

Highlights
► rMV vector immunization primes cellular responses to an adenovirus vector heterologous boost. ► rMV priming is highly effective for suboptimal doses of the boosting vector. ► Recombinant MV retains robust measles-specific humoral and cellular immunogenicity.
Keywords: Measles virus, Adenovirus, Mucosal immunization, Heterologous prime-boost
Abstract
Licensed live attenuated virus vaccines capable of expressing transgenes from other pathogens have the potential to reduce the number of childhood immunizations by eliciting robust immunity to multiple pathogens simultaneously. Recombinant attenuated measles virus (rMV) derived from the Edmonston Zagreb vaccine strain was engineered to express simian immunodeficiency virus (SIV) Gag protein for the purpose of evaluating the immunogenicity of rMV as a vaccine vector in rhesus macaques. rMV-Gag immunization alone elicited robust measles-specific humoral and cellular responses, but failed to elicit transgene (Gag)-specific immune responses, following aerosol or intratracheal/intramuscular delivery. However, when administered as a priming vaccine to a heterologous boost with recombinant adenovirus serotype 5 expressing the same transgene, rMV-Gag significantly enhanced Gag-specific T lymphocyte responses following rAd5 immunization. Gag-specific humoral responses were not enhanced, however, which may be due to either the transgene or the vector. Cellular response priming by rMV against the transgene was highly effective even when using a suboptimal dose of rAd5 for the boost. These data demonstrate feasibility of using rMV as a priming component of heterologous prime-boost vaccine regimens for pathogens requiring strong cellular responses.
1. Introduction
Vaccination against measles was implemented in the WHO Expanded program of Immunization in 1974; since then millions of children have been safely immunized and protected from the disease [1], [2], [3], [4], [5], [6]. All currently registered vaccines are based on live attenuated measles viruses, generated by extensive propagation and attenuation in cell culture [4], [7], [8], [9], [10]. Although different measles vaccines differ in the way they were attenuated, they all share a durable efficacy and safety record [4]. The protective immunity against measles is efficiently induced with 1 to 2 measles vaccine immunizations [6], [11], which strongly stimulate both innate and adaptive immune responses. Measles-specific humoral responses are detectable for as long as 33 years after vaccination [4], possibly due to boosting by natural exposure.
Measles vaccines have an excellent safety record not only after injection but also after mucosal (aerosol) delivery. Mucosal delivery mimics natural infection, which is known to be highly immunogenic [12], [13]. A number of vaccine campaigns demonstrated superior immunogenicity of aerosol delivered Edmonston Zagreb (EZ) measles vaccine compared to the injected vaccine, either as a primary or a boosting immunization [14], [15], [16]. The safety of attenuated measles vaccine has been proven in numerous clinical trials and with post-marketing surveillances [3]. Good safety profile of attenuated measles vaccine is due to high stability of the viral genome that prevents reversion to highly virulent species. In addition, measles virus, which is a non-segmented single-stranded negative sense RNA virus, replicates exclusively in the cytoplasm of infected cells [17], [18]; thus the viral genome cannot integrate into the host chromosome. Due to straightforward manufacturing, the measles vaccine can be produced in large scale and at low cost. The safety record, immunogenicity and manufacturability make live attenuated measles virus an attractive candidate to develop as a recombinant vaccine vector.
In a recombinant measles vaccine (rMV) vector, an antigen from another pathogen is incorporated into the measles genome. During the replication and transcription of rMV in a cell, the transgene is expressed together with viral proteins and presented to the host immune system, inducing a transgene-specific immune response. Thus a multivalent vaccine vector would induce not only strong immunity and protection against measles but also against another pathogen. A number of different transgenes, including genes from human papilloma virus, SARS coronavirus, West Nile virus, and human and simian immunodeficiency viruses (HIV/SIV), have been stably incorporated into the recombinant measles genome, with demonstrated transgene protein expression [19], [20], [21], [22], [23], [24], [25]. In vivo studies with measles virus and recombinant measles vectors have traditionally been performed in immunocompromised (IFNα receptor −/−) mice transgenic for human CD46 receptor, a measles virus receptor [19], [22], [24], [26]. Immunogenicity studies in these animals demonstrated that recombinant measles vectors induce not only strong immune response against measles but also against the transgene [21], [22], [24], [25]. In immunocompetent mice, however, the immune response against the transgene is generally very low even though measles-specific responses are well induced (unpublished data), possibly reflecting the inability of measles vector to efficiently replicate in these animals. Our initial immunogenicity study performed in non-human primates using rMV vector based on the licensed EZ vaccine strain as the backbone and expressing SIV gag (rMV-Gag) failed to demonstrate transgene specific responses while animals developed measles specific immune responses (data not shown) [23]. We assume that this is due to the highly attenuated nature of the EZ strain, which is one of the safest measles vaccines currently in use [4], [8], and would argue against the use of recombinant measles vector as a stand-alone vaccine against other pathogens.
In the current study, we evaluated whether recombinant measles vector could enhance immune response when combined with a heterologous Ad5-based vaccine vector. Heterologous prime-boost immunization regimens are highly promising vaccination approaches and are currently being evaluated for different diseases, including proven difficult pathogens such as malaria parasites, Mycobacterium tuberculosis and HIV [27]. Heterologous prime-boost regimens may improve not only the magnitude but also the quality of the immune response, by increasing epitope coverage and functionality of the response, due to the different mechanisms of uptake and processing of different vaccine vectors in vivo [28], [29].
Experimental intratracheal (IT) and aerosol (AE) deliveries have been used successfully in several animal experiments. The advantage of IT inoculation is that it ensures delivery of the complete viral dose to the lungs. However, attenuated measles Edmonston tag strain inoculated IT did not replicate in the upper respiratory tract, while it did following AE inhalation [30]. Since the EZ vaccine strain may behave differently, we pursued both mucosal delivery routes to increase the possibility of viral replication at the upper and lower respiratory tract and to efficiently elicit a mucosal immune response against the viral vector and the inserted transgene.
