

Abstract
A series of eighteen 1,3,4-oxadiazole derivatives have been synthesized by treating aromatic acid hydrazides with carbon disulfide in ethanolic potassium hydroxide yielding potassium salts of 1,3,4-oxadiazoles. Upon neutralization with 1 N hydrochloric acid yielded crude crystals of 1,3,4-oxadiazoles, which were purified by recrystallization in boiling methanol. The synthesized 1,3,4-oxadiazoles derivatives were evaluated in vitro for their urease inhibitory activities, most of the investigated compounds were potent inhibitors of Jack bean urease. The molecular docking studies were performed by docking them into the crystal structure of Jack bean urease to observe the mode of interaction of synthesized compounds. The synthesized compounds were also tested for antibacterial and antioxidant activities and some derivatives exhibited very promising results.
1. Introduction
Urease (urea amidohydrolase; E.C. 3.5.1.5) is a nickel containing enzyme that catalyzes the hydrolysis of urea to the formation of ammonia and carbon dioxide [1]. It plays a pivotal role in nitrogen metabolism of plants during the germination process [2]. A variety of ureases have been isolated from bacteria, algae, fungi, and plants [1–3]. Irrespective of structural differences of plant and microbial originated urease, it follows same catalysis pattern. It is mainly because of similar sequence of amino acids and presence of Ni+2 ions in active site of this multimeric enzyme which indicates emergence from a common ancestry [2, 4–6].
The primary physiological role of urease is to provide nitrogen for organisms in the form of ammonia for their growth. However, high urease activity is responsible for release of abnormally large amounts of ammonia into atmosphere which may lead to environmental and economic problems [1, 2] Human and animal pathogenicity of hepatic encephalopathy, hepatic coma urolithiasis, gastric and peptic ulcers, pyelonephritis, and urinary catheter encrustation are caused by ammonia produced by ureases [1, 2, 7, 8]. The urease activity of Helicobacter pylori plays an important role in the pathogenesis of gastric and peptic ulcer [2]. Therefore, urease inhibitors have the potential to be used as anti-ulcer drugs. For the said infections caused by the bacterial ureases, more effective and potent compounds are required with a whole new level of safety and specificity.
Urease has diverse functions and its inhibition has received special attention over the past few years and many antiurease agents have been reported. Among these are hydroxamic acid derivatives [9], hydroxyurea [10], hydroxamic acids [11], phosphorodiamidates [12, 13], imidazoles such as rabeprazole, [14] lansoprazole, [15] omeprazole, [16] quinines, [17] thiol-compounds, and [18] plaunotol and its thiourea derivatives [19]. Very recently we have investigated schiff base derivatives, which were most active inhibitors of Jack bean urease [20]. Through molecular modeling simulations and high-throughput virtual screening new derivatives of coumarin and triazoles were also found as urease inhibitors [21].
In the current paper, we present the synthesis of 1,3,4-oxadiazoles derivatives and their evaluation for inhibitory activity against Jack bean urease. It is notable that most of the compounds were more potent inhibitors of the enzyme as compared to standard inhibitor (thiourea). One of the compounds (4j) has potent urease inhibitory activity with IC50 value of 1.15 μM, which is 20-fold more active than the standard. Molecular docking study is also carried out to gain an understanding of urease inhibitory activity of 1,3,4-oxadiazoles derivatives. Newly synthesized compounds were also investigated on pathogenic bacterial strains and it was observed that most of the compounds also exhibited potent antibacterial activities.
2. Experimental
2.1. Synthesis
All the common solvents and chemicals were of analytical grade or dry distilled. Reaction progress was determined by thin layer chromatography (TLC) analysis and Rf values were determined by employing precoated silica gel aluminium plates, Kieslgel 60 F254 from Merck (Germany), using petroleum ether : ethyl acetate (8 : 2) as an eluent and TLC was visualized under UV lamp. Melting points were determined on a Stuart melting point apparatus (SMP3) and are uncorrected. The IR spectra were recorded on Bruker Optics Alpha FT-IR spectrophotometer. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker Avance 300 MHz spectrometer with TMS as an internal standard. Chemical shift are reported as δ values (ppm) downfield from internal tetramethylsilane of the indicated organic solution. Peak multiplicities are expressed as follows: s, singlet; d, doublet; t, triplet; q, quartet; dt, doublet of triplets. Coupling constants (J values) are given in hertz (Hz). Mass spectra were recorded on Agilent Technologies 6890 N gas chromatograph and an inert mass selective detector 5973 mass spectrometer. The elemental analysis was performed on Leco CHNS-932 Elemental Analyzer, Leco Corporation (USA). Abbreviations are used as follows: DMSO-d6, dimethyl sulfoxide-d6; FT-IR spectroscopy, fourier transform infrared spectroscopy; KDa, Kilo Dalton.
2.1.1. Synthesis of Substituted Aromatic Esters 2 (a–e) and Aromatic Acid Hydrazides 3 (a–e)
Substituted aromatic acid 1 was esterified 2 by refluxing in methanol and in the presence of catalytic amount of sulfuric acid. Substituted aromatic ester 2 was converted into their corresponding acid hydrazide 3 by refluxing in hydrazine hydrate and methanol was used as solvent through reported literature procedure [22–24].
2.1.2. Synthesis of 1,3,4-oxadiazole-2-thiones
Acid hydrazides 3 was treated with carbon disulfide in ethanolic potassium hydroxide under reflux to give 5-(substituted) 1,3,4-oxadiazole-2-thione. The recrystallization with ethanol afforded pure oxadiazoles.
5-(2,3,4-Trimethoxyphenyl)-1,3,4-oxadiazole-2(3H)-thione (4a) —
Light yellow solid; yield: 74%; mp 132–134°C; Rf: 0.72 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3178, 3032, 2918, 2867, 1568, 1526, 1479; 1H NMR (300 MHz, DMSO-d6) δ 13.23 (s, 1H, NH), 7.34 (d, 1H, J = 7.8 Hz, Ar-H), 7.21 (d, 1H, J = 7.8 Hz, Ar-H), 3.73 (s, 3H, OCH3) 3.65 (s, 3H, OCH3), 3.58 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6) δ 178.25, 163.74, 160.03, 160.20, 159.93, 134.66, 132.43, 127.63, 56.23, 55.56, 55.34; Anal. Calcd for C11H12N2O4S: C, 49.24; H, 4.51; N, 10.44; O, 23.85; S, 11.95; Found: C, 49.23; H, 4.52; N, 10.43; O, 23.86; S, 11.94.
