

The PaOrf2 and γ-toxin subunits of Pichia acaciae toxin (PaT) and Kluyveromyces lactis zymocin are tRNA anticodon nucleases encoded by cytoplasmic DNA plasmids. Toxicity can be recapitulated conveniently by induced intracellular expression of PaOrf2 or γ-toxin in Saccharomyces cerevisiae. Mutational analysis of γ-toxin has identified amino acids required for ribotoxicity in vivo and RNA transesterification in vitro. Here, the authors report that PaOrf2 residues Glu9 and His287 (putative counterparts of γ-toxin Glu9 and His209) are essential for toxicity in vivo. The results suggest a similar basis for RNA transesterification by PaOrf2 and γ-toxin, despite their dissimilar primary structures and distinctive tRNA target specificities. PaOrf2 makes two sequential incisions in tRNA, the first of which occurs 3′ from the mcm5s2U wobble nucleoside and depends on the mcm5 modification. A second incision of the phosphodiester two nucleotides upstream results in the net excision of a di-nucleotide. These results highlight the logic of a two-incision mechanism of tRNA anticodon damage that evades productive repair by tRNA ligases.
Keywords: RNA repair, anticodon nuclease, mcm5, tRNA, toxin
Abstract
PaOrf2 and γ-toxin subunits of Pichia acaciae toxin (PaT) and Kluyveromyces lactis zymocin are tRNA anticodon nucleases. These secreted ribotoxins are assimilated by Saccharomyces cerevisiae, wherein they arrest growth by depleting specific tRNAs. Toxicity can be recapitulated by induced intracellular expression of PaOrf2 or γ-toxin in S. cerevisiae. Mutational analysis of γ-toxin has identified amino acids required for ribotoxicity in vivo and RNA transesterification in vitro. Here, we report that PaOrf2 residues Glu9 and His287 (putative counterparts of γ-toxin Glu9 and His209) are essential for toxicity. Our results suggest a similar basis for RNA transesterification by PaOrf2 and γ-toxin, despite their dissimilar primary structures and distinctive tRNA target specificities. PaOrf2 makes two sequential incisions in tRNA, the first of which occurs 3′ from the mcm5s2U wobble nucleoside and depends on mcm5. A second incision two nucleotides upstream results in the net excision of a di-nucleotide. Expression of phage and plant tRNA repair systems can relieve PaOrf2 toxicity when tRNA cleavage is restricted to the secondary site in elp3 cells that lack the mcm5 wobble U modification. Whereas the endogenous yeast tRNA ligase Trl1 can heal tRNA halves produced by PaOrf2 cleavage in elp3 cells, its RNA sealing activity is inadequate to complete the repair. Compatible sealing activity can be provided in trans by plant tRNA ligase. The damage-rescuing ability of tRNA repair systems is lost when PaOrf2 can break tRNA at both sites. These results highlight the logic of a two-incision mechanism of tRNA anticodon damage that evades productive repair by tRNA ligases.
INTRODUCTION
Infliction of tRNA damage by site-specific endoribonucleases (“ribotoxins”) underlies cellular stress responses and self-nonself discrimination in a wide range of microbial taxa. tRNA ribotoxins incise the tRNA anticodon loop to leave 2′,3′-cyclic phosphate and 5′-OH ends at the break. Anticodon breakage inhibits protein synthesis via depletion of the functional pool of specific tRNA isoacceptors, which, in turn, results in growth arrest or cell death.
The secreted eukaryal tRNA ribotoxins zymocin and PaT are elaborated by cytoplasmic linear DNA plasmids resident in the fungi Kluyveromyces lactis and Pichia acaciae, respectively (Klassen et al. 2004, 2008a; Lu et al. 2005). Zymocin is a heterotrimer of α, β, and γ subunits; PaT is a heterodimer of PaOrf1 and PaOrf2 subunits (Stark and Boyd 1986; McCracken et al. 1994; Klassen et al. 2004). Zymocin and PaT arrest growth of the nonself yeast species Saccharomyces cerevisiae by exposure from without. The α and β subunits of zymocin and the PaOrf1 subunit of PaT interact with the target cell surface to effect the transport of the γ subunit and the PaOrf2 subunit (the anticodon endonuclease enzymes) into the cytoplasm of the target cell (Jablonowski et al. 2004; Jablonowski and Schaffrath 2007; Meinhardt and Klassen 2009). Toxicity can also be achieved from within by induced intracellular expression in S. cerevisiae of just the γ-toxin or PaOrf2 subunits (minus their N-terminal signal peptides) (Butler et al. 1991; Klassen et al. 2004).
γ-toxin is an anticodon nuclease that cleaves its in vivo target tRNAGlu at a single phosphodiester 3′ from the modified wobble base mcm5s2U (5-methoxycarbonylmethyl-2-thiouridine) of the UUC anticodon, leaving 2′,3′-cyclic phosphate and 5′-OH ends at the break. γ-toxin can also cleave the two other yeast tRNAs (tRNAGln and tRNALys) that have an mcm5s2U wobble base, albeit less efficiently than it incises tRNAGlu. The wobble modification is required for tRNA cleavage by γ-toxin; yeast mutants that lack any of the enzymes responsible for synthesizing the mcm5 moiety are resistant to zymocin (Frohloff et al. 2001; Huang et al. 2005; Lu et al. 2005; Jablonowski et al. 2006; Huang et al. 2008). The latter includes the Elongator subunits (Elp1-6) and Elongator interacting proteins Kti11, Kti12, and Kti13, as well as the methylase complex Trm9-Trm112 and the phosphatase Sit4 and kinase Kti14 (Jablonowski et al. 2004; Jablonowski and Schaffrath 2007; Huang et al. 2008; Studte et al. 2008; Mehlgarten et al. 2009). Individual loss of the wobble uridine 2-thio modification of tRNAGlu provides only partial zymocin resistance and weak cleavage stimulation in vitro (Huang et al. 2008; Lu et al. 2005), indicating the mcm5 side chain, but not the 2-thio group of mcm5s2U, to be crucial for tRNA cleavage by γ-toxin.