We report the humoral and cellular immunogenicity of recombinant live attenuated measles virus (rMV) expressing SIVgag when used primarily as a mucosal vaccine vector in rhesus macaques. rMV failed to elicit robust immune responses against the recombinant insert, despite inducing robust responses to the measles virus. However, when administered as a priming immunization for recombinant adenovirus serotype 5 (rAd5) expressing the same immunogen, rMV significantly enhanced SIVgag specific T-cell responses, in particular CD8+ T cells. This was true even for an otherwise inert, suboptimal dose of rAd5 used as a boosting vaccine. These findings reveal a potential role for rMV as a priming component in heterologous prime-boost vaccine regimens against other pathogens while simultaneously immunizing against measles.
2. Results
2.1. Experimental schema
In vitro replication of rMV encoding SIV gag was similar to the parent EZ vaccine strain, transgene expression was confirmed by western blot, and transgene immunogenicity was observed in immunocompetent CD46-transgenic mice but not rhesus macaques (data not shown). To determine the immunogenicity of rMV as a heterologous prime-boost vaccine vector component, we immunized rhesus macaques with rMV-Gag as either a prime or boost to rAd5 using a highly immunogenic AE delivery platform (Fig. 1A, Study A) [31]. Two doses were compared: either 5 × 104 pfu (approximately a single human dose, 1×) or twenty-fold greater, 106 pfu (20×), administered twice as a prime to a single 1010 PU rAd5 AE immunization or the 20× dose administered twice as a boost to rAd5 priming. All immunizations were spaced 8 weeks apart.
Fig. 1.

Experimental schema for the rMV immunization regimens. (A) In Study A, rhesus macaques were immunized with 5 × 104 pfu (1× dose; group 1, n = 3) or 106 pfu (20× dose; group 2, n = 3) of rMV-Gag twice by aerosol as a prime to rAd5-Gag, at 1010 PU, also by aerosol. A third group (n = 3) received a single rAd5 prime followed by two boosting immunizations with the 20× rMV-Gag, all by aerosol. Immunizations were administered eight weeks apart. (B) In Study B, animals received either rMV-null (n = 6) or rMV-Gag (n = 16) delivered twice: intratracheally (IT) at week 0 and intramuscularly (IM) at week 12. Half of animals received the 1× dose and half the 20×. A heterologous boost of 107 PU of rAd5-Gag was administered IM to all animals 20 weeks after the second rMV.
In a separate experiment, to further study the ability of rMV to prime for a systemic rAd5 boost, a mucosal IT and IM priming regimen was subsequently boosted with a suboptimal dose (107 PU) of rAd5 (Fig. 1B, Study B). The same two doses of rMV used in Study A were also compared in Study B, and animals received either rMV-Gag or rMV without insert (rMV-null).
2.2. Immunogenicity of aerosolized rMV
To assess the immunogenicity of rMV vector alone and as a vaccine prime or boost, we first employed a highly immunogenic aerosol vaccine delivery system (Study A). We previously observed T cell responses to rAd5-encoded immunogens ranging from 10 to 60% of CD4 and CD8 subsets in the BAL following aerosol vaccination [31]. Since five of the nine Study A animals were MV seropositive at the start (presumably due to natural MV exposure), these animals were divided into the rAd5 prime group (n = 3) and one into each of the 1× and 20× rMV prime groups.
Following aerosol rMV-Gag immunization, all naïve animals mounted significant serum IgG responses to the rMV vector, regardless of the rMV dose (Fig. 2A). Of note, the vaccine-induced responses were similar in magnitude to the IgG levels in the five animals that were seropositive at the study start, indicating a robust humoral response to the vaccine. In addition, CD4+ T cells specific for the MV N protein were also detected in the BAL of all the animals four weeks after the homologous rMV boost except for one of the MV seropositive animals, ranging from 1 to 16% of the subset, as measured by intracellular cytokine staining following ex vivo peptide stimulation (ICS, Fig. 2B). There was no significant difference between the 1× and 20× groups with respect to the magnitude of the T cell response. Moreover, one of the animals with preexisting MV titers had a vigorous N-specific CD4+ T cell response (7%), presumably elicited by the vaccine since MV-seropositive animals that did not receive rMV (rAd5 prime group) were all <1%. These data are consistent with previous observations that aerosolized vaccine vectors are resistant to neutralization by preexisting serum antibodies to the vector [31]. Low-frequency CD8+ T cell responses (<1%) were also observed for half of the animals. Thus aerosol delivery of rMV alone elicited robust systemic IgG and local mucosal T cell responses to the MV vector.
Fig. 2.
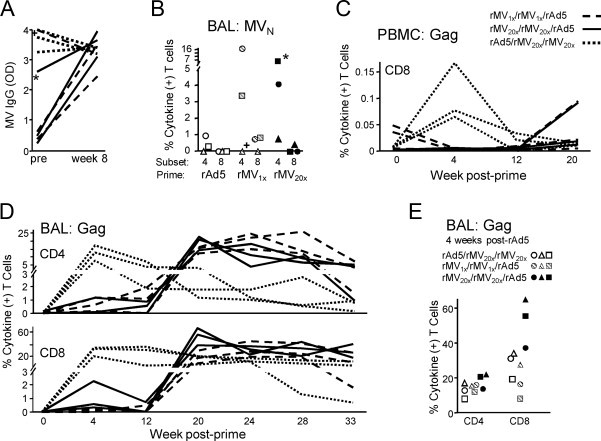
Immunogenicity of aerosolized rMV alone and as a prime or boost for rAd5. (A) Serum IgG responses to MV were measured by ELISA and are reported in optical density units (OD). Pre-immune and week 8 responses are shown for each animal with lines coded according to vaccine group assignment. The two MV-seropositive animals assigned to rMV-Gag prime groups are indicated (+, 1×; *, 20×). Line patterns correspond to groups as shown in the legend at far right. (B) BAL MV N-specific CD4+ and CD8+ T cell responses were measured by in vitro peptide pool stimulation and ICS for IFNγ, IL-2, and TNF four weeks after the second immunization. The total percent of each subset making any combination of these cytokines is plotted for each animal, depicted by a unique symbol. (C) PBMC CD8+ T cell Gag-specific responses were measured as in (B) and shown over time for each animal. (D) BAL Gag-specific T cell responses were measured as in (B). (E) BAL Gag-specific T cell responses from (D) are shown for all animals at four weeks after rAd5 immunization. Symbols correspond to the same animals in (B).