5-(1H-Indol-2-yl)-1,3,4-oxadiazole-2(3H)-thione (4b) —
Brown solid; yield: 76%; mp 122–124°C; Rf: 0.74 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3167, 3029, 2947, 2849, 1581, 1537, 1486; 1H NMR (300 MHz, DMSO-d6) δ 13.23 (s, 1H, NH), 8.71 (s, 1H, NH), 7.62 (m, 1H, Ar-H), 7.47 (m, 1H, Ar-H), 6.92 (s, 1H, Ar-H), 6.80 (m, 2H, Ar-H); 13C NMR (75 MHz, DMSO-d6) δ 178.25, 162.26, 161.28, 152.20, 146.34, 134.66, 132.43, 127.63, 126.36, 124.12; Anal. Calcd for C10H7N3OS: C, 55.29; H, 3.25; N, 19.34; O, 7.36; S, 14.76; Found: C, 55.30; H, 3.26; N, 19.32; O, 7.34; S, 14.77.
5-(4-Bromobenzyl)-1,3,4-oxadiazole-2(3H)-thione (4c) —
White solid; yield: 71%; mp: 126–128°C; Rf: 0.74 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3205, 3087, 2953, 2826, 1569, 1517, 1511, 1495, 1484; 1H NMR (300 MHz, DMSO-d6) δ 14.26 (s, 1H, NH), 7.57–7.46 (m, 2H, Ar-H), 7.23–7.16 (m, 2H, Ar-H), 3.99 (s, 2H, CH2); 13C NMR (75 MHz, DMSO-d6) δ 178.25, 160.23, 136.55, 131.54, 129.32, 127.34, 123.52, 118.37, 30.63; Anal. Calcd for C9H7BrN2OS: C, 39.87; H, 2.60; N, 10.33; S, 11.83; Found: C, 39.80; H, 2.54; N, 10.28; S, 11.77.
5-(4-Methylbenzyl)-1,3,4-oxadiazole-2(3H)-thione (4d) —
Light yellow solid; yield: 74%; mp 125–127°C; Rf: 0.68 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3184, 3045, 2939, 2849, 1593, 1522, 1489; 1H NMR (300 MHz, DMSO-d6) δ 13.25 (s, 1H, NH), 7.18–7.11 (m, 2H, Ar-H), 6.85–6.78 (m, 2H, Ar-H), 4.17 (s, 2H, CH2), 2.51(s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ 178.35, 161.88, 142.03, 132.13, 128.25, 124.76, 123.75, 118.19, 31.47, 26.55; Anal. Calcd for C10H10N2OS: C, 58.23; H, 4.89; N, 13.58; O, 7.76; S, 15.55; Found: C, 58.24; H, 4.89; N, 13.57; O, 7.75; S, 15.56.
5-(3,4-Dichlorobenzyl)-1,3,4-oxadiazole-2(3H)-thione (4e) —
Light yellow solid; yield: 76%; mp: 123–125°C; Rf: 0.73 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3201, 3019, 2936, 2859, 1593, 1536, 1489; 1H NMR (300 MHz, DMSO-d6) δ 14.17 (s, 1H, NH), 7.65–7.56 (m, 1H, Ar-H), 7.24–7.38 (m, 2H, Ar-H), 4.31 (s, 2H, CH2); 13C NMR (75 MHz, DMSO-d6) δ 178.21, 162.21, 149.12, 147.99, 134.65, 132.47, 129.41, 126.37, 27.88; Anal. Calcd for C9H6Cl2N2OS: C, 41.40; H, 2.32; N, 10.73; S, 12.28; Found: C, 41.30; H, 2.25; N, 10.65; S, 12.19.
5-(2-Fluorobenzyl)-1,3,4-oxadiazole-2(3H)-thione (4f) —
Dull white solid; yield: 69%; mp: 131–133°C; Rf: 0.73 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3191, 3057, 2915, 2881, 1590, 1521, 1502, 1485; 1H NMR (300 MHz, DMSO-d6) δ 14.34 (s, 1H, NH), 7.48–7.35 (m, 2H, Ar-H), 7.29–7.18 (m, 2H, Ar-H), 4.18 (s, 2H, CH2); 13C NMR (75 MHz, DMSO-d6) δ 178.29, 162.52, 159.98, 132.18, 130.50, 125.27, 120.93, 116.13, 25.50; Anal. Calcd for C9H7FN2OS: C, 51.42; H, 3.36; N, 13.33; S, 15.25; Found: C, 51.34; H, 3.28; N, 13.23; S, 15.17.
5-(4-Methoxyphenethyl)-1,3,4-oxadiazole-2(3H)-thione (4 g) —
Light yellow solid; yield: 89%; mp 76–78°C; Rf: 0.78 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3233, 3072, 2937, 2812, 1583, 1509, 1483; 1H NMR (300 MHz, DMSO-d6) δ 14.31 (s, 1H, NH), 7.28–7.15 (m, 2H, Ar-H), 6.88–6.72 (m, 2H, Ar-H), 2.91 (t, 2H, J = 7.8 Hz, CH2), 2.88 (t, 2H, J = 7.8 Hz, CH2), 3.72 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6) δ 178.14, 163.98, 159.91, 140.47, 130.92, 127.94, 121.40, 118.36, 55.66, 31.24, 26.89; Anal. Calcd for C11H12N2O2S9: C, 55.91; H, 5.12; N, 11.86; S, 13.57; Found: C, 55.86; H, 5.07; N, 11.77; S, 13.47.
5-(2,4-Dichlorobenzyl)-1,3,4-oxadiazole-2(3H)-thione (4h) —
White solid; yield: 70%; mp 119–121°C; Rf: 0.72 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3214, 3014, 2915, 2839, 1569, 1521,1482; 1H NMR (300 MHz, DMSO-d6) δ 14.19 (s, 1H, NH), 7.73–7.63 (m, 1H, Ar-H), 7.26–7.17 (m, 2H, Ar-H), 4.29 (s, 2H, CH2); 13C NMR (75 MHz, DMSO-d6) δ 178.33, 161.32, 147.76, 147.19, 136.45, 133.25, 129.41, 126.37, 27.88; Anal. Calcd for C9H6Cl2N2OS: C, 41.40; H, 2.32; N, 10.73; S, 12.28; Found: C, 41.32; H, 2.21; N, 10.65; S, 12.19.
