

Abstract
Objectives:
While plasma biomarkers have been proposed to aid in the clinical diagnosis of Alzheimer disease (AD), few biomarkers have been validated in independent patient cohorts. Here we aim to determine plasma biomarkers associated with AD in 2 independent cohorts and validate the findings in the multicenter Alzheimer's Disease Neuroimaging Initiative (ADNI).
Methods:
Using a targeted proteomic approach, we measured levels of 190 plasma proteins and peptides in 600 participants from 2 independent centers (University of Pennsylvania, Philadelphia; Washington University, St. Louis, MO), and identified 17 analytes associated with the diagnosis of very mild dementia/mild cognitive impairment (MCI) or AD. Four analytes (apoE, B-type natriuretic peptide, C-reactive protein, pancreatic polypeptide) were also found to be altered in clinical MCI/AD in the ADNI cohort (n = 566). Regression analysis showed CSF Aβ42 levels and t-tau/Aβ42 ratios to correlate with the number of APOE4 alleles and plasma levels of B-type natriuretic peptide and pancreatic polypeptide.
Conclusion:
Four plasma analytes were consistently associated with the diagnosis of very mild dementia/MCI/AD in 3 independent clinical cohorts. These plasma biomarkers may predict underlying AD through their association with CSF AD biomarkers, and the association between plasma and CSF amyloid biomarkers needs to be confirmed in a prospective study.
The clinical diagnosis of mild cognitive impairment (MCI) and probable Alzheimer disease (AD) is increasingly aided by biomarkers predictive of underlying pathology.1,2 These include CSF biomarkers reflecting the plaque and tangle pathology underlying AD such as β-amyloid 1–42 (Aβ42), total tau (t-tau), and tau phosphorylated at threonine 181 (p-tau181)3; substrate-specific brain imaging such as 11C and 18F PET imaging4,5; and structural MRI findings such as hippocampal volume.6 Each modality has a different sensitivity-specificity profile, and additional technical barriers and patient preferences may dictate the successful implementation of any biomarker into clinical practice, including aversion to having a lumbar puncture for CSF biomarkers and cost for advanced imaging. Thus, a blood-based test is an appealing alternative because of its simplicity and cost-effectiveness for widespread clinical use as well as in specialty centers.7,8 A multi-analyte profiling approach to plasma proteins and peptides can also yield biologically important signatures of disease and endophenotypes to allow for prognostication and novel therapeutic development.
A number of recent studies generated additional enthusiasm for a blood-based test predictive of underlying AD pathology.7,9 Using clinically diagnosed patients, 1 study identified plasma analyte combinations that predicted AD diagnosis and subsequent conversion to AD among subjects with MCI.7 However, results from biomarker studies such as these often suffer from failed replication due to a combination of causes including lack of methodologic standardization, insufficient sample size, and overtraining of imprecise data,10 and the 2 published studies may overestimate the clinical utility of blood-based tests given the single-batch nature and reliance on internal cross-validation within each study. Recently, serum analytes predictive of AD were found to have only modest accuracy in plasma.8 Here we attempt to focus on the overlap between plasma analytes associated with MCI/AD in 2 independently recruited and characterized cohorts,3,11–14 and then validate these findings in an independent cohort of 566 participants from the multicenter Alzheimer's Disease Neuroimaging Initiative (ADNI)15,16 while exploring whether plasma analytes associated with MCI/AD correlated with CSF AD biomarker levels.
METHODS
Standard protocol approvals, registrations, and patient consents.
The study protocol involving University of Pennsylvania (Penn) subjects and samples was approved by the Penn Institutional Review Board. The study protocol involving Washington University (WU) subjects and samples was approved by the Human Studies Committee at Washington University. Written and verbal informed consent was obtained at enrollment at each center.
Participants.
Subjects in the 2 discovery sets were recruited and longitudinally followed at Penn and WU (table 1), while subjects in ADNI were previously described.16,17 At Penn, participants (n = 267) were community-dwelling healthy volunteers and patients evaluated at subspecialty clinics dedicated to the evaluation of neurodegenerative disorders including MCI,18 AD,19,20 and related dementia.21,22 Cognitively normal subjects were recruited through all subspecialty clinics to participate in biofluid studies. APOE genotyping was performed for 235 out of 267 Penn subjects.
Table 1.
Demographic features of subjects included in plasma multianalyte profiling from the University of Pennsylvania, Washington University, and Alzheimer's Disease Neuroimaging Initiative