To assess the immune response to the SIV Gag immunogen, we measured serum IgG and both blood and BAL T cell responses. Significant Gag-specific IgG was not observed at any time point during the study, including after rAd5 immunization (data not shown). This was surprising since a single aerosol immunization of rAd5 encoding an EnvHIV transgene at the same dose was previously shown to elicit serum IgG responses [31], but may be due to greater immunogenicity of Env relative to Gag. Gag-specific blood T cell responses to the rMV prime or boost were also undetectable (Fig. 2C and data not shown). By contrast, rAd5 induced demonstrable but not statistically significant CD8+ T cell responses 4 weeks post-prime (Fig. 2C, p = 0.09), which were transient and therefore consistent with no boosting by 20× rMV [31].
Since aerosol immunization typically results in exceptional T cell responses in BAL, we considered BAL to be the most sensitive site for detecting responses. We measured Gag-specific responses by ICS at weeks 4 and 12, following each of the rMV priming immunizations. Four of six animals primed with rMV had small but notable T cell responses in the BAL 4 weeks after the first prime: two from each of the 1× and 20× groups (Fig. 2D). These responses ranged from 0.6 to 2.3% of the CD4+ or CD8+ T cell subsets and declined after the second rMV immunization for all but one animal. By contrast, the rAd5 prime elicited robust and durable Gag-specific BAL T cell responses in all animals: peaking at 8–17% of CD4+ and 20–35% of CD8+ T cells four weeks after immunization. There was no evidence that rMV administered 8 and 16 weeks after rAd5 boosted these responses; however, without matched rAd5 only controls we cannot exclude this or the possibility of enhanced durability. We did observe a trend in which 20× rMV primed for a greater magnitude CD8+ T cell response 4 weeks following rAd5 immunization, compared to unprimed rAd5 (Fig. 2D and E, p = 0.07). Thus aerosolized rMV alone is a weak vector platform, but it has the potential to enhance responses elicited by a subsequent rAd5 boosting immunization.
2.3. Humoral immunogenicity of suboptimal rAd5 dose primed by rMV
To elaborate on the ability of rMV to prime an rAd5 immunization seen in Study A, we immunized a second group of rhesus macaques with rMV followed by a low dose of rAd5 that ordinarily would not elicit robust responses (Study B, Fig. 1B). Using the same doses as in Study A, rMV was administered IT and then IM, to first assess rMV mucosal priming of a homologous systemic boost. The IM rAd5 boost was given 20 weeks after the second rMV (IM) immunization (week 32).
Serum IgG was elicited to the measles virus vector within two weeks of IT delivery in Study B (Fig. 3A), indicating successful vaccine take. Responses to MV were similar for both the null and SIV gag-encoding rMV, ranging from 102 to 103 U/ml at both week two and week four, with no apparent effect of dose. Titers increased slightly by week eight and peaked two weeks after the IM rMV boost (week 14). These titers were sufficient to mediate MV neutralization (Fig. 3B).
Fig. 3.

Humoral immunogenicity of intratracheal and intramuscular rMV primes, suboptimal rAd5 boost. (A) Serum IgG antibodies to MV lysate are plotted for each animal grouped by vaccine regimen at the indicated weeks after the first rMV immunization in Study B. The ELISA results are shown as units (U) per ml, with 1 U/ml roughly equal to 1 ng/ml, as described in Materials and Methods. (B) MV 50% neutralization titers are shown for each animal as in (A) with rMV-null and -Gag groups combined by rMV dose. Protective titer of 120 mIU/ml is indicated.
By contrast, most animals did not mount a systemic IgG response to the Gag insert following rMV immunization, as measured by ELISA (data not shown). Gag-specific B cells were likely elicited by rMV in two animals, one in each of the 1× (396) and 20× (339) groups, as these were the only animals that responded to the rAd5 boost. IgG titers in animal 339 increased 6.2-fold from pre-immune levels at week 14, and then 6.3-fold from week 32 to week 34. Animal 396 underwent a 10.3-fold increase in titer from week 0 to 34, with undetectable responses to rMV alone. Mucosal IgA responses were largely undetectable, with no significant Gag-specific responses in the bronchoalveolar lavage (BAL), saliva, or rectum (data not shown). Together, these data demonstrate robust immunogenicity elicited by rMV-Gag to the measles virus vector but weak humoral responses to the SIV Gag insert, consistent with AE delivery.
2.4. Cellular immunogenicity of suboptimal rAd5 dose primed by rMV
To determine if rMV primed cellular responses to rAd5 as suggested in Study A, we measured T cell responses by ICS and ELISpot following each rMV prime and the limiting dose rAd5 boost. The IT rMV immunization failed to induce robust T cell responses in peripheral blood at any time point up to week 12, with no significant difference from pre-immune levels for either the 1× or 20× vaccine group (Fig. 4A and data not shown). Low-level responses were observed for some animals within each group by ELISpot, but these generally did not exceed 250 antigen-specific cells per million cells. Notably, two of the animals that mounted a significant serum Gag IgG response following rMV, 339 and 399, also had modest but durable T cell responses (data not shown). Similarly, Gag ELISpot responses after the homologous IM rMV boost were low, with evidence of a boost only in the 20× group at week 16 (p = 0.02, relative to week 12). Following the rAd5 boost, however, significant PBMC T cell responses were detected in both the 1× and 20× rMV-Gag primed groups at both two (week 34) and four (week 36) weeks after rAd5 (p = 0.002 and p = 0.0008, respectively, versus week 32, Fig. 4A). By contrast, T cell responses to this dose of rAd5 were undetectable without priming (MV-null).
Fig. 4.