5-(2-Methoxyphenethyl)-1,3,4-oxadiazole-2(3H)-thione (4i) —
Yellow solid; yield: 71%; mp 85–87°C; Rf: 0.78 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3195, 3052, 2936, 2845, 1592, 1522, 1485; 1H NMR (300 MHz, DMSO-d6) δ 14.36 (s, 1H, NH), 7.26–7.12 (m, 2H, Ar-H), 7.01–6.92 (m, 1H, Ar-H), 6.90–6.85 (m, 1H, Ar-H), 2.94 (t, 2H, J = 7.8 Hz, CH2), 2.49 (t, 2H, J = 7.8 Hz, CH2), 3.75 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6) δ 178.19, 162.79, 159.63, 141.43, 130.91, 122.34, 119.48, 114.96, 55.51, 29.36, 26.89; Anal. Calcd for C11H12N2O2S9: C, 55.91; H, 5.12; N, 11.86; S, 13.57; Found: C, 55.78; H, 5.02; N, 11.86; S, 13.57.
5-(4-Chlorobenzyl)-1,3,4-oxadiazole-2(3H)-thione (4j) —
Yellow solid; yield: 69%; mp 112–114°C; Rf: 0.70 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3223, 3049, 2915, 2856, 1582, 1537, 1498; 1H NMR (300 MHz, DMSO-d6) δ 14.31 (s, 1H, NH), 7.25–7.18 (m, 2H, Ar-H), 7.09–6.95 (m, 2H, Ar-H), 4.21 (s, 2H, CH2); 13C NMR (75 MHz, DMSO-d6) δ 178.37, 162.19, 158.87, 133.55, 132.43, 130.07,126.97, 120.12, 31.41; Anal. Calcd for C9H7ClN2OS: C, 47.69; H, 3.11; Cl, 15.64; N, 12.36; O, 7.06; S, 14.15; Found: C, 47.69; H, 3.11; Cl, 15.64; N, 12.36; O, 7.06; S, 14.15.
5-(2-Chlorobenzyl)-1,3,4-oxadiazole-2(3H)-thione (4k) —
Pink solid; yield: 68%; mp 112–114°C; Rf: 0.71 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3182, 3035, 2952, 2856, 1569, 1488, 1466, 1437; 1H NMR (300 MHz, DMSO-d6) δ 14.44 (s, 1H, NH), 7.59–7.46 (m, 2H, Ar-H), 7.42–7.38 (m, 2H, Ar-H), 4.26 (s, 2H, CH2); 13C NMR (75 MHz, DMSO-d6) δ 178.26, 151.24, 133.85, 132.36, 131.65, 130.18, 128.13, 124.12, 29.87; Anal. Calcd for C9H7ClN2OS: C, 39.87; H, 2.60; N, 10.33; S, 11.83; Found: C, 39.79; H, 2.47; N, 10.25; S, 11.65.
5-(4-Methoxybenzyl)-1,3,4-oxadiazole-2(3H)-thione (4l) —
Light yellow solid; yield: 70%; mp 107–109°C; Rf: 0.72 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3181, 3011, 2914, 2876, 1575, 1508, 1488; 1H NMR (300 MHz, DMSO-d6) δ 13.29 (s, 1H, NH), 7.29–7.19 (m, 2H, Ar-H), 6.97–6.86 (m, 2H, Ar-H), 4.05 (s, 2H, CH2), 3.73 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6) δ 178.25, 163.74, 160.03, 130.64, 127.05, 125.66, 120.43, 114.63, 55.56, 30.64; Anal. Calcd for C10H10N2O2S: C, 54.04; H, 4.53; N, 12.60; S, 14.43; Found: C, 54.01; H, 4.41; N, 12.45; S, 14.35.
5-(2-Methoxybenzyl)-1,3,4-oxadiazole-2(3H)-thione (4m) —
White solid; yield: 62%; mp 112–114°C; Rf: 0.72 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3199, 3014, 2936, 2871, 1574, 1512, 1489, 1480; 1H NMR (300 MHz, DMSO-d6) δ 14.34 (s, 1H, NH), 7.38–7.23 (m, 2H, Ar-H), 7.08–7.09 (m, 1H, Ar-H), 6.99-6.91 (m, 1H, Ar-H), 4.04 (s, 2H, CH2), 3.77 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6) δ 178.16, 163.33, 157.52, 131.11, 129.68, 121.72, 121.00, 111.64, 56.02, 26.56; Anal. Calcd for C10H10N2O2S: C, 54.04; H, 4.53; N, 12.60; S, 14.43; Found: C, 53.92; H, 4.42; N, 12.43; S, 14.23.
5-(3-Methoxybenzyl)-1,3,4-oxadiazole-2(3H)-thione (4n) —
Light pink solid; yield: 75%; mp 93–95°C; Rf: 0.69 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3202, 3002, 2935, 2839, 1568, 1531, 1489, 1473; 1H NMR (300 MHz, DMSO-d6) δ 14.43 (s, 1H, NH), 7.33–7.24 (m, 1H, Ar-H), 6.93–6.84 (m, 3H, Ar-H), 4.10 (s, 2H, CH2), 3.74 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6) δ 178.30, 163.32, 159.92, 135.32, 130.32, 121.61, 115.31, 113.29, 55.53, 31.45; Anal. Calcd for C10H10N2O2S: C, 54.04; H, 4.53; N, 12.60; S, 14.43; Found: C, 53.94; H, 4.44; N, 12.52; S, 14.22.
5-(3-Methoxyphenethyl)-1,3,4-oxadiazole-2(3H)-thione (4o) —
Dull white solid; yield: 77%; mp 78–80°C; Rf: 0.76 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3190, 3071, 2979, 2836, 1581, 1508, 1491; 1H NMR (300 MHz, DMSO-d6) δ 14.29 (s, 1H, NH), 7.21–7.11 (m, 1H, Ar-H), 6.92–6.66 (m, 3H, Ar-H), 2.86 (t, 2H, J = 7.6 Hz, CH2), 2.77 (t, 2H, J = 7.6 Hz, CH2), 3.78 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6) δ 178.19, 163.99, 160.17, 141.46, 130.91, 122.34, 119.48, 114.96, 55.51, 29.36, 26.12; Anal. Calcd for C11H12N2O2S9: C, 55.91; H, 5.12; N, 11.86; S, 13.57; Found: C, 55.43; H, 5.01; N, 11.80; S, 13.49.