PaOrf2 cleaves the same three yeast mcm5s2U-containing tRNAs in vitro, though its toxicity in vivo is attributed to specific cleavage and depletion of tRNAGln (Klassen et al. 2008a). Whereas PaOrf2 and γ-toxin are functionally analogous and have overlapping tRNase activities, PaOrf2’s cytotoxicity in S. cerevisiae is not stringently dependent on the presence of the mcm5-modified wobble uridine in the target tRNA (Klassen et al. 2008a). In the absence of mcm5, PaOrf2 can cleave tRNAGln(UUG) at an alternative phosphodiester upstream of the wobble U34 nucleoside, a property that is not shared with γ-toxin (Klassen et al. 2008a). Interestingly, PaOrf2 is unable to cleave its substrates when the wobble uridine methylase Trm9 (part of a complex with Trm112) is lacking (Klassen et al. 2008a). Since Trm9-Trm112 catalyzes the last step of mcm5/mcm5s2 biosynthesis (Kalhor and Clarke 2003; Mazauric et al. 2010; Chen et al. 2011), and trm9 mutants accumulate ncm5s2 (5-carbamoylmethyl-2-thiouridine) instead of mcm5s2 (Chen et al. 2011), it appears that this moiety is inhibitory for PaT.
A remarkable feature of the two known fungal tRNA anticodon nucleases is that, at first glance, they have no discernible primary structure similarity to one another (via BLAST queries), to any of the bacterial tRNA ribotoxins (PrrC, colicin D, colicin E5, VapC), or to any known ribonucleases or phosphotransferases. Alignment of the protein sequences in search of a structural or evolutionary connection between PaOrf2 and γ-toxin is now aided by the identification of two other putative plasmid-encoded fungal ribotoxins (PiT of Pichia inositovora and DrT of Debaryomyces robertsiae) and by the results of an extensive mutational analysis of γ-toxin that identified an ensemble of amino acids required for toxicity in vivo and anticodon nuclease activity in vitro (Jablonowski et al. 2006; Keppetipola et al. 2009; Jain et al. 2011).
The PiT toxin subunit (PiOrf4) shows weak primary structure similarity to γ-toxin (Klassen and Meinhardt 2003) but none to PaOrf2. The DrT toxin subunit (DrOrf3) shows strong similarity to PaOrf2 but none to γ-toxin (Klassen et al. 2004). An alignment of all four fungal polypeptides revealed only eight positions of side chain identity (exclusive of the N-terminal methionine of the signal peptide) (Fig. 1). Two of these correspond to γ-toxin Glu9 and His209, which are essential for toxicity and are the imputed general acid-base catalysts of RNA transesterification at the wobble uridine (Jablonowski et al. 2006; Jain et al. 2011). Here, we used the alignment in Figure 1 to guide a mutational analysis of PaOrf2 and thereby identified five side chains that are critical for toxicity in vivo, including the conserved glutamate and histidine residues. These results hint at a common chemical mechanism of RNA incision by the fungal anticodon nucleases.
FIGURE 1.

Alignment of putative ACNase subunits of killer toxins from Pichia acaciae, Debaryomyces robertsiae, Kluyveromyces lactis, and Pichia inositovora. For the multiple alignment, ClustalV (PAM250) was applied. Positions which have the same amino acid in at least two of the four proteins are shaded gray. Positions of side chain identity in all four proteins are shown in boxes. Negative positions represent the predicted signal peptide of each protein; position +1 accounts for the methionine added to achieve intracellular expression of variants lacking the signal peptide. Numbering of conserved residues refers to the intracellular version of PaOrf2-(1-319). NCBI GenBank accession numbers: P. acaciae pPac1-2 ORF2, CAE84960.1; D. robertsiae pWR1A ORF3, CAE84956.1; K. lactis pGKL1 ORF4, YP_001648056.1; P. inositovora pPin1-3 ORF4, CAD91887.1.
We also queried the rationale for the capacity of PaOrf2 to break its tRNA target at more than one site in the anticodon loop. A growing body of evidence highlights the existence of RNA repair systems in diverse taxa that are capable of sealing broken RNAs with 2′,3′-cyclic phosphate and 5′-OH ends (Amitsur et al. 1987; Martins and Shuman 2004, 2005; Nandakumar et al. 2008; Chan et al. 2009; Tanaka et al. 2011). In some cases, it is clear that the biological role of RNA repair is to “cure” ribotoxic tRNA damage. For example, during bacteriophage T4 infection of Escherichia coli, the viral enzymes Pnkp and Rnl1 repair a 2′,3′-cyclic phosphate/5′-OH “nick” in the anticodon loop of tRNALys, inflicted by virus-activation of the host ribotoxin PrrC (Amitsur et al. 1987). The budding yeast S. cerevisiae has an RNA repair enzyme, Trl1, that is responsible for joining the 2′,3′-cyclic phosphate and 5′-OH exons (broken in the anticodon loop) during tRNA splicing. Trl1 executes RNA repair via end-healing and end-sealing steps catalyzed by its three enzymatic domains (Apostol et al. 1991; Sawaya et al. 2003). Therefore, the toxicity of the fungal tRNA anticodon nucleases to S. cerevisiae must rely on either (1) an innate inability of Trl1 to repair the broken tRNA ends generated by the fungal ribotoxin, or (2) the rate and extent of tRNA damage greatly exceeding those of the endogeneous tRNA repair enzymes. There is good evidence in favor of the first scenario, insofar as (1) S. cerevisiae cells are susceptible to γ-toxin because the sealing domain of yeast Trl1 is unable to seal a break at the modified wobble base of tRNAGlu, and (2) the cytotoxicity of γ-toxin in S. cerevisiae can be ameliorated by expressing heterologous RNA repair enzymes from plant or bacteriophage T4 (Nandakumar et al. 2008). These findings underscored how the K. lactis ribotoxin exploits an Achilles’ heel in the target cell’s tRNA repair system by specifically nicking its target flanking a “lesion” (the modified wobble base) that impedes the endogenous repair system.
What about the Pichia acaciae ribotoxin? An appealing hypothesis is that dual cleavage of a tRNA molecule by PaOrf2 would result in the excision of a di-nucleotide, thereby rendering the damage functionally irreversible, because repair of such ends (if it occurred) would yield an inactive tRNA with a 2-nt deletion in the anticodon loop. Here, we flesh out this model by (1) showing that PaOrf2 does perform sequential cleavages at two sites in a tRNA and (2) testing the ability of Trl1 and plant and phage RNA repair enzymes to protect against PaOrf2 toxicity under conditions that favor nicking versus excision of the anticodon loop.
