the α-methyl-L-aspartate external aldimine structure in Figure 3B. Except for the absence of Lys258, the structural and catalytic properties of K258A are intact, and inferences drawn from K258A BIEs should be applicable to wild type AAT. An additional advantage of K258A here is that it binds amino acid substrates very tightly, simplifying experimental design.16
Figure 3.

(A) Top. Active site electron density for the K258A/deazaPLP/L-aspartate external aldimine. (PDB ID 4DBC) (B) Bottom. Overlay of the active sites of K258A/deazaPLP/L-aspartate with AAT/PLP/α-methyl-L-aspartate (PDB ID 1ART).
BIEs for K258A and L-aspartate were measured as follows (see SI for details). The ratio of K258A to total amino acid was varied to give different fractional conversions, with the initial concentrations of protiated and [2,3,3-2H]-L-aspartate approximately equal. The K258A-bound and free amino acids were separated by ultrafiltration and the H/D ratios of the initial (unreacted) mixture and the free amino acid were determined by ESI-MS. Plots of ln(D/H)free against ln(1-fractional reaction) were fitted to Eq. 1, where ln(D/H)0 is the initial ratio of the isotopic peaks.
| (1) |
The BIE for K258A/PLP with L-aspartate is 1.34 ± 0.05 based on three independent determinations. This BIE implies that the Cα-H bond in the external aldimine of AAT is considerably weakened by hyperconjugation. For comparison, the equilibrium IE due to hyperconjugation in the tert-butyl carbocation is 2.0 ± 0.2 for protiated vs. perdeuterated.18
The equilibrium IE corresponding to external aldimine formation was modeled computationally at the B3LYP/631g level employing the PCM water model in Gaussian09.19 The structures of (1) the alanine zwitterion, and (2) the aldimine formed between alanine and 3-hydroxy-pyridine-4-aldehyde were optimized, frequencies calculated, and ISOEFF98 was used to calculate equilibrium IEs.20 A series of aldimine structures, in which the Cα-H bond was fixed perpendicular to the ring plane (to mimic the external aldimine conformation; Figure 3) and its bond length was incremented, was analyzed. The Cα-H bond lengths were converted to changes in Pauling bond order and plotted against the calculated IE (Figure 4).19
Figure 4.

Calculated IE vs. change in Cα-H Pauling bond order (c = 0.3) for alanine combining with 3-hydroxy-pyridine-4-aldehyde to form an aldimine.
The K258A/PLP/L-aspartate BIE of 1.34 ± 0.05 for a single deuterium atom is large and corresponds to a change in bond order of 0.2 for the model compound. This remarkable result implies that ground state active site interactions of L-aspartate in the external aldimine intermediate, including hyperconjugation from the Cα-H bond into the electrophilic π system, weaken the Cα-H bond by ~20% compared to free substrate in water. It is expected that this electronic ground state destabilization plays a major role in catalysis of proton transfer by AAT via raising the energy of the reactant state toward that of the transition state, and that it contributes to reaction specificity as originally proposed by Dunathan.7
We recently synthesized a carbocyclic analogue of PLP (deazaPLP) that allows the effect of the pyridine ring to be explored.21 The BIE for [2,3,3-2H]-L-aspartate reacting with K258A/deazaPLP is 1.15 ± 0.04 based on three independent determinations, indicating that, as expected, in AAT the pyridine nitrogen is important to enforcing ground state destabilization. This is additionally supported by the reported BIE of 1.26 for alanine racemase.15 AAT enforces protonation of the pyridine nitrogen by juxtaposing Asp222, while in alanine racemase Arg219 hydrogen bonds to the pyridine nitrogen, preventing protonation.22
In conclusion, ground state destabilization appears to play a major role in catalysis by PLP enzymes, and in controlling reaction specificity. The measurement of BIEs with catalytically inactive mutant enzymes may provide a general means for characterizing the extent of ground state destabilization in many enzymes. Lastly, the BIE observed here is a substantial fraction of the KIE on kcat/KM (~2),23 and must be taken into account in quantitative analyses of these effects.24
Supplementary Material
Acknowledgments
This work was supported by Grant GM54779 from the National Institutes of Health.