5-(Pyridin-3-yl)-1,3,4-oxadiazole-2(3H)-thione (4p) —
Light yellow solid; yield: 766%; mp 94–96°C; Rf: 0.73 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3218, 3079, 2958, 2847, 1549, 1526, 1492, 1488; 1H NMR (300 MHz, DMSO-d6) δ 14.17 (s, 1H, NH), 8.98 (s, 1H, Ar-H), 8.40 (m, 1H, Ar-H), 8.27 (m, Ar-H), 7.54 (m, 1H, Ar-H); 13C NMR (75 MHz, DMSO-d6) δ 178.32, 161.12, 154.56, 152.13, 134.56, 130.32, 121.31; Anal. Calcd for C7H5N3OS: C, 46.92; H, 2.81; N, 23.45; O, 8.93; S, 17.89; Found: C, 46.93; H, 2.80; N, 23.47; O, 8.91; S, 17.5.
5-(2,6-Dichlorobenzyl)-1,3,4-oxadiazole-2(3H)-thione (4q) —
White solid; yield: 74%; mp 127–129°C; Rf: 0.73 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3235, 3041, 2951, 2874, 1582, 1545, 1498; 1H NMR (300 MHz, DMSO-d6) δ 14.21 (s, 1H, NH), 7.65–7.57 (m, 2H, Ar-H), 7.31–7.26 (m, 1H, Ar-H), 4.34 (s, 2H, CH2); 13C NMR (75 MHz, DMSO-d6) δ 178.37, 160.28, 158.17, 157.26, 135.48, 131.41, 129.92, 123.21, 30.24; Anal. Calcd for C9H6Cl2N2OS: C, 41.40; H, 2.32; N, 10.73; S, 12.28; Found: C, 41.32; H, 2.21; N, 10.65; S, 12.19.
5-(4-Chloro-2-fluorophenyl)-1,3,4-oxadiazole-2(3H)-thione (4r) —
White solid; yield: 69%; mp 98–100°C; Rf: 0.75 (chloroform : methanol, 9 : 1); IR (ν/cm−1): 3199, 3042, 2939, 2832, 1572, 1512, 1488, 1476; 1H NMR (300 MHz, DMSO-d6) δ 14.21 (s, 1H, NH), 7.72–7.67 (m, 1H, Ar-H), 7.42–7.38(m, 2H, Ar-H); 13C NMR (75 MHz, DMSO-d6) δ 178.31, 162.22, 150.01, 136.29, 133.41, 131.65, 130.18, 119.21; Anal. Calcd for C8H4ClFN2OS: C, 39.87; H, 2.60; N, 10.33; S, 11.83; Found: C, 39.78; H, 2.55; N, 10.27; S, 11.67.
2.2. Urease Assay and Inhibition
Indophenols method was used for the quantification of ammonia and the enzyme activity was determined by measuring its absorbance [25]. In brief, 40 μL buffer (100 mM urea, 1 mM EDTA, 0.01 M K2HPO4, 0.01 M LiCl2, pH 8.2), 10 μL of test compound and 10 μL of enzyme (5 U/mL) were incubated in a 96 well plate for 10 minutes at 37°C. In addition, solutions of 40 μL of phenol reagent (1%, w/v phenol, 0.005%, w/v sodium nitroprusside) and 40 μL of alkali reagent (0.5%, w/v NaOH, 0.1% active chloride NaOCl) were introduced to each well. Experiments were performed in a triplicate fashion and thiourea was used as standard inhibitor. Microplate reader (Bio-TekELx 800, Instruments, Inc., USA) was used to read the absorbance at 625 nm. The percentage inhibition was calculated using the following equation 100 − (ODtest well/ODcontrol) × 100. The results were calculated using PRISM 5.0 (GraphPad, San Diego, CA, USA).
2.3. Measurement of Antibacterial Activity
The antibacterial activity data is represented in Table 2. Some derivatives of 1,3,4-oxadiazole have shown even more potency than the standard drug ciprofloxacin while some of them have comparable potency against different strains. Ciprofloxacin was used as standard drug. The compounds were found active against bacterial strains (2 Gram positive bacteria, namely, Staphylococcus aureus, Bacillus subtilis, and 2 Gram negative bacteria namely Escherichia coli, Shigella flexneri) in variable concentration. The antibacterial activities of 1,3,4-oxadiazole derivatives were evaluated in vitro by serial tube dilution method. The compounds and standard drug ciprofloxacin were dissolved in N,N-dimethylformamide (DMF) to give a concentration of 5 μg/mL (stock solution). Necessary apparatus and test tubes set of capacity 5 mL was washed cleaned and completely dried. For the bacterial culture double strength nutrient broth was used. The culture media was made by dissolving 15 g of nutrient broth in 1 L of distilled water. Approximately, 1 mL of culture media was prepared and transferred to each test tube by micropipette and capped with non-adsorbent cotton plugs. The test tubes containing 1 mL culture media was sterilized in an autoclave at 121°C for 20 min at 15 psi pressure. Sub-culturing of bacteria was done by transferring a loopful of particular bacterial strain from standard bacterial agar to 10 mL sterilized nutrient broth aseptically in a laminar air flow cabinet. It was then incubated for a period of 24 h at 37°C in a B. O. D. incubator. Bacterial strain suspension was prepared, after 24 h incubation, by aseptically inoculating 0.2 mL of revived bacterial colony into 100 mL of 0.9% m/v saline. A series of 5 assay tubes for a single derivative against each bacterial strain was employed. A stock solution of each test derivative at concentration 5 μg/mL (containing 1 mL nutrient broth) was serially diluted to achieve concentrations of 2.5, 1.25, 0.625, 0.313, and 0.156 μg/mL. Then, 0.1 mL of normal saline suspension of revived bacteria was added to each test tube. The inoculated tubes were incubated at 37°C for 24 h. The MIC (minimum inhibitory concentration) values were determined by subsequently checking for the absence of visual turbidity.
Table 2.
Antibacterial activity of 1,3,4-oxadiazoles derivatives by micro dilution method (MICa values μg/mL).
| Compound | E. coli | B. subtilus | S. aureus | S. flexneri |
|---|---|---|---|---|
| 4a | 0.313 | 0.156 | 1.25 | 0.313 |
| 4b | 0.625 | 0.313 | 0.156 | 0.313 |
| 4c | 2.50 | 0.156 | 0.625 | 0.156 |
| 4e | 0.156 | 0.625 | 0.313 | 0.313 |
| 4f | 0.313 | 0.156 | 0.156 | 1.25 |
| 4g | 0.156 | 0.625 | 2.50 | 0.313 |
| 4h | 1.25 | 0.156 | 0.625 | 0.156 |
| 4i | 0.156 | 0.625 | 0.313 | 0.313 |
| 4j | 0.313 | 1.25 | 0.313 | 0.156 |
| 4k | 0.156 | 0.313 | 0.156 | 0.156 |
| 4l | 0.313 | 2.50 | 0.625 | 0.156 |
| 4m | 0.625 | 0.156 | 1.25 | 0.313 |
| 4n | 0.625 | 0.156 | 0.156 | 0.156 |
| 4o | 0.313 | 0.625 | 2.50 | 0.156 |
| 4r | 1.25 | 0.313 | 0.156 | 0.313 |
| Ciprofloxacin | 0.156 | 0.625 | 0.156 | 0.31 |
aValues are the average of three reading.