RESULTS
Phylogenetically guided mutational analysis of the PaOrf2 ribotoxin
Alignment of the γ-toxin, PaOrf2, PiOrf4, and DrOrf3 polypeptides highlighted eight positions of side chain identity, corresponding to Glu9, Asn74, Lys97, Leu165, Ile168, His287, Ile291, and Cys317 in the PaOrf2 protein (Fig. 1). To check the relevance of these eight conserved residues for PaOrf2 toxicity in vivo, we mutated them individually to alanine and conditionally expressed mutant and wild-type PaORF2 in S. cerevisiae under the control of a GAL promoter, which is repressed by glucose and induced by galactose. Whereas cells carrying the wild-type PaORF2 plasmid grew as well on glucose-containing medium as control cells carrying the empty vector, induced expression of wild-type PaORF2 inhibited cell growth on galactose medium (Fig. 2). Induced expression of PaORF2-Ala variants revealed that the E9A, L165A, I168A, H287A, and I291A variants lost their toxicity, while N74A, K97A, and C317A remained toxic (Fig. 2). The equivalents of PaOrf2 Glu9 and His287 in γ-toxin (Glu9 and His209) are imputed to be active site residues, at which even conservative substitutions led to a loss of toxicity in vivo and ablation of anticodon RNase activity in vitro (Jain et al. 2011). Here, we substituted PaOrf2 Glu9 with glutamine and aspartate and His287 with glutamine and asparagine and checked the effect on the in vivo activity of PaOrf2. The E9Q, E9D, H287Q, and H287N mutants were nontoxic (Fig. 2). Thus, five of the eight residues targeted, including those directly involved in the proposed transesterification mechanism of γ-toxin (Jain et al. 2011), were essential for PaOrf2 function, suggesting similarities in the γ-toxin and PaOrf2 active sites.
FIGURE 2.

Mutational effects on PaOrf2 toxicity. (A) Alanine substitutions of residues conserved among putative fungal ACNases. (B) Conservative substitutions of Glu9 and His287 (putative counterparts of γ-toxin Glu9 and His209). Expression vectors carrying the indicated PaORF2 alleles were transformed into S. cerevisiae. Serial dilutions of the plasmid-bearing yeast cells were spotted on agar medium containing 2% glucose or galactose. All mutant variants except C317A were expressed from 2μ plasmids. The C317A variant was expressed from a CEN plasmid. The plates were photographed after incubation at 30°C for 3 d.
Analysis of in vitro tRNA cleavage by PaOrf2
We showed previously that PaOrf2 cleaves tRNAGln(UUG), tRNAGlu(UUC), and tRNALys(UUU) in vitro, even though tRNAGln(UUG) is the main target in vivo (Klassen et al. 2008a). Loss of the mcm5 modification at U34 in an elp3 mutant strain confers resistance to γ-toxin, which correlates with a lack of detectable tRNA cleavage in vivo in elp3 cells expressing γ-toxin and with a ∼100-fold reduction in γ-toxin-induced cleavage in vitro of tRNA lacking the mcm5 wobble modification (Huang et al. 2005; Lu et al. 2005). In contrast, PaOrf2 is able to cleave hypomodified tRNA from elp3 yeast in vivo and in vitro. Mapping the site of in vitro cleaved tRNAGln from elp3 cells revealed the incision to occur at a position 2 nt upstream of the wobble nucleoside (Klassen et al. 2008a). PaOrf2 cleavage of tRNA from wild-type cells yields two different 5′ cleavage products, corresponding to N1–32 and N1–34 of tRNAGln, suggesting that there are two alternative sites of incision, U34 and U32 (Klassen et al. 2008a). It was unclear if one of these sites is cleaved preferentially and whether PaT is capable of catalyzing an excision of 2 nt from the anticodon loop by double cleavage of the same tRNA molecule.
Here, we analyzed the temporal order of the incisions of the anticodon loop. Total yeast tRNA was incubated with recombinant PaOrf2 (Klassen et al. 2008a), and aliquots were removed and quenched at intervals of 30 min. tRNAGln 5′ half-molecules were detected by Northern analysis using a probe that hybridizes to the 5′ end of tRNAGln (Fig. 3A). (In vitro transcribed tRNAGln half-molecules corresponding to nt 1–37, 1–35, and 1–33 served as standards.) After 30 min, there was a reduction in signal strength for full-length tRNAGln, concomitant with the appearance of signals for the N1–32 and N1–34 cleavage products. By 60 and 90 min, full-length tRNAGln had declined further, while the N1–34 to N1–32 products persisted (Fig. 3A).
FIGURE 3.

Northern blot analysis of tRNA cleavage by PaOrf2. Total yeast tRNA was incubated with PaOrf2His6, and aliquots were withdrawn and quenched at indicated times. Detection of tRNAGln (A) and tRNAGlu (B) and their 5′ half-cleavage products N1–32 (N32) and N1–34 (N34) was carried out by probing the membrane with DIG-labeled oligonucleotides complementary to tRNAGln or tRNAGlu. A set of in vitro transcribed tRNAGln half-molecules comprising nucleotide 1–37, 1–35, and 1–33 served as standards.
Northern blot analysis of the kinetics of PaOrf2 incision tRNAGlu in vitro revealed that the larger N1–34 cleavage product appeared prior to the smaller N1–32 product, indicating a preference for PaOrf2 to cleave tRNAGlu at the wobble U34 position. The appearance of the N1–32 product was concomitant with the disappearance of the N1–34 species, suggesting a precursor-product relationship. These results suggest that PaOrf2 incises tRNAGln initially at either U32 or U34, whereas it breaks tRNAGlu first at U34 and subsequently at U32. Thus, PaOrf2 can cleave one of its target tRNAs twice, resulting in a net excision of 2 nt.
Alleviation of PaOrf2 ribotoxicity by RNA repair enzymes
Growth arrest of S. cerevisiae by exogenous zymocin or intracellular γ-toxin can be “cured” by intracellular expression of eukaryotic and phage-encoded tRNA repair enzymes (Nandakumar et al. 2008). In Eukarya, tRNA repair is an essential component of tRNA splicing (Abelson et al. 1998). Bacteriophage T4 exploits tRNA repair to evade a ribotoxin-mediated host antiviral response (Amitsur et al. 1987). Ribotoxic tRNA damage results in 2′,3′-cyclic phosphate and 5′-OH ends, which need to be converted to 5′-PO4 and 3′-OH ends (the “end-healing” phase of tRNA repair) before they can be sealed by a classic ATP-dependent RNA ligase.