Cellular immunogenicity of intratracheal and intramuscular rMV primes, suboptimal rAd5 boost. Rhesus macaques primed with rMV-null, rMV-Gag1×, or rMV-Gag20× in Study A were boosted with 107 PU rAd5 IM at week 32. Bars depict the interquartile range for each group; (#) and (+) indicate significant difference (p<0.05) from the specified time point within each group by Wilcoxon rank-sum and student's t-test, respectively. (A) Gag-specific PBMC T cell responses measured by ELISpot. Statistical comparison to week 32 pre-rAd5 values is indicated. (B) PBMC CD4+ (left) and CD8+ (right) T cell responses to SIV Gag measured by ICS as in Fig. 2C before (week 14) and after (week 36) the rAd5 boost. Statistical comparison to week 14 pre-rAd5 values is indicated. (C) Gag-specific T cell responses in BAL measured by ICS as in (B) at week 36, 4 weeks after the rAd5 boost, for CD4+ (left) and CD8+ (right) subsets. Not all animals are represented as insufficient BAL T cell subset populations were detected.
ELISpot PBMC responses were corroborated by ICS on individual T cell subsets. As expected, 20× rMV-Gag immunizations primed CD8+ T cell responses for rAd5 (p = 0.01 relative to pre-rAd5, Fig. 4B). The 1× prime also trended towards significance (p = 0.06), with elevated CD8+ T cell responses in half of the animals. By contrast, PBMC CD4+ T cell responses were not significantly primed by rMV, as measured by ICS. Large Gag-specific responses were also measured in BAL following the rAd5 boost for some animals primed with rMV-Gag, particularly within the CD8+ T cell compartment (Fig. 4C). Combined, these data demonstrate a distinct ability of rMV to prime T cell responses.
3. Discussion
In this study, we assessed the immunogenicity of recombinant live attenuated Edmonston Zagreb (EZ) measles strain expressing SIVgag transgene (rMV-Gag) as a priming vaccine vector in a heterologous prime-boost regimen with rAd5. When delivered either by aerosol or a combination of intratracheal and intramuscular routes, rMV-Gag increased the magnitude of Gag-specific peripheral blood and BAL T cell responses following the rAd5-Gag boost, particularly among CD8+ T cells. By contrast, humoral responses to the transgene were not significantly enhanced by rMV, despite elicitation of strong functional antibodies against the MV vector. rMV on its own elicited robust measles-specific immunity but failed to induce transgene-specific responses. This finding is consistent with results from initial tests of the vector in rhesus macaques, in which rMV-Gag was administered intramuscularly as a homologous prime-boost (data not shown). rMV derived from the EZ strain is thus a potent vaccine against measles but not an immunogenic vector against a transgene unless combined with another vector, such as adenovirus, in a heterologous prime-boost regimen. In this context, rMV can prime for strong cellular responses against the transgene even with suboptimal boosting.
The reason transgene immunogenicity was weak when rMV was administered by itself, despite robust measles-specific responses, is probably due to limited replication of the attenuated vaccine strain. Replication of measles virus, a single-stranded negative-sense RNA virus, is necessary for high-level transgene protein synthesis. While measles proteins are present within the vaccine and prime the immune response for a boost with small quantities of measles protein produced upon low-level viral replication, high-level transgene protein availability and immune response elicitation is dependent on the extent of vector replication. The EZ measles strain used for generation of the rMV-gag vector is highly attenuated, as demonstrated by its superb safety in infants, and may thus explain the poor immunogenicity of rMV-Gag for the transgene when used as a stand-alone vaccine. Although we confirmed the Gag protein expression in vitro by western blot (data not shown), it is possible that the amount of Gag protein produced by the measles vector is insufficient to elicit detectable immune response but adequate to prime T cells for a subsequent boost. We also cannot exclude potential defects in antigen processing and presentation of the rMV-encoded transgene, but why this would occur for the transgene but not for MV proteins is not evident.
Improved transgene expression from this attenuated measles virus vector can be achieved by inserting the transgene upstream of the N gene (so-called position 1) as there is a transcript expression gradient from N to L with N being expressed highest [23], [24]. However, this approach might negatively impact vector genetic stability and replication kinetics [23]. Alternatively, strategies might be developed in which the transgene protein is packaged as an integral component of the measles virion possibly allowing the induction of potent transgene-specific humoral responses.
T cell priming was observed in both of our non-human primate studies, although the effect was more pronounced in Study B compared to Study A. This difference may be due to several factors. First, rMV was administered exclusively in the airway mucosa in Study A, whereas one of the rMV primes in Study B was administered IM. A systemic rMV prime may enhance blood cellular responses to a greater extent than AE delivery. In addition, rAd5 was administered by different routes. Aerosol rAd5 delivery (Study A) is less immunogenic for peripheral blood T cell responses than IM delivery (Study B) [32], limiting the magnitude of the PBMC response to rAd5 in Study A. Furthermore, different doses of rAd5 were used in the studies, although it is not clear why a suboptimal dose of rAd5 (107 PU; Study B) would induce a larger response than an optimal dose (1010 PU; Study A) primed by the same dose of rMV. Finally, the interval between the rMV prime and the rAd5 boost was much greater in Study B (20 weeks) than in Study A (8 weeks). This may have allowed more time for rMV-Gag replication, transgene synthesis, and immune presentation. Although we cannot conclude at this time which factor is crucial for the observed differences, the fact that we observe T cell priming by rMV in two independent and different heterologous prime-boost studies underscores the robustness of this mechanism. We hypothesize that the priming ability of rMV is not limited to boosting with rAd5. Likely any other live viral vector would be a suitable boosting vaccine for rMV in a heterologous prime-boost regimen.
A significant Gag-specific antibody response was not observed at any time point during the study, suggesting weak priming of CD4+ T cells and B cell help as compared to CD8+ T cells. However, Gag-specific antibodies were not induced after rAd5 immunization either (data not shown). Lack of Gag antibody responses might be due to relatively poor immunogenicity of Gag, since we previously demonstrated that a single aerosol immunization with rAd5 encoding an EnvHIV transgene at the same dose elicits serum IgG responses [31]. In another study, a recombinant version of Edmonston B MV induced robust EnvHIV transgene- and MV-specific antibody responses in non-human primates [22], suggesting Env is more immunogenic than Gag. However, we cannot exclude the possibility that these transgene-specific responses were due to higher replication (i.e. less attenuation) of the measles strain employed relative to our rMV.