2.4. Measurement of Antioxidant Activity
The free radical scavenging capacity of the compounds was measured by 1,1-diphenyl-2-picrylhydrazyl. Test compounds were allowed to react with stable free radical, 1,1-diphenyl-2-picrylhydrazyl radical (DPPH) for half an hour at 37°C. The concentration of DPPH was kept as 300 μM. The test samples were dissolved in DMSO while the DPPH solution was prepared in ethanol. After incubation, decrease in absorption was measured at 515 nm using microplate reader. Percent radical scavenging activity of samples was determined in comparison with a DMSO treated as control. Propyl gallate and 3-tert-Butyl-4-hydroxyanisole were used as standards.
2.5. Enzyme and Compounds Preparation for Docking
Docking study was performed by using the available crystal structure of Jack bean urease from Protein Data Bank (PDB code: 3LA4). It is very crucial to carefully prepare the protein and small molecule structures before using them in the docking calculations. The enzyme structure was prepared using MOE program. Protonation was performed using the Protonate3D algorithm implemented in MOE. Force-field-based parameterization and energy minimization was carried out by choosing Amber99 force-field library. Correct protonation and metal atoms states for the active site histidine residues and two Ni+2 ions were assigned. By using Protonate3D algorithm, the four histidine residues surrounding the Ni+2 ions in the active site pocket of the enzyme were protonated according to the bound state of the two Ni+2 ions. Wrong protonation states of the active site histidine residues can lead to a drastic effect on the binding modes of the ligands during docking. After protonation and force-field-based parameterization setup, the enzyme structure was energy minimized. During energy minimization, the protein heavy atoms were restrained to avoid changes in the active site pocket and to allow only the relaxation in protein side chains and added hydrogens. After energy minimization, the cocrystallized bound compounds and water molecules were stripped off from the crystal structure.
Similarly, the compounds structures were also prepared before carrying out docking calculations. It is also very important to prepare the correct protonation and ionization states of the small molecule structures. 3D conformations were generated for the compounds followed by energy minimization by choosing MMFF94x force-field and using the “wash” module in MOE.
2.6. Docking Calculations
The docking calculations were carried out using FlexX [26] program. The enzyme's active site pocket was defined by setting 10 Å spacing around the cocrystallized bound PO4. The nonstandard protein residues and single metal ions were included in the binding site definition. The two Ni+2 metal atoms were selected as metal pharmacophores. The geometry parameters for the two metal atoms were set up as automated spheres. The default docking and scoring parameters were used for docking calculations and the top 10 best conformations that fulfilled the metal pharmacophore criteria were retained for further analysis.
3. Results and Discussion
3.1. General Procedure for Synthesis
Synthesis for target compounds, 1,3,4-oxadiazole 4(a–r) is illustrated in Scheme 1. Substituted aromatic esters 2(a–r) were synthesized by the reaction of corresponding substituted aromatic acids 1(a–r) in the presence of catalytic amount of sulfuric acid, the esters 2(a–r) were converted into corresponding aromatic acid hydrazides 3(a–r) by refluxing with hydrazine hydrate (80%) in methanol. Treatment of the aromatic acid hydrazides 3(a–r) with carbon disulfide in the presence of potassium hydroxide and ethanol under reflux afford corresponding oxadiazole 4(a–r).
Scheme 1.
Synthesis of oxadiazoles (4a–r). Reagents and conditions: (a) H2SO4 (conc.), methanol, reflux, 8–12 h; (b) NH2NH2·H2O (80%), ethanol, reflux, 8–12 h; (c) (1) CS2/KOH, ethanol, reflux, 12 h; (2) HCl pH 5-6.
3.2. Urease Inhibition Assay
The synthesized compounds were tested for their in vitro urease inhibition against Jack bean urease. Thiourea was used as a standard inhibitor in assay having IC50 value of 22.3 ± 12 μM. Most of the compounds of this series showed promising urease inhibitory potency. The results indicated that ortho and para substituted benzene in the vicinity of parent oxadiazole core is important to obtain the potent activity. It is observed from the results that methoxy substituted benzene next to oxadiazole ring and halo-substituted compounds showed excellent urease inhibition.
Potent compounds have their activities in the range of 1.15 μM to 42.42 μM (Table 1). Among investigated compounds, 4j bearing a 4-chlorobenzyl ring, was found to be the most active urease inhibitor with an IC50 value of 1.15 ± 0.2 μM. Compounds 4a bearing methoxy group at 2,3,4 positions and 4g, 4i, 4l, 4m also showed strong inhibitory activities in the range of 5.6 μM to 6.22 μM. These compounds bear electron donating methoxy groups at o- and p-positions and were more active than compounds 4n and 4o having electron donating groups at m-position. Compound 4c having bromo substitution at para position and compound 4e having chloro substitution at meta and para position showed slightly less activity than methoxy substituted 1,3,4-oxadiazoles. Compound 4b and standard thiourea had nearly same activity against urease. The exceptions were compounds 4h and 4r having 2,4-dichlorobenzyl group exhibited weak inhibitory activities.
Table 1.
Inhibitory activity of 1,3,4-oxadiazoles derivatives against Jack bean urease.
| Compound | IC50 ± SEM (μM) (or % inhibition) |
|---|---|
| 4a | 5.79 ± 0.3 |
| 4b | 21.3 ± 0.7 |
| 4c | 11.8 ± 0.4 |
| 4e | 11.3 ± 0.6 |
| 4f | 3.27 ± 0.3 |
| 4g | 5.61 ± 0.3 |
| 4h | 42.4 ± 1.2 |
| 4i | 6.22 ± 0.4 |
| 4j | 1.15 ± 0.2 |
| 4k | 12.9 ± 0.6 |
| 4l | 5.83 ± 0.08 |
| 4m | 5.60 ± 0.6 |
| 4n | 12.2 ± 0.05 |
| 4o | 15.1 ± 0.8 |
| 4r | (39)a |
| Thiourea (standard) | 22.3 ± 1.2 |
a% age inhibition was evaluated using inhibitor at a concentration of 100 μM.