The yeast tRNA ligase Trl1 and the orthologous plant tRNA ligase AtRNL have three discrete enzymatic activities within three modular catalytic domains (see Fig. 5, below). A carboxyl-terminal 2′,3′ cyclic phosphodiesterase (CPD) hydrolyzes the 2′,3′-cyclic phosphate end to a 2′-PO4, 3′-OH end. A central polynucleotide kinase domain transfers the γ-phosphate from an NTP donor to the broken 5′-OH end to yield a 5′-PO4. An amino-terminal ATP-dependent ligase domain then joins the healed ends to form a 3′-5′ phosphodiester, 2′-PO4 repair junction (Apostol et al. 1991; Sawaya et al. 2003; Wang et al. 2006). The 2′-PO4 is subsequently removed by the phosphotransferase Tpt1 (Spinelli et al. 1997). Bacteriophage T4 encodes a two-protein RNA repair system that performs related end-healing and end-sealing reactions. T4 polynucleotide kinase-phosphatase (Pnkp) is a bifunctional healing enzyme. The carboxyl-terminal phosphatase domain removes the 2′,3′-cyclic phosphate to yield a 2′-OH, 3′-OH end and the amino-terminal kinase domain phosphorylates the 5′-OH end. The healed ends are then joined by T4 RNA ligase 1 (Rnl1) to generate a 3′-5′ phosphodiester repair junction (Amitsur et al. 1987).
FIGURE 5.

The ScTrl1 healing domain inhibits the rescue of PaOrf2 toxicity by Pnkp and Rnl1. Serial 10-fold dilutions of S. cerevisiae cells expressing PaOrf2 (PaT) from a CEN vector or an empty vector or in the presence of an additional 2μ plasmid encoding full-length ScTrl1 or its component end-healing (Kin-CPD) or end-sealing (LIG) domains were spotted on –Ura–His agar plates with 2% glucose or galactose. The plates were photographed after incubation at 30°C for 2 d (glucose) or 3 d (galactose).
Expression of the phage or plant tRNA repair enzymes in yeast confers γ-toxin resistance, whereas the endogenous ScTrl1 is apparently unable to carry out repair of γ-toxin-induced damage even when ScTrl1 is overexpressed (Nandakumar et al. 2008). ScTrl1 is able to heal the ends of γ-toxin cleaved tRNAGlu halves, but not seal them. It was suggested that the presence of the bulky mcm5s2U nucleobase on the 3′-OH side of the break inhibits sealing by ScTrl1 but is tolerated by AtRNL or T4 Rnl1 (Nandakumar et al. 2008).
Because PaOrf2 can cleave its substrate at two alternative positions, one of which (U32) is not mcm5-modified, we asked whether these sites are accessible to tRNA repair by plant and phage enzymes. PaOrf2 was conditionally expressed from a CEN plasmid under control of the GAL promoter in the presence or absence of plasmids constitutively expressing AtRNL or T4 Pnkp+Rnl1. In wild-type TRL1 cells, neither plant nor phage RNA repair enzymes could alleviate the galactose-dependent toxicity of PaOrf2 (Fig. 4). However, in trl1Δ cells, in which the essential tRNA splicing activity was provided by AtRNL or Pnkp+Rnl1 (Schwer et al. 2004), we observed that the toxicity of PaOrf2 was ameliorated by the phage tRNA repair enzymes but not by AtRNL (Fig. 4). We surmise that (1) AtRNL is unable to repair PaOrf2-mediated tRNA damage, and (2) the capacity of T4 Pnkp and Rnl1 to repair tRNAs damaged by PaOrf2 is inhibited by the endogenous ScTrl1.
FIGURE 4.

Rescue of PaOrf2 toxicity by Pnkp and Rnl1 in a trl1Δ strain background. Serial 10-fold dilutions of S. cerevisiae cells bearing a CEN PaORF2 plasmid (PaT) or an empty vector in the presence or absence of an additional plasmid encoding AtRNL (2μ) or T4 Pnkp and Rnl1 were spotted on selective agar plates with 2% glucose or galactose. The plates were photographed after incubation at 30°C for 2 d (glucose) or 3 d (galactose).
We queried which activity of ScTrl1 is inhibitory to the repair of PaOrf2-mediated damage by individually expressing Trl1’s sealing (ligase) or healing (kinase-CPD) domains in the trl1Δ Pnkp Rnl1 yeast strain (Fig. 5). Whereas the Trl1 ligase domain did not diminish the protective effects of phage tRNA repair, the kinase-CPD domain rendered the trl1Δ Pnkp Rnl1 strain sensitive to galactose-dependent growth arrest by PaOrf2 (Fig. 5). Thus, healing of PaOrf2-damaged tRNA by endogenous ScTrl1 prevented completion of tRNA repair by the phage pathway. Because the kinase modules of the phage and yeast end-healing enzymes are structurally similar and the reaction products are identical (i.e., a 5′-PO4 RNA end), we ascribe the repair-defeating effect of the ScTrl1 kinase-CPD domain to the distinctive 3′-OH, 2′-PO4 end configuration generated by yeast CPD versus the 3′-OH, 2′-OH end produced by Pnkp. In other words, the yeast CPD diverts the processing of the 3′ end of the PaOrf2-damaged tRNAs to a 3′-OH, 2′-PO4 structure that is not sealed as effectively by T4 Rnl1 as is a 3′-OH, 2′-OH terminus. This inhibitory effect of CPD on the T4 repair system is apparently particular to PaOrf2-damaged tRNAs and is either inapplicable or relatively benign with respect to physiologically spliced yeast tRNAs, insofar as ScTrl1 kinase-CPD expression itself did not affect the growth of the trl1Δ Pnkp Rnl1 yeast strain (Fig. 5).