Our main finding that rMV efficiently primes CD8+ T cells in a heterologous prime-boost regimen with a suboptimal boosting dose of an adenovirus vector has important implications for vaccine design. In general, heterologous prime-boost regimens are appealing for their potential to increase and broaden responses as well as to evade neutralizing antibodies against the priming vector. Employing rMV as a vaccine vector achieves sterilizing immunity against measles in addition to priming responses against another pathogen in a single immunization, which would help to reduce the number of immunizations given to children. Moreover, the specific regimen tested here offers an additional advantage of dose sparing for the boosting vaccine and, as such, increasing available dosages and decreasing costs. Future studies should examine rMV priming in combination with other boosting vaccine vectors and delivery via different routes to further enhance transgene immunogenicity, particularly for humoral responses. In addition, incorporation of other, possibly more immunogenic transgenes will be important to assess the full potential of rMV to elicit transgene-specific immune responses. Evaluation of immunogenicity in MV naïve and seropositive animals, followed by protection studies in an established animal challenge model, should ultimately prove the suitability of the technology and justify initiation of human clinical trials.
4. Materials and methods
4.1. Plasmids and viruses
To create rMV vectors expressing foreign proteins, cDNA corresponding to the antigenome of Edmonston Zagreb vaccine strain was cloned with additional transcription units to insert exogenous genes encoding foreign antigens into the viral genome [24]. In this study two measles vectors were used, rMV empty (rMVEZ-null, or rMV-null) and rMV containing SIVmac239 gag gene at position 2, between P and M genes (rMVEZb2.SIVgag, or rMV-Gag). Viruses were rescued as previously described [33]. Vaccine batches were prepared on MRC-5 cells as described with few modifications [13]. In brief, recombinant measles vectors were grown in MRC-5 cells in roller bottles at 35 °C/5%CO2 and viruses were harvested at several time points post infection. Viral titers were determined by standard plaque assay on Vero cells. Presence of the transgene was confirmed by RT-PCR and sequencing and protein expression was confirmed by western blotting (anti-SIV Gag p27 antibody 2F12, NIH AIDS Research & Reference Reagent Program). Recombinant E1/E3/E4-deleted rAd5 construct expressing GagPolSIV and virus stocks (rAd5-Gag) were generated as previously described [34], [35].
4.2. Animals and immunizations
Colony-bred Indian-origin rhesus macaques were immunized as described in the Results section. Briefly, rMV was administered at either a single human dose (5 × 104 pfu, 1×) or 106 pfu (20×), and rAd5-Gag at either 1010 PU (Study A) or 107 PU (Study B). In Study A, five animals were seropositive at study start and assigned one per rMV-Gag prime group (1× and 20×, n = 3 each) and three into the rAd5 prime group (n = 3). Aerosol immunizations were delivered in 1.0 ml by the e-Flow® Nebulizer System (PARI Pharma, Germany) [31]. In Study B, standard IT and IM immunizations were conducted using 1.0 ml of vaccine. The rMV-null groups (1× and 20×, n = 3 each) and rMV-Gag groups (1× and 20×, n = 8 each) were all measles naïve. Study A animals were housed at Bioqual, Inc., Rockville, MD; Study B at New England Primate Research Center of Harvard Medical School, Southborough, MA. All animals were maintained in accordance with National Institutes of Health and Harvard Medical School guidelines.
4.3. Adenovirus neutralization assay
Neutralizing antibodies against Adenovirus serotype 5 were detected by luciferase transgene expression inhibition as previously described [36]. Briefly, heat inactivated serum samples were twofold serially diluted in medium (Dulbecco's modified Eagle's medium containing 10% FBS) and in duplicate. Serum dilutions ranged from 1/16 to 1/32,768 in an end volume of 50 μl of medium in a 96-well plate. Fifty μl of a fixed amount of Adenovirus (Ad5 Adapt Luc, 1 × 108 PU/ml) was added to each serum dilution and incubated for 30 min at room temperature. Then 104 A549 cells in 100 μl were added to every well and plates were incubated for 24 h at 37 °C and 10% CO2. The medium was discarded, 50 μl of phosphate-buffered saline (PBS) was added and one freeze-thaw cycle was made. Next, 50 μl of Steady-Lite luciferase assay system reagent (PerkinElmer) was added to every well and incubated for 15 min at room temperature. Fifty μl from each well was transferred to a black and white isoplate and luminescence counts were measured on a 1450 MicroBeta Trilux. As negative control, no serum was added resulting in the maximum luciferase activity. The minimum luciferase activity was obtained from wells where no virus was added. As positive control, serum from immunized mice was used. The 90% neutralizing titers were calculated by a non-linear curve fit through the sample data using the minimum luciferase activity control (no virus) as baseline (0%), and the maximum luciferase activity control (no serum) as plateau (100%).
4.4. Measles virus antibody responses
Enzyme immunoassays were used to measure MV-specific IgG in Study A as previously described with some modifications [37]. Briefly, sera were diluted 1:100 and incubated overnight at 4 °C with MV-infected Vero cell lysate (1.1 μg/well; Advanced Biotechnologies) coating a Maxisorp 96-well plate (Nalge Nunc International). Plates were washed 4 times with PBS containing 0.05% Tween-20 (PBST). Alkaline phosphatase-conjugated rabbit anti-monkey IgG (BIOMAKOR; Accurate Chemicals) was added to each well (1:1500, 100 μl/well) and plates were incubated for 2 h at 37 °C followed by four washes in PBST. Plates were developed using the substrate para-nitrophenyl phosphate (SIGMAFAST, Sigma) and the absorbance was read at 405 nm (SoftMax Pro Software v3.1.1, Molecular Devices) with the average of the three samples reported as optical density. Three negative controls using plasma from naïve monkeys were included in this assay (negative if average optical density ≤0.565).