It is clear from the SAR of synthesized derivatives of 1,3,4-oxadiazoles that 2 and 4 positions of terminal benzene ring is favorable site for high activity. However, a single parameter is insufficient to explain the pattern and mechanism by which 1,3,4-oxadiazoles exhibited the urease inhibition.
3.3. Antibacterial Activity
The synthesized derivatives were screened for antibacterial activity against Gram-positive bacteria (Staphylococcus aureus, Bacillus subtilis) and Gram-negative bacteria (Escherichia coli, and Shigella flexneri). The results of antibacterial activity of 1,3,4-oxadiazoles are presented in Table 2. Ciprofloxacin was used as standard and the minimum inhibitory concentrations (MICs) were determined in vitro by using serial tube dilution method.
Some of the synthesized derivatives were found to have more potent antibacterial activity then standard ciprofloxacin against the tested strains. In particular, derivatives 4e, 4 g, 4i, 4k, and 4r which possess a 3,4-dichlorobenzyl, 4-methoxyphenethyl, 2,4-dichlorobenzyl, 2-methoxy, 2-chloro and 2-F, 4-Cl phenyl groups respectively, on the 1,3,4-oxadiazole ring, were found to have potent activities (MIC: 0.156 μg/mL) against E. coli and were equipotent in vitro as standard drug ciprofloxacin (MIC: 0.156 μg/mL). Compounds 4c, 4 h, and 4r were considerably less active. The compounds demonstrated significant antibacterial activity against B. subtilus. The derivatives 4a, 4c, 4f, 4h, 4m, and 4n exhibited 4-fold activity. The exceptions were 4j and 4l with para chloro and para methoxy group, respectively. For bacterial strain S. Aureus, compounds 4b, 4f, 4k, 4n, and 4r showed equipotent activity as compared to standard ciprofloxacin. These compounds were obtained by the substitution of halogen at various positions of benzene ring. Compounds 4c, 4h, 4j, 4k, 4l, 4n, and 4o showed two- fold activities against S. flexneri strain. Compounds 4a, 4b, 4e, 4g, 4i, 4m, and 4r showed equipotent activity. However, 4f showed no activity as it has floro group at ortho position. The high potency of discussed analogues may be attributed to the F, Cl and OCH3 at 2- or 4-positions which rationally correlates with SAR of urease inhibition activity.
3.4. Antioxidant Studies
The 2,2-diphenyl-1-picryl-hydrazyl (DPPH) radical scavenging activity assessment is a standard assay in antioxidant activity measurements. For comparison purpose, the well-defined antioxidant propyl gallate and 3-tert-Butyl-4-hydroxyanisole were used in assay as positive control. The antioxidant activities of the compounds are shown in Table 3.
Table 3.
DPPH radical scavenging activity of 1,3,4-oxadiazoles derivatives.
| Compounds | IC50 (μM) |
|---|---|
| 4 a | 122.6 ± 5.1 |
| 4b | 107.2 ± 3.4 |
| 4c | 37.98 ± 4.1 |
| 4e | 58.97 ± 6.3 |
| 4f | 46.63 ± 4.6 |
| 4g | 50.78 ± 2.1 |
| 4h | 57.82 ± 1.8 |
| 4i | 40.20 ± 3.3 |
| 4j | 51.61 ± 2.1 |
| 4k | 34.40 ± 3.2 |
| 4l | 88.18 ± 4.1 |
| 4m | 42.74 ± 3.7 |
| 4n | 60.28 ± 4.3 |
| 4o | 43.59 ± 3.1 |
| 4r | 10.83 ± 0.2 |
| Propyl gallate | 40.80 ± 1.2 |
| 3-tert-Butyl-4-hydroxyanisole | 28.20 ± 1.1 |
The most interesting activity was observed in 4r having 2-F, 4-Cl phenyl groups which showed four-fold DPPH radical scavenging activity as compared to standard propyl gallate. Two other derivatives 4k and 4c were also more effective than the propyl gallate having 2-chloro and 4-bromo substitutions on benzene ring next to parent core. Equipotent radical scavenging activity was found in 4i having 2-methoxy phenyl group. Among methoxy substituted phenyl rings, the derivatives 4m, 4o, 4g, and 4n exhibited good potentials having IC50 values 42.74 μM to 60.28 μM. Similarly, 4f, 4j, 4k, and 4e which have halogen substituted phenyl rings showed potency in the range of 46.63 μM to 58.97 μM. 4a having trimethoxy phenyl ring and 4b were relatively less active derivatives of the series.