Rescue of PaOrf2 ribotoxicity in elp3 yeast cells by RNA repair
Ablation of the yeast ELP3 gene eliminates the mcm5 modification of the wobble uridine (U34) of the anticodon in the tRNAs targeted by zymocin and PaT (Huang et al. 2005). Whereas elp3 mutants are resistant to growth arrest by zymocin, PaT retains its toxicity to elp3 cells, albeit at higher PaT concentrations compared to ELP3 cells (Klassen et al. 2004, 2008a; Lu et al. 2005). PaOrf2’s ability to cleave tRNAs isolated from elp3 cells exclusively at the secondary site (U32) (Klassen et al. 2008a) raises the question of whether tRNA restriction at a single site flanking the anticodon affects the ability of tRNA repair enzymes to protect elp3 cells from PaOrf2 toxicity.
Galactose-induced expression of PaORF2 from a CEN vector in elp3 cells did not cause growth arrest (data not shown). However, when PaORF2 was expressed in elp3 TRL1 cells at high gene dosage from a 2μ plasmid, growth arrest was observed on galactose medium (Fig. 6). When phage or plant tRNA repair enzymes were expressed in elp3 TRL1 cells, the toxicity of 2μ PaORF2 was partially alleviated, resulting in cell growth under inducing conditions (albeit more slowly that control cells, as judged by colony size) (Fig. 6). Neither Pnkp/Rnl1 nor AtRNL could rescue 2μ PaORF2 toxicity in wild-type ELP3 cells (Fig. 6). We infer that AtRNL and T4 Pnkp/Rnl1 (but not the endogenous ScTrl1) were able to repair the PaOrf2 damage when the target tRNAs were broken uniquely at U32.
FIGURE 6.

Rescue of high-copy PaOrf2 toxicity by plant T4-encoded tRNA repair in the absence of U34 mcm5 modification. Serial 10-fold dilutions of S. cerevisiae cells bearing a 2μ PaORF2 plasmid (PaT) or an empty vector plus an additional plasmid encoding AtRNL (2μ) or T4 Pnkp and Rnl1 were spotted on selective agar plates with 2% glucose or galactose. The plates were photographed after incubation at 30°C for 2 d (glucose) or 3 d (galactose).
In order to gauge whether the inability of ScTrl1 to rescue toxicity in the elp3 background was due to limitations of its healing or sealing activities, we expressed ligase-dead (K152A), kinase-dead (S701A), or CPD-dead (T1001A) mutants of AtRNL (Wang et al. 2006) in elp3 TRL1 cells bearing the 2μ PaORF2 plasmid. The kinase-dead AtRNL S701A mutant conferred resistance to PaOrf2 comparable to that of wild-type AtRNL (Fig. 7), signifying that the endogenous ScTrl1 kinase activity was competent for repair of the PaOrf2 damage. The CPD-dead T1001A mutant also conferred rescue, albeit not as well as wild-type AtRNL, as judged from the smaller colony size on galactose medium (Fig. 7). In contrast, the ligase-dead K152A AtRNL mutant was unable to ameliorate PaOrf2 toxicity (Fig. 7). Thus, we infer that ScTrl1 could heal tRNA ends broken at position 32 in elp3 cells but was unable to seal them. This deficiency may reflect a fastidiousness of the ScTrl1 ligase with respect to the position of the break within the tRNA anticodon loop (spliced yeast tRNAs are incised 3′ from the anticodon at position 37) that is not shared by AtRNL or Rnl1.
FIGURE 7.
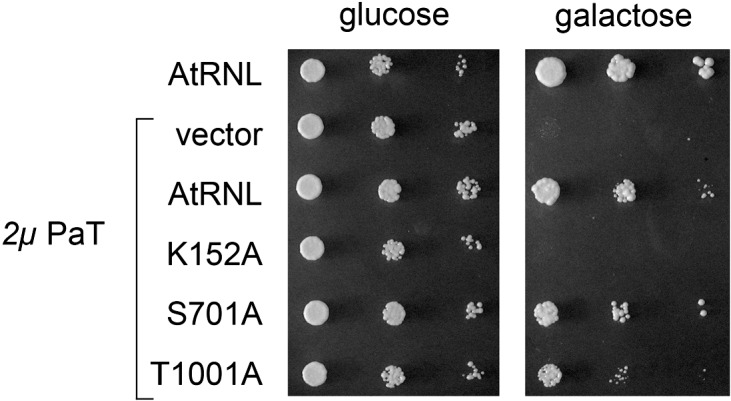
ScTrl1 can heal U32 breaks but requires AtRNL sealing activity for rescuing PaOrf2 toxicity in elp3. Serial 10-fold dilutions of S. cerevisiae cells bearing a 2μ PaORF2 plasmid (PaT) or an empty vector plus 2μ plasmids encoding full-length AtRNL or ligase-dead (K152A), kinase-dead (S701A), and CPD-dead (T1001A) variants as specified were spotted on –Ura–His agar plates with 2% glucose or galactose. The plates were photographed after incubation at 30°C for 2 d (glucose) or 4 d (galactose).
DISCUSSION
Two distinct eukaryotic ACNase toxins have been identified in the yeasts K. lactis and P. acaciae (Lu et al. 2005; Klassen et al. 2008a). Both toxins are encoded by cytoplasmic linear plasmids that are phylogenetically related and share a common receptor-binding and trans-membrane passage apparatus, consisting of chitin-binding and hydrophobic domains located on the same (in PaT) or different (in zymocin) subunits (Jablonowski et al. 2001; Klassen et al. 2004). Yet, the anticodon nuclease subunits PaOrf2 and γ-toxin are seemingly unrelated (Klassen et al. 2004). An expanded comparison to putative ribotoxins encoded by related linear plasmids from P. inositovora and D. robertsiae led us to identify a small number of conserved residues, two of which—a glutamate and a histidine—are known to be essential for γ-toxin activity (Keppetipola et al. 2009; Jain et al. 2011). We find that the corresponding Glu and His residues of PaOrf2 are essential for its toxicity in vivo. We suggest that the Glu and His side chains conserved in the primary structure of the four fungal ribotoxins are constituents of the endonuclease active site. Leu165, Ile168, and Ile291 of PaOrf2 are also conserved among all four fungal ribotoxins and are essential for the in vivo toxicity of PaOrf2. It is possible that the fungal ribotoxins adopt related tertiary structures, notwithstanding their sparse primary structure similarity.