Footnotes
Supporting Information Supporting information (table of crystallographic data, and experimental procedures) is available via the web at http://pubs.acs.org/jacs.
References
- 1.Eliot AC, Kirsch JF. Annu Rev Biochem. 2004;73:383. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 2.Hayashi H. J Biochem. 1995;118:463. doi: 10.1093/oxfordjournals.jbchem.a124931. [DOI] [PubMed] [Google Scholar]
- 3.Christen PM, DE . Transaminases. J. Wiley & Sons; New York, NY: 1985. [Google Scholar]
- 4.Kuramitsu S, Hiromi K, Hayashi H, Morino Y, Kagamiyama H. Biochemistry. 1990;29:5469. doi: 10.1021/bi00475a010. [DOI] [PubMed] [Google Scholar]
- 5.Richards JPA, Crugeiras TL, Rios JA. Curr Opin Chem Biol. 2009;13:475. doi: 10.1016/j.cbpa.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernasconi CF. Adv Phys Org Chem. 2010;44:223. [Google Scholar]
- 7.Dunathan HC. Proc Natl Acad Sci U S A. 1966;55:712. doi: 10.1073/pnas.55.4.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okamoto A, Higuchi T, Hirotsu K, Kuramitsu S, Kagamiyama H. J Biochem. 1994;116:95. doi: 10.1093/oxfordjournals.jbchem.a124509. [DOI] [PubMed] [Google Scholar]
- 9.Stamper GF, Morollo AA, Ringe D. Biochemistry. 1999;38:6714. doi: 10.1021/bi995075y. [DOI] [PubMed] [Google Scholar]
- 10.Anderson VE. Arch Biochem Biophys. 2005;433:27. doi: 10.1016/j.abb.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 11.Schramm VL. Curr Opin Chem Biol. 2007;11:529. doi: 10.1016/j.cbpa.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schramm VL. Annu Rev Biochem. 2011;80:703. doi: 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LaReau RD, Wan W, Anderson VE. Biochemistry. 1989;28:3619. doi: 10.1021/bi00434a070. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Schramm VL. Biochemistry. 2011;50:4813. doi: 10.1021/bi200638x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spies MA, Toney MD. J Am Chem Soc. 2007;129:10678. doi: 10.1021/ja067643k. [DOI] [PubMed] [Google Scholar]
- 16.Toney MD, Kirsch JF. Biochemistry. 1993;32:1471. doi: 10.1021/bi00057a010. [DOI] [PubMed] [Google Scholar]
- 17.Toney MD, Kirsch JF. Protein Sci. 1992;1:107. doi: 10.1002/pro.5560010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mautner M. J Am Chem Soc. 1987;109:7947. [Google Scholar]
- 19.Singh V, Luo M, Brown RL, Norris GE, Schramm VL. J Am Chem Soc. 2007;129:13831. doi: 10.1021/ja0754204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anisimov V, Paneth P. J Math Chem. 1999;26:75. [Google Scholar]
- 21.Griswold WR, Toney MD. Bioorg Med Chem Lett. 2010;20:1352. doi: 10.1016/j.bmcl.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaw JP, Petsko GA, Ringe D. Biochemistry. 1997;36:1329. doi: 10.1021/bi961856c. [DOI] [PubMed] [Google Scholar]
- 23.Gloss LM, Kirsch JF. Biochemistry. 1995;34:3999. doi: 10.1021/bi00012a018. [DOI] [PubMed] [Google Scholar]
- 24.Ruszczycky MW, Anderson VE. J Theor Biol. 2006;243:328. doi: 10.1016/j.jtbi.2006.06.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.