3.5. Molecular Docking Studies
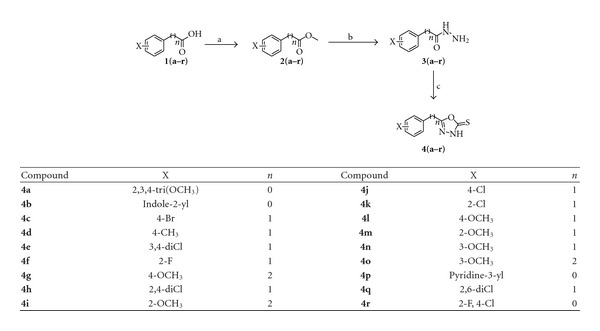
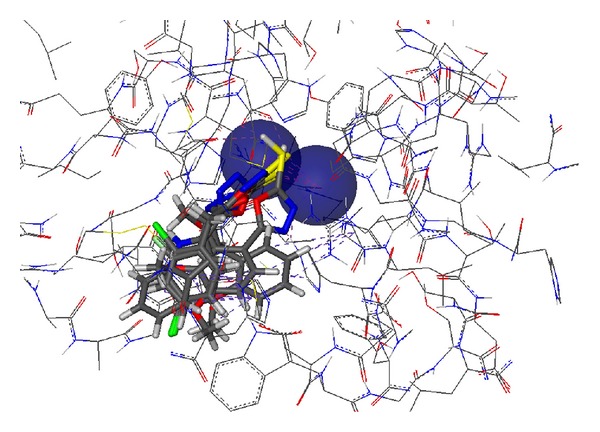
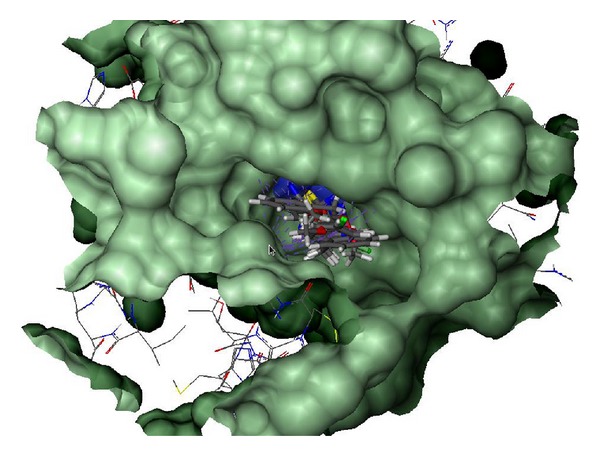
The compounds were studied by docking them into the crystal structure of Jack bean urease to observe the common behavior of interaction of these compounds with the enzyme. The top 18 predicted docked solutions (conformations) generated by FlexX were retained for analyzing the binding modes of the compounds. It was observed that all of the compounds have a similar binding mode in the first out of 18 ranked predictions. The docking scores varied from −11.09 to −26.84, which are given in Table 4. The docking results showed that all of these compounds interact with the bi-nickel center of the enzyme. The S group of the compounds tends to interact with the two nickel atoms (Figure 1) while the aromatic moieties of the compounds adopt flexible conformations in the large hydrophobic opening of the active site pocket (Figure 2). With a special pharmacophore module, FlexX-Pharm [27], FlexX offers an automated pharmacophore query building for the metal atoms. The two Nickel atoms were chosen to be used as essential part of the pharmacophoric constraints for filtering the predicted docking solutions. It was observed that all compounds bind in the same mode fulfilling that metal pharmcophoric constraints. The aromatic rings of the compounds make similar stack of interactions with HIS492, HIS593, ARG439, and ALA440 residues which form a hydrophobic cavity in the opening of the active site pocket and allow greater flexibility to the compounds to adopt different conformations in that area. The aromatic interactions with histidine ring of HIS593 on one side and ARG439 and ALA440 on other side in the hydrophobic pocket is common to the majority of predicted binding poses of the compounds. The docked conformations of the compounds 4a and 4b are shown in Figures 3 and 4, respectively.
Table 4.
FlexX docking scores and ranks for the docked compounds.
| Compound | Rank | Score |
|---|---|---|
| 4 a | 1 | −14.09 |
| 4b | 1 | −19.35 |
| 4c | 1 | −16.55 |
| 4d | 1 | −11.09 |
| 4e | 1 | −13.05 |
| 4f | 1 | −16.96 |
| 4g | 1 | −20.95 |
| 4h | 1 | −14.64 |
| 4i | 1 | −12.46 |
| 4j | 1 | −14.63 |
| 4k | 1 | −13.03 |
| 4l | 3 | −23.49 |
| 4m | 1 | −13.61 |
| 4n | 1 | −26.26 |
| 4o | 1 | −18.46 |
| 4p | 1 | −13.35 |
| 4q | 1 | −13.35 |
| 4r | 1 | −26.84 |
Figure 1.
Predicted conformations of the docked compounds inside the binding pocket of Jack bean urease. The large blue spheres indicate the metal pharmacophores around the two nickels (Ni+2) which shows that the metal atoms can interact in all directions. The dotted lines indicate various types of interactions of the compounds atoms with the active site residues including hydrogen bonding and aromatic interactions.
Figure 2.
Surface representation of the active site pocket of the Jack bean urease with the bound ligands shown inside the pocket in CPK model. The wide opening of the binding site pocket allows the compounds to adopt flexible conformations in this area.
Figure 3.
Interaction diagram of the docked conformation of compound 4a with the active site residues of the enzyme. The dotted lines show the interactions between the compound and residues atoms.
Figure 4.
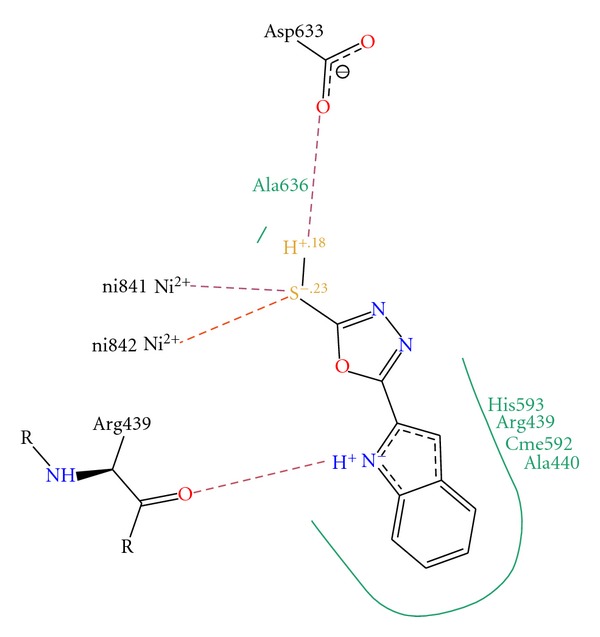
Interaction diagram of the docked conformation of compound 4b with the active site residues of the enzyme. The dotted lines show the interactions between the compound and residues atoms.
Acknowledgment
This work was financially supported by the Higher Education Commission (HEC), Pakistan, under the National Research Support Program for Universities and German-Pakistani Research Collaboration Program to J. Iqbal.