PaOrf2 and γ-toxin have distinctive substrate specificities. Whereas γ-toxin cleaves tRNA exclusively at the mcm5s2-modified wobble base, PaOrf2 cleaves at the same position and at a secondary site 2 nt upstream (Lu et al. 2005; Klassen et al. 2008a). Time-resolved monitoring of PaOrf2 cleavage products revealed that the tRNAGlu substrate is initially cleaved at position 34, while cleavage at position 32 occurs with a delay, implying that PaOrf2 can excise a di-nucleotide in vitro. tRNAGln appears to be cleaved at either position 34 or 32. At least one other anticodon nuclease toxin, Geobacillus kaustrophilus RloC, a homolog of the bacterial single-incision Escherichia coli PrrC ribotoxin (Davidov and Kaufmann 2008), also exhibits anticodon “excisase” activity. RloC breaks tRNALys at two positions, immediately 3′ from and 5′ of the wobble uridine nucleoside, resulting in net excision of the wobble mononucleotide. Thus, the advancement from an incisase to an excisase mode of ribotoxins action may have occurred repeatedly during evolution and provides an increased benefit to the ribotoxin producer. It has been suggested that evasion of RNA repair is one such benefit (Davidov and Kaufmann 2008).
Our studies of the effect of RNA repair enzymes on PaOrf2 ribotoxicity provide new insights into this issue (Fig. 8). First, heterologous plant and phage T4 RNA repair pathways, but not endogenous yeast Trl1, can counteract tRNA damage when cleavage is restricted to U32 in an elp3 mutant that lacks the mcm5 wobble uridine modification. ScTrl1 can provide the end-healing activities to counteract PaOrf2 ribotoxicity in elp3 cells (when supplemented by the AtRNL ligase activity), but it is apparently unable to carry out the ligation step (Fig. 8A). The ScTrl1 ligase activity in vivo appears to be sensitive to the position of the break within the anticodon loop (U32 “rejected”) and/or the presence of a modified nucleobase on the 3′-OH side of the break (mcm5U34 “rejected”). In contrast, AtRNL sealing activity is not thwarted by these variations.
FIGURE 8.

Summary model of eukaryal and phage T4 RNA repair pathways operating on PaOrf2-cleaved tRNA. (A) Single cleavage site action of PaOrf2 in the absence of mcm5 wobble uridine modification in elp3 cells. (B) Dual cleavage site action of PaOrf2 in ELP3 cells. (P) Phosphate, (cycloP) cyclo-phosphate. See text for details.
The ability of plant and T4 tRNA repair pathways to antagonize PaOrf2 toxicity changes in ELP3 cells when dual incisions of tRNA are allowed. Since both pathways can operate, in general, on single U32 and U34 tRNA breaks but fail under conditions that allow PaT to access both sites, we conclude that PaT’s toxicity in vivo involves excision cleavage, even though in vitro, double cleavage was restricted to tRNAGlu. It is clear, however, that target specificities of PaT are modulated by additional, yet unknown factors in vivo, as the highly specific targeting tRNAGln observed in vivo cannot be recapitulated in vitro. Results of tRNA repair accessibility strongly support our model (Fig. 8B), in which double cleavage of a toxicity-relevant target tRNA does occur in PaT-challenged wild-type cells. In this context, AtRNL is ineffective, presumably because PaOrf2 can cleave enough of its targets twice before the plant enzymes can act, so that any break sealing that occurs thereafter would be nonproductive. Pnkp and Rnl1 can rescue PaOrf2 ribotoxicity in ELP3 cells when the endogenous Trl1 activity is ablated. The implication here is that the phage enzymes can access and repair enough of the tRNAs incised singly at U34 before a second incision at position 32 renders them irreparable (Fig. 8B). Our speculation that the rates of tRNA repair in vivo by plant and phage repair enzymes may differ, with the phage pathway acting more effectively than AtRNL, is in keeping with the findings that the specific activity of the AtRNL ligase domain in tRNA splicing in vitro is ∼10-fold lower than that of T4 Rnl1 (Nandakumar et al. 2008). The ill effect of yeast Trl1 on the repair of PaOrf2-inflicted RNA damage by T4 Rnl1 signifies that the healed 3′-OH, 2′-PO4 ends generated by the CPD domain of ScTrl1 are poor substrates for Rnl1. Such 2′-PO4 ends are also less than optimal substrates for yeast tRNA splicing by Rnl1, insofar as a trl1Δ RNL1 TRL1(389–827) yeast strain that relies on the combination of the phage ligase and the yeast kinase-CPD domain to perform tRNA splicing is viable at 25° and 30°C but not at 37°C (Schwer et al. 2004).
Because PaT and zymocin are functional analogs produced by related fungal plasmids, it is reasonable to think that the second tRNA incision inflicted by PaOrf2 represents an acquired advantage over the single-cutting endonuclease γ-toxin. Given that AtRNL is able to repair single-site tRNA damage flanking a modified wobble base, and that all fungi have tRNA ligases homologous to Trl1 and AtRNL, we envision that some fungal tRNA ligases will resemble the plant enzyme in their ability to repair ribotoxic tRNA incision. The excisional mode of action of a secreted anticodon nuclease toxin potentially expands the spectrum of self-nonself discrimination by overriding target cell resistance in taxa that have an AtRNL-like repair capacity. Additionally, excision cleavage may represent an adaptation of the toxin-antitoxin system encoded by the parasitizing virus-like DNAs to counteract the potential occurrence of an AtRNL-like repair capacity within the host species and prevent the establishment of autoimmunity via RNA repair.