In Study B, anti-MV IgG antibodies were measured using Fisherbrand high protein-binding microtiter plates coated for 5 h at room temperature with 60 ng/well of beta-propiolactone-inactivated measles virus (Edmonston strain ATCC VR-24; Virion-Serion, Wurzburg, Germany) in 0.05 M carbonate buffer, pH 9.4. Plates were washed with PBST, then blocked for 30 min with 2% Goat Serum (GS) in PBST. Pooled serum from 3 MV-immunized macaques was arbitrarily assigned 1000 units/ml of anti-MV IgG antibody and used as a standard. Individual serum samples were tested at eight serial 3-fold dilutions using a starting dilution of 1/100. After an overnight reaction at 4˚C, the plates were washed with PBST and treated with 20 ng/well biotinylated goat anti-human IgG (SouthernBiotech, Birmingham, AL) for 1 h at 37˚C. Plates were washed, then reacted for 30 min at room temperature with 50 ng/well of streptavidin-peroxidase (Sigma). Final development was with tetramethylbenzidine (SouthernBiotech) for 30 min and 2 N sulfuric acid stop solution. Absorbance was recorded at 450 nm in a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA). Serum was considered positive for IgG antibody if the post-immune level was 3.4-fold greater than the pre-immune level.
Plaque reduction neutralization assay was used to measure neutralizing antibody titer following immunization [38]. The monkey serum samples were two-fold serially diluted from 1:4 to 1:16,384 and standardized to the 3rd WHO international anti-measles standard serum (1.0 IU/ml, NIBSC code 97/648, NIBSC, Potters Bar Hertfodshire EN6 3QG, UK) serially diluted 1:8 to 1:256. Two hundred μl of each serum dilution was incubated with 200 μl of a fixed amount of rMVb2EZ (approximately 70 pfu/ml) for 1 h at 37 °C. Vero cells seeded in 6-well plates were infected with 200 μl of the serum-MV mix in a humid chamber for 1 h at 25 °C then overlaid with a semi-solid medium (MEM and 1.2% Methocel) and incubated for 6 days at 35 °C and 5% CO2. Cells were fixed and stained (7.4% formaldehyde and 0.4 g/L crystal violet in PBS pH 7.4) and plaques counted. The 50% neutralizing end-point titers were calculated using the Spearman and Kärber method. One hundred % neutralization was defined as no plaques and 0% as the geometric mean plaque count of the no serum negative control. Normalized titer was calculated as the ratio between the estimated 50% neutralizing end-point and the WHO standard multiplied by the WHO titer (1 IU/ml): 10(log sample–log WHO) × 1 IU/ml. Fifty % neutralizing end-point values less 4 (i.e. first dilution) were considered to be negative. Titers ≥120 mIU/ml were considered protective [39].
4.5. SIV Gag antibody measurements
Pre-immune and post-immunization serum, BAL and rectal sponge eluates were analyzed for humoral responses by ELISA as previously described [40]. Rectal secretions were sampled by a modified wick method using Weck-Cel Spears (Windsor Biomedical, Newton, NH) as previously described [41]. SIV-specific IgA and IgG antibodies to Gag were measured using microtiter plates coated respectively with SIVmac251 viral lysate (125 ng total protein/well, Advanced Biotechnologies Inc, Columbia, MD). Total IgA or IgG was measured using plates coated with goat anti-monkey IgA (Rockland, Gilbertsville, PA) or IgG (MP BioMedicals, Solon, OH). Pooled macaque serum containing previously calibrated amounts of the relevant antibody or immunoglobulin was used for the standards. Plates were developed with biotinylated goat anti-monkey IgA (25 ng/ml, OpenBiosystems, Huntsville, AL) or anti-human IgG as described above. For secretions, the concentration of antigen-specific IgA was divided by the concentration of total IgA in each sample to obtain the specific activity. Results were considered to contain significant antibody if the specific activity was ≥mean + 3 standard deviations of negative controls and 3.4-fold above the pre-immune specific activity.
4.6. T cell intracellular cytokine staining
Peripheral blood and BAL were collected from animals following immunization. Single cell suspensions were stimulated with overlapping peptide pools of MV N-protein or SIV Gag at 2.0 μg/ml for 16 h. Following stimulation, cells were labeled with cell surface markers (CD4-Alexa700APC and CD8-QDot655; unconjugated monoclonal antibodies from Becton Dickenson; conjugations in house) and ViViD (LIVE/DEAD® stain, Invitrogen), then fixed and permeabilized (BD Cytofix/Cytoperm, Becton Dickenson) for intracellular cytokine staining with IFNγ-FITC, TNFα-Cy7PE, IL-2-PE, and CD3-Cy7APC (Becton Dickenson). Background from co-stimulation alone (anti-CD28 and anti-CD49d) was subtracted to determine antigen-specific responses.
4.7. T cell ELISpot measurements
Multiscreen ninety-six well plates were coated overnight with 100 μl per well of 5 μg/ml anti-human interferon-γ (IFN-γ) (B27; BD Pharmingen) in endotoxin-free Dulbecco's-PBS (D-PBS). The plates were then washed three times with D-PBS containing 0.1% Tween-20, blocked for 1–4 h with RPMI containing 10% FBS to remove the Tween-20, and incubated with peptide pools at 1 μg/ml and 2 × 105 PBMCs in triplicate in 100 μl reaction volumes. Following an 18 h incubation at 37 °C, the plates were washed nine times with D-PBS containing 0.1% Tween-20 and once with distilled water. The plates were then incubated with 2 μg/ml biotinylated rabbit anti-human IFN-γ (Biosource) for 2 h at room temperature, washed six times with D-PBS containing 0.1% Tween-20 and incubated for 2.5 h with a 1:500 dilution of streptavidin-AP (Southern Biotechnology). After five washes with D-PBS containing 0.1% Tween-20 and one with D-PBS, the plates were developed with NBT/BCIP chromogen (Pierce), stopped by washing with tap water, air dried, and read with an ELISpot reader using Immunospot software (version 5.0) (Cellular Technology Ltd., Shaker Heights, OH).
Acknowledgments
We thank B. Lau and Dr. Diane Griffin of the Johns Hopkins School of Public Health for performing MV antibody assays. We thank J.P. Todd, A. Ault, A. Carville (NEPRC) for assisting with animal immunizations, challenges, and sampling scheduling as well as T. Jenkins and A. Dodson of Bioqual, Inc. for animal care and handling. We thank the flow cytometry facility of the Division of Viral Pathogenesis, BIDMC. Adenovirus viral stocks were kindly provided by J. Gall of GenVec, Inc.
Funding: This work was supported in part by NIH grant AI058896 and the Louisiana Vaccine Center and the South Louisiana Institute for Infectious Disease Research sponsored by the Louisiana Board of Regents, by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, NIH, by Harvard University CFAR and CAVD, and by the NIH (NIAID) contract NO1AI60018.
References
- 1.Global routine vaccination coverage, 2009 MMWR Morb Mortal Wkly Rep. 2010;59(October (42)):1367–1371. [PubMed] [Google Scholar]
- 2.Ten great public health achievements – United States, 1900–1999 MMWR Morb Mortal Wkly Rep. 1999;48(April (12)):241–243. [PubMed] [Google Scholar]
- 3.Global reductions in measles mortality 2000–2008 and the risk of measles resurgence Wkly Epidemiol Rec. 2009;84(December (49)):509–516. [PubMed] [Google Scholar]
- 4.Measles vaccines: WHO position paper Wkly Epidemiol Rec. 2009;84(August (35)):349–360. [PubMed] [Google Scholar]
- 5.Clemens J., Holmgren J., Kaufmann S.H., Mantovani A. Ten years of the global alliance for vaccines and immunization: challenges and progress. Nat Immunol. 2010;11(December (12)):1069–1072. doi: 10.1038/ni1210-1069. [DOI] [PubMed] [Google Scholar]
- 6.Hilleman M.R. Current overview of the pathogenesis and prophylaxis of measles with focus on practical implications. Vaccine. 2001;20(December (5–6)):651–665. doi: 10.1016/s0264-410x(01)00384-x. [DOI] [PubMed] [Google Scholar]
- 7.Enders J.F., Katz S.L., Milovanovic M.V., Holloway A. Studies on an attenuated measles-virus vaccine. I. Development and preparations of the vaccine: techniques for assay of effects of vaccination. N Engl J Med. 1960;263(July):153–159. doi: 10.1056/NEJM196007282630401. [DOI] [PubMed] [Google Scholar]
- 8.Ikic D., Juzbasic M., Beck M., Hrabar A., Cimbur-Schreiber T. Attenuation and characterisation of Edmonston–Zagreb measles virus. Ann Immunol Hung. 1972;16:175–181. [PubMed] [Google Scholar]
- 9.Schwarz A.J. Immunization against measles: development and evaluation of a highly attenuated live measles vaccine. Ann Paediatr. 1964;202:241–252. [PubMed] [Google Scholar]
- 10.Tillieux S.L., Halsey W.S., Sathe G.M., Vassilev V. Comparative analysis of the complete nucleotide sequences of measles, mumps, and rubella strain genomes contained in Priorix-Tetra and ProQuad live attenuated combined vaccines. Vaccine. 2009;27(April (16)):2265–2273. doi: 10.1016/j.vaccine.2009.01.112. [DOI] [PubMed] [Google Scholar]
- 11.Griffin D.E. Measles virus. In: Knipe D.H.P., editor. Field's Virology. 4th ed. Lippincott-Raven; Philadelphia, PA: 2001. pp. 1401–1441. [Google Scholar]
- 12.Hiremath G.S., Omer S.B. A meta-analysis of studies comparing the respiratory route with the subcutaneous route of measles vaccine administration. Hum Vaccin. 2005;1(January–Februay (1)):30–36. doi: 10.4161/hv.1.1.1423. [DOI] [PubMed] [Google Scholar]
- 13.Liniger M., Zuniga A., Morin T.N., Combardiere B., Marty R., Wiegand M. Recombinant measles viruses expressing single or multiple antigens of human immunodeficiency virus (HIV-1) induce cellular and humoral immune responses. Vaccine. 2009;27(May (25–26)):3299–3305. doi: 10.1016/j.vaccine.2009.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennett J.V., Fernandez de Castro J., Valdespino-Gomez J.L., Garcia-Garcia Mde L., Islas-Romero R., Echaniz-Aviles G. Aerosolized measles and measles-rubella vaccines induce better measles antibody booster responses than injected vaccines: randomized trials in Mexican schoolchildren. Bull World Health Organ. 2002;80(10):806–812. [PMC free article] [PubMed] [Google Scholar]
- 15.Dilraj A., Sukhoo R., Cutts F.T., Bennett J.V. Aerosol and subcutaneous measles vaccine: measles antibody responses 6 years after re-vaccination. Vaccine. 2007;25(May (21)):4170–4174. doi: 10.1016/j.vaccine.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Fernández-de Castro J., K-RJ, Sepúlveda J., Ramírez-Isunza J.M., Valdespino-Gómez J.L. Measles vaccination by the aerosol method in Mexico. Salud Publica Mex. 1997;39(January–February (1)):53–60. [PubMed] [Google Scholar]
- 17.Duprex W.P., McQuaid S., Hangartner L., Billeter M.A., Rima B.K. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. J Virol. 1999;73(November (11)):9568–9575. doi: 10.1128/jvi.73.11.9568-9575.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rima B.K., Duprex W.P. The measles virus replication cycle. Curr Top Microbiol Immunol. 2009;329:77–102. doi: 10.1007/978-3-540-70523-9_5. [DOI] [PubMed] [Google Scholar]
- 19.Cantarella G., Liniger M., Zuniga A., Schiller J.T., Billeter M., Naim H.Y. Recombinant measles virus-HPV vaccine candidates for prevention of cervical carcinoma. Vaccine. 2009;27(May (25–26)):3385–3390. doi: 10.1016/j.vaccine.2009.01.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Despres P., Combredet C., Frenkiel M.P., Lorin C., Brahic M., Tangy F. Live measles vaccine expressing the secreted form of the West Nile virus envelope glycoprotein protects against West Nile virus encephalitis. J Infect Dis. 2005;191(January (2)):207–214. doi: 10.1086/426824. [DOI] [PubMed] [Google Scholar]