References
- 1.Mobley HLT, Hausinger RP. Microbial ureases: significance, regulation, and molecular characterization. Microbiological Reviews. 1989;53(1):85–108. doi: 10.1128/mr.53.1.85-108.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mobley HLT, Island MD, Hausinger RP. Molecular biology of microbial ureases. Microbiological Reviews. 1995;59(3):451–480. doi: 10.1128/mr.59.3.451-480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vassiliou S, Grabowiecka A, Kosikowska P, Yiotakis A, Kafarski P, Berlicki L. Design, synthesis, and evaluation of novel organophosphorus inhibitors of bacterial ureases. Journal of Medicinal Chemistry. 2008;51(18):5736–5744. doi: 10.1021/jm800570q. [DOI] [PubMed] [Google Scholar]
- 4.Ha NC, Oh ST, Sung JY, et al. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nature Structural & Molecular Biology. 2001;8(6):505–509. doi: 10.1038/88563. [DOI] [PubMed] [Google Scholar]
- 5.Jabri E, Carr MB, Hausinger RP, Karplus PA. The crystal structure of urease from Klebsiella aerogenes. Science. 1995;268(5213):998–1004. [PubMed] [Google Scholar]
- 6.Benini S, Rypniewski WR, Wilson KS, Miletti S, Ciurli S, Mangani S. A new proposal for urease mechanism based on the crystal structures of the native and inhibited enzyme from Bacillus pasteurii: why urea hydrolysis costs two nickels. Structure. 1999;7(2):205–216. doi: 10.1016/S0969-2126(99)80026-4. [DOI] [PubMed] [Google Scholar]
- 7.Gripenberg-Lerche C, Zhang L, Ahtonen P, Toivanen P, Skurnik M. Construction of urease-negative mutants of Yersinia enterocolitica serotypes O:3 and O:8: role of urease in virulence and arthritogenicity. Infection and Immunity. 2000;68(2):942–947. doi: 10.1128/iai.68.2.942-947.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Mobley HLT. Vaccines for Proteus mirabilis in urinary tract infection. International Journal of Antimicrobial Agents. 2002;19(6):461–465. doi: 10.1016/s0924-8579(02)00102-4. [DOI] [PubMed] [Google Scholar]
- 9.Amtul Z, Rahman AU, Siddiqui RA, Choudhary MI. Chemistry and mechanism of urease inhibition. Current Medicinal Chemistry. 2002;9(14):1323–1348. doi: 10.2174/0929867023369853. [DOI] [PubMed] [Google Scholar]
- 10.Uesato S, Hashimoto Y, Nishino M, Nagaoka Y, Kuwajima H. N-substituted hydroxyureas as urease inhibitors. Chemical and Pharmaceutical Bulletin. 2002;50(9):1280–1282. doi: 10.1248/cpb.50.1280. [DOI] [PubMed] [Google Scholar]
- 11.Odake S, Morikawa T, Tsuchiya M, Imamura L, Kobashi K. Inhibition of Helicobacter pylori urease activity by hydroxamic acid derivatives. Biological and Pharmaceutical Bulletin. 1994;17(10):1329–1332. doi: 10.1248/bpb.17.1329. [DOI] [PubMed] [Google Scholar]
- 12.Faraci WS, Yang BV, O’Rourke D, Spencer RW. Inhibition of Helicobacter pylori urease by phenyl phosphorodiamidates: mechanism of action. Bioorganic and Medicinal Chemistry. 1995;3(5):605–610. doi: 10.1016/0968-0896(95)00043-g. [DOI] [PubMed] [Google Scholar]
- 13.Pope AJ, Toseland CDN, Rushant B, Richardson S, Mcvey M, Hills J. Effect of potent urease inhibitor, fluorofamide, on Helicobacter sp. in vivo and in vitro. Digestive Diseases and Sciences. 1998;43(1):109–119. doi: 10.1023/a:1018884322973. [DOI] [PubMed] [Google Scholar]
- 14.Park JB, Imamura L, Kobashi K. Kinectic studies of Helicobacter pylori urease inhibition by a novel proton pump inhibitor, rabeprazole. Biological and Pharmaceutical Bulletin. 1996;19(2):182–187. doi: 10.1248/bpb.19.182. [DOI] [PubMed] [Google Scholar]
- 15.Nagata K, Satoh H, Iwahi T, Shimoyama T, Tamura T. Potent inhibitory action of the gastric proton pump inhibitor lansoprazole against urease activity of Helicobacter pylori: unique action selective for H. pylori cells. Antimicrobial Agents and Chemotherapy. 1993;37(4):769–774. doi: 10.1128/aac.37.4.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuhler TC, Fryklund J, Bergman NA, Weilitz J, Lee A, Larsson H. Structure-activity relationship of omeprazole and analogues as Helicobacter pylori urease inhibitors. Journal of Medicinal Chemistry. 1995;38(25):4906–4916. doi: 10.1021/jm00025a008. [DOI] [PubMed] [Google Scholar]
- 17.Bundy LG, Bremner JM. Effects of substituted p-benzoquinones on urease activity in soils. Soil Biology and Biochemistry. 1973;5(6):847–853. [Google Scholar]
- 18.Todd MJ, Hausinger RP. Competitive inhibitors of Klebsiella aerogenes urease. Mechanisms of interaction with the nickel active site. The Journal of Biological Chemistry. 1989;264(27):15835–15842. [PubMed] [Google Scholar]
- 19.Kogen H, Tago K, Arai M, Minami E, Masuda K, Akiyama T. A highly stereoselective synthesis of plaunotol and its thiourea derivatives as potent antibacterial agents against Helicobacter pylori. Bioorganic and Medicinal Chemistry Letters. 1999;9(10):1347–1350. doi: 10.1016/s0960-894x(99)00203-6. [DOI] [PubMed] [Google Scholar]
- 20.Aslam MA, Mahmood SU, Shahid M, et al. Synthesis, biological assay in vitro and molecular docking studies of new Schiff base derivatives as potential urease inhibitors. European Journal of Medicinal Chemistry. 2011;46(11):5473–5479. doi: 10.1016/j.ejmech.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 21.Abid O-U-R, Babar TM, Ali FI, et al. Identification of novel urease inhibitors by high-throughput virtual and in vitro screening. ACS Medicinal Chemistry Letters. 2010;1(4):145–149. doi: 10.1021/ml100068u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moise M, Sunel V, Profire L, Popa M, Desbrieres J, Peptu C. Synthesis and biological activity of some new 1,3,4-thiadiazole and 1,2,4-triazole compounds containing a phenylalanine moiety. Molecules. 2009;14(7):2621–2631. doi: 10.3390/molecules14072621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi W, Qian X, Zhang R, Song G. Synthesis and quantitative structure-activity relationships of new 2,5-disubstituted-1,3,4-oxadiazoles. Journal of Agricultural and Food Chemistry. 2001;49(1):124–130. doi: 10.1021/jf0007941. [DOI] [PubMed] [Google Scholar]
- 24.Amir M, Shikha K. Synthesis and anti-inflammatory, analgesic, ulcerogenic and lipid peroxidation activities of some new 2-[(2,6-dichloroanilino) phenyl]acetic acid derivatives. European Journal of Medicinal Chemistry. 2004;39(6):535–545. doi: 10.1016/j.ejmech.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Weatherfourn MW. PhenoI-hypochlorite reaction for determination of ammonia. Analytical Chemistry. 1967;39(8):971–974. [Google Scholar]
- 26.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. Journal of Molecular Biology. 1996;261(3):470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 27.Hindle SA, Rarey M, Buning C, Lengauer T. Flexible docking under pharmacophore type constraints. Journal of Computer-Aided Molecular Design. 2002;16(2):129–149. doi: 10.1023/a:1016399411208. [DOI] [PubMed] [Google Scholar]