MATERIALS AND METHODS
Expression plasmids and S. cerevisiae strains
PaOrf2 was expressed in haploid S. cerevisiae in a galactose-inducible manner. The strains used are derivatives of W303a or CEN.PK2-1c and have been described (van Dijken et al. 2000; Sawaya et al. 2003; Huang et al. 2005; Jablonowski et al. 2006; Nandakumar et al. 2008). The expression constructs (encoding for PaOrf2 lacking the N-terminal 12-aa signal peptide) pPACTOXLF (ARS-CEN LEU2, in pLF16), pPACBX (2μ LEU2 URA3), and pRS415-Gal1-PaT (CEN LEU2) are described (Klassen et al. 2004, 2008a; Tanaka et al. 2011). An additional construct (pPACTOXEP) containing the GAL1-promotor-PaORF2 fusion in YEplac181 was made by subcloning via PstI-PvuII from pPACTOXLF. We found that the cloned gene in pPACTOXLF and pPACTOXEP differs at two nucleobase positions from the sequence in GenBank (NCBI accession number: CAE84960.1). The resulting coding changes N82K and K199I do not affect PaOrf2 toxicity. Missense mutations were introduced into the PaORF2 gene in pPACTOXEP or pPACTOXLF via site-directed mutagenesis using two partially overlapping primers. The mutated genes were sequenced to verify that no unwanted coding changes were acquired. The RNA repair plasmids pRS423-TPI-AtRNL (2μ HIS3), pRS423-TPI-Trl1 (2μ HIS3), and pRS413-Pnkp/Rnl1 and plasmids for separate expression of the yeast Trl1 healing (1–388) and sealing (389–827) domains were described previously (Nandakumar et al. 2008; Meineke and Shuman 2012).
PaORF2 toxicity assay
The indicated S. cerevisiae strains were transformed with plasmid DNA by the lithium acetate method (Schiestl and Gietz 1989). Transformants were selected on appropriate minimal synthetic media on 2% (w/v) bacto agar plates. Cells derived from single transformants were grown at 30°C in liquid culture in selective media containing 2% glucose. The cultures were adjusted to A600 of 0.1 and then serially diluted in 10-fold steps in water. Aliquots (3 μL) of the dilutions were spotted in parallel on selective agar plates containing either 2% glucose or 2% galactose. The plates were photographed after incubation at 30°C for 2 d (glucose) or 3 or 4 d (galactose) as indicated.
Analysis of tRNA cleavage in vitro
Recombinant PaOrf2 protein was produced in E. coli as a C-terminal His6 fusion and isolated from a soluble extract by Ni-affinity chromatography as described previously (Klassen et al. 2008a). tRNA was isolated from wild-type yeast cells (S. cerevisiae CEN.PK2-1c) (van Dijken et al. 2000) as described (Klassen et al. 2008b). Five μg of tRNA was incubated at 30°C in a total volume of 100 μL in 10 mM Tris-HCl pH 7.5, 10 mM MgCl2, 50 mM NaCl, 1 mM dithiothreitol, containing 50 ng of PaOrf2His6. At time intervals of 30 min, 10-μL aliquots (containing 0.5 μg of tRNA) were removed, mixed with an equal volume of formamide loading buffer (95% w/v formamide, 0.025% w/v SDS, 0.025% w/v bromophenol blue, 0.025% xylene cyanol), then incubated at 65°C for 30 min and analyzed by electrophoresis through a 12% polyacrylamide gel containing 7.5 M urea in TBE. Synthetic tRNAGln half-molecules comprising nucleotides 1–37, 1–35, and 1–33 were produced by in vitro transcription as described (Klassen et al. 2008a). The gel contents were transferred to positively charged nylon membranes by semidry electroblotting. The membrane was incubated overnight at 37°–40°C in hybridization buffer (Roche, Mannheim) containing 50% formamide and 10 pmol μL−1 digoxygenin-labeled oligonucleotides specific for tRNAGln(UUG) or tRNAGlu(UUC) (Klassen et al. 2008a). Detection of the annealed primers was carried out using phosphatase-conjugated anti-DIG antibodies and chemiluminescent alkaline phosphatase substrate CDPstar as recommended by the manufacturer (Roche, Mannheim).
ACKNOWLEDGMENTS
This research was supported by NIH grant GM42498 (S.S.). S.S. is an American Cancer Society Research Professor.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.034132.112.
REFERENCES
- Abelson J, Trotta CR, Li H 1998. tRNA splicing. J Biol Chem 273: 12685–12688 [DOI] [PubMed] [Google Scholar]
- Amitsur M, Levitz R, Kaufman G 1987. Bacteriophage T4 anticodon nuclease, polynucleotide kinase, and RNA ligase reprocess the host lysine tRNA. EMBO J 6: 2499–2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostol BL, Westaway SK, Abelson J, Greer CL 1991. Deletion analysis of a multifunctional yeast tRNA ligase polypeptide: Identification of essential and dispensable functional domains. J Biol Chem 266: 7445–7455 [PubMed] [Google Scholar]
- Butler AR, Porter M, Stark MJ 1991. Intracellular expression of Kluyveromyces lactis toxin γ subunit mimics treatment with exogenous toxin and distinguishes two classes of toxin-resistant mutant. Yeast 7: 617–625 [DOI] [PubMed] [Google Scholar]
- Chan CM, Zhou C, Huang R 2009. Reconstituting bacterial RNA repair and modification in vitro. Science 326: 247 doi: 10.1126/science.1179480 [DOI] [PubMed] [Google Scholar]
- Chen C, Huang B, Anderson JT, Byström AS 2011. Unexpected accumulation of ncm5U and ncm5S2U in a trm9 mutant suggests an additional step in the synthesis of mcm5U and mcm5S2U. PLoS ONE 6: e20783 doi: 10.1371/journal.pone.0020783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidov E, Kaufmann G 2008. RloC: A wobble nucleotide-excising and zinc-responsive bacterial tRNase. Mol Microbiol 69: 1560–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohloff F, Fichtner L, Jablonowski D, Breunig KD, Schaffrath R 2001. Saccharomyces cerevisiae Elongator mutations confer resistance to the Kluyveromyces lactis zymocin. EMBO J 20: 1993–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Johansson MJO, Byström AS 2005. An early step in wobble tRNA modification requires the Elongator complex. RNA 11: 424–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Lu J, Byström AS 2008. A genome-wide screen identifies genes required for formation of the wobble nucleoside 5-methoxycarbonylmethyl-2-thiouridine in Saccharomyces cerevisiae. RNA 14: 2183–2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonowski D, Schaffrath R 2007. Zymocin, a composite chitinase and tRNase killer toxin from yeast. Biochem Soc Trans 35: 1533–1537 [DOI] [PubMed] [Google Scholar]