- 21.Liniger M., Zuniga A., Tamin A., Azzouz-Morin T.N., Knuchel M., Marty R.R. Induction of neutralising antibodies and cellular immune responses against SARS coronavirus by recombinant measles viruses. Vaccine. 2008;26(April (17)):2164–2174. doi: 10.1016/j.vaccine.2008.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lorin C., Mollet L., Delebecque F., Combredet C., Hurtrel B., Charneau P. A single injection of recombinant measles virus vaccines expressing human immunodeficiency virus (HIV) type 1 clade B envelope glycoproteins induces neutralizing antibodies and cellular immune responses to HIV. J Virol. 2004;78(January (1)):146–157. doi: 10.1128/JVI.78.1.146-157.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tangy F., Naim H.Y. Live attenuated measles vaccine as a potential multivalent pediatric vaccination vector. Viral Immunol. 2005;18(2):317–326. doi: 10.1089/vim.2005.18.317. [DOI] [PubMed] [Google Scholar]
- 24.Zuniga A., Wang Z., Liniger M., Hangartner L., Caballero M., Pavlovic J. Attenuated measles virus as a vaccine vector. Vaccine. 2007;25(April (16)):2974–2983. doi: 10.1016/j.vaccine.2007.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brandler S., Ruffie C., Najburg V., Frenkiel M.P., Bedouelle H., Despres P. Pediatric measles vaccine expressing a dengue tetravalent antigen elicits neutralizing antibodies against all four dengue viruses. Vaccine. 2010;28(September (41)):6730–6739. doi: 10.1016/j.vaccine.2010.07.073. [DOI] [PubMed] [Google Scholar]
- 26.Mrkic B., Pavlovic J., Rulicke T., Volpe P., Buchholz C.J., Hourcade D. Measles virus spread and pathogenesis in genetically modified mice. J Virol. 1998;72(September (9)):7420–7427. doi: 10.1128/jvi.72.9.7420-7427.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radosevic K., Rodriguez A., Lemckert A., Goudsmit J. Heterologous prime-boost vaccinations for poverty-related diseases: advantages and future prospects. Expert Rev Vaccines. 2009;8(May (5)):577–592. doi: 10.1586/erv.09.14. [DOI] [PubMed] [Google Scholar]
- 28.Liu J., O‘Brien K.L., Lynch D.M., Simmons N.L., La Porte A., Riggs A.M. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature. 2009;457(January (7225)):87–91. doi: 10.1038/nature07469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seder R.A., Darrah P.A., Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8(April (4)):247–258. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- 30.de Vries R.D., Lemon K., Ludlow M., McQuaid S., Yuksel S., van Amerongen G. In vivo tropism of attenuated and pathogenic measles virus expressing green fluorescent protein in macaques. J Virol. 2010;84(May (9)):4714–4724. doi: 10.1128/JVI.02633-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song K., Bolton D.L., Wei C.J., Wilson R.L., Camp J.V., Bao S. Genetic immunization in the lung induces potent local and systemic immune responses. Proc Natl Acad Sci U S A. 2010;107(December (51)):22213–22218. doi: 10.1073/pnas.1015536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolton D.L., Song K., Wilson R.L., Kozlowski P.A., Tomaras G.D., Keele B.F. Comparison of systemic and mucosal vaccination: impact on intravenous and rectal SIV challenge. Mucosal Immunol. 2011;(October) doi: 10.1038/mi.2011.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radecke F., Spielhofer P., Schneider H., Kaelin K., Huber M., Dotsch C. Rescue of measles viruses from cloned DNA. EMBO J. 1995;14(December (23)):5773–5784. doi: 10.1002/j.1460-2075.1995.tb00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brough D.E., Lizonova A., Hsu C., Kulesa V.A., Kovesdi I. A gene transfer vector-cell line system for complete functional complementation of adenovirus early regions E1 and E4. J Virol. 1996;70(September (9)):6497–6501. doi: 10.1128/jvi.70.9.6497-6501.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gall J.G., Lizonova A., EttyReddy D., McVey D., Zuber M., Kovesdi I. Rescue and production of vaccine and therapeutic adenovirus vectors expressing inhibitory transgenes. Mol Biotechnol. 2007;35(March (3)):263–273. doi: 10.1007/BF02686012. [DOI] [PubMed] [Google Scholar]
- 36.Sprangers M.C., Lakhai W., Koudstaal W., Verhoeven M., Koel B.F., Vogels R. Quantifying adenovirus-neutralizing antibodies by luciferase transgene detection: addressing preexisting immunity to vaccine and gene therapy vectors. J Clin Microbiol. 2003;41(November (11)):5046–5052. doi: 10.1128/JCM.41.11.5046-5052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin W.H., Griffin D.E., Rota P.A., Papania M., Cape S.P., Bennett D. Successful respiratory immunization with dry powder live-attenuated measles virus vaccine in rhesus macaques. Proc Natl Acad Sci U S A. 2011;108(February (7)):2987–2992. doi: 10.1073/pnas.1017334108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Albrecht P., Herrmann K., Burns G.R. Role of virus strain in conventional and enhanced measles plaque neutralization test. J Virol Methods. 1981;3(December (5)):251–260. doi: 10.1016/0166-0934(81)90062-8. [DOI] [PubMed] [Google Scholar]
- 39.Chen R.T., Markowitz L.E., Albrecht P., Stewart J.A., Mofenson L.M., Preblud S.R. Measles antibody: reevaluation of protective titers. J Infect Dis. 1990;162(November (5)):1036–1042. doi: 10.1093/infdis/162.5.1036. [DOI] [PubMed] [Google Scholar]
- 40.Letvin N.L., Rao S.S., Dang V., Buzby A.P., Korioth-Schmitz B., Dombagoda D. No evidence for consistent virus-specific immunity in simian immunodeficiency virus-exposed, uninfected rhesus monkeys. J Virol. 2007;81(November (22)):12368–12374. doi: 10.1128/JVI.00822-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kozlowski P.A., Lynch R.M., Patterson R.R., Cu-Uvin S., Flanigan T.P., Neutra M.R. Modified wick method using Weck-Cel sponges for collection of human rectal secretions and analysis of mucosal HIV antibody. J Acquir Immune Defic Syndr. 2000;24(August (4)):297–309. doi: 10.1097/00126334-200008010-00001. [DOI] [PubMed] [Google Scholar]