- Jablonowski D, Fichtner L, Martin VJ, Klassen R, Meinhardt F, Stark MJR, Schaffrath R 2001. Saccharomyces cerevisiae cell wall chitin, the potential Kluyveromyces lactis zymocin receptor. Yeast 18: 1285–1299 [DOI] [PubMed] [Google Scholar]
- Jablonowski D, Fichtner L, Stark MJ, Schaffrath R 2004. The yeast Elongator histone acetylase requires Sit4-dependent dephosphorylation for toxin-target capacity. Mol Biol Cell 15: 1459–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonowski D, Zink S, Mehlgarten C, Daum G, Schaffrath R 2006. tRNAGlu wobble uridine methylation by Trm9 identifies Elongator’s key role for zymocin-induced cell death in yeast. Mol Microbiol 59: 677–688 [DOI] [PubMed] [Google Scholar]
- Jain R, Poulos MG, Gros J, Chakravarty AK, Shuman S 2011. Substrate specificity and mutational analysis of Kluyveromyces lactis γ-toxin, a eukaryal tRNA anticodon nuclease. RNA 17: 1336–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalhor HR, Clarke S 2003. Novel methyltransferase for modified uridine residues at the wobble position of tRNA. Mol Cell Biol 23: 9283–9292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppetipola N, Jain R, Meineke B, Diver M, Shuman S 2009. Structure-activity relationships in Kluyveromyces lactis γ-toxin, a eukaryal tRNA anticodon nuclease. RNA 15: 1036–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen R, Meinhardt F 2003. Structural analysis of the killer element pPin1-3 from Pichia inositovora. Mol Genet Genomics 270: 190–199 [DOI] [PubMed] [Google Scholar]
- Klassen R, Teichert S, Meinhardt F 2004. Novel yeast killer toxins provoke S-phase arrest and DNA damage checkpoint activation. Mol Microbiol 53: 263–273 [DOI] [PubMed] [Google Scholar]
- Klassen R, Paluszynski J, Wemhoff S, Pfeiffer A, Fricke J, Meinhardt F 2008a. The primary target of the killer toxin from Pichia acaciae is tRNAGln. Mol Microbiol 69: 681–697 [DOI] [PubMed] [Google Scholar]
- Klassen R, Fricke J, Pfeiffer A, Meinhardt F 2008b. A modified DNA isolation protocol for obtaining pure RT-PCR grade RNA. Biotechnol Lett 30: 1041–1044 [DOI] [PubMed] [Google Scholar]
- Lu J, Huang B, Esberg A, Johansson MJ, Byström AS 2005. The Kluyveromyces lactis γ-toxin targets tRNA anticodons. RNA 11: 1648–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins A, Shuman S 2004. Characterization of a baculovirus enzyme with RNA ligase, polynucleotide 5′ kinase and polynucleotide 3′ phosphatase activities. J Biol Chem 279: 18220–18231 [DOI] [PubMed] [Google Scholar]
- Martins A, Shuman S 2005. An end-healing enzyme from Clostridium thermocellum with 5′ kinase, 2′,3′ phosphatase, and adenylyltransferase activities. RNA 11: 1271–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazauric MH, Dirick L, Purushothaman SK, Bjork GR, Lapeyre B 2010. Trm112p is a 15-kDa zinc finger protein essential for the activity of two tRNA and one protein methyltransferases in yeast. J Biol Chem 285: 18505–18515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken DA, Martin VJ, Stark MJR, Bolen PL 1994. The linear-plasmid-encoded toxin produced by the yeast Pichia acaciae: Characterization and comparison with the toxin of Kluyveromyces lactis. Microbiology 140: 425–431 [DOI] [PubMed] [Google Scholar]
- Mehlgarten C, Jablonowski D, Breunig KD, Stark MJ, Schaffrath R 2009. Elongator function depends on antagonistic regulation by casein kinase Hrr25 and protein phosphatase Sit4. Mol Microbiol 73: 869–881 [DOI] [PubMed] [Google Scholar]
- Meineke B, Shuman S 2012. Determinants of the cytotoxicity of PrrC anticodon nuclease and its amelioration by tRNA repair. RNA 18: 145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinhardt F, Klassen R 2009. Yeast killer toxins: Fundamentals and applications. In The Mycota (ed. T Anke, D Weber), pp. 107–130. Springer, New York, NY [Google Scholar]
- Nandakumar J, Schwer B, Schaffrath R, Shuman S 2008. RNA repair: An antidote to cytotoxic eukaryal RNA damage. Mol Cell 31: 278–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya R, Schwer B, Shuman S 2003. Genetic and biochemical analysis of the functional domains of yeast tRNA ligase. J Biol Chem 278: 43928–43938 [DOI] [PubMed] [Google Scholar]
- Schiestl RH, Gietz RD 1989. High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr Genet 16: 339–346 [DOI] [PubMed] [Google Scholar]
- Schwer B, Sawaya R, Ho CK, Shuman S 2004. Portability and fidelity of RNA-repair systems. Proc Natl Acad Sci 101: 2788–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli SL, Consaul SA, Phizicky EM 1997. A conditional lethal yeast phosphotransferase (tpt1) mutant accumulates tRNAs with a 2′-phosphate and an undermodified base at the splice junction. RNA 3: 1388–1400 [PMC free article] [PubMed] [Google Scholar]
- Stark MJ, Boyd A 1986. The killer toxin of Kluyveromyces lactis: Characterization of the toxin subunits and identification of the genes which encode them. EMBO J 5: 1995–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studte P, Zink S, Jablonowski D, Bär C, von der Haar T, Tuite MF, Schaffrath R 2008. tRNA and protein methylase complexes mediate zymocin toxicity in yeast. Mol Microbiol 69: 1266–1277 [DOI] [PubMed] [Google Scholar]
- Tanaka N, Meineke B, Shuman S 2011. RtcB, a novel RNA ligase, can catalyze tRNA splicing and HAC1 mRNA splicing in vivo. J Biol Chem 286: 30253–30257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijken JP, Bauer J, Brambilla L, Duboc P, Francois JM, Gancedo C, Giuseppin ML, Heijnen JJ, Hoare M, Lange HC, et al. 2000. An interlaboratory comparison of physiological and genetic properties of four Saccharomyces cerevisiae strains. Enzyme Microb Technol 26: 706–714 [DOI] [PubMed] [Google Scholar]
- Wang LK, Schwer B, Englert M, Beier H, Shuman S 2006. Structure–function analysis of the kinase-CPD domain of yeast tRNA ligase (Trl1) and requirements for complementation of tRNA splicing by a plant Trl1 homolog. Nucleic Acids Res 34: 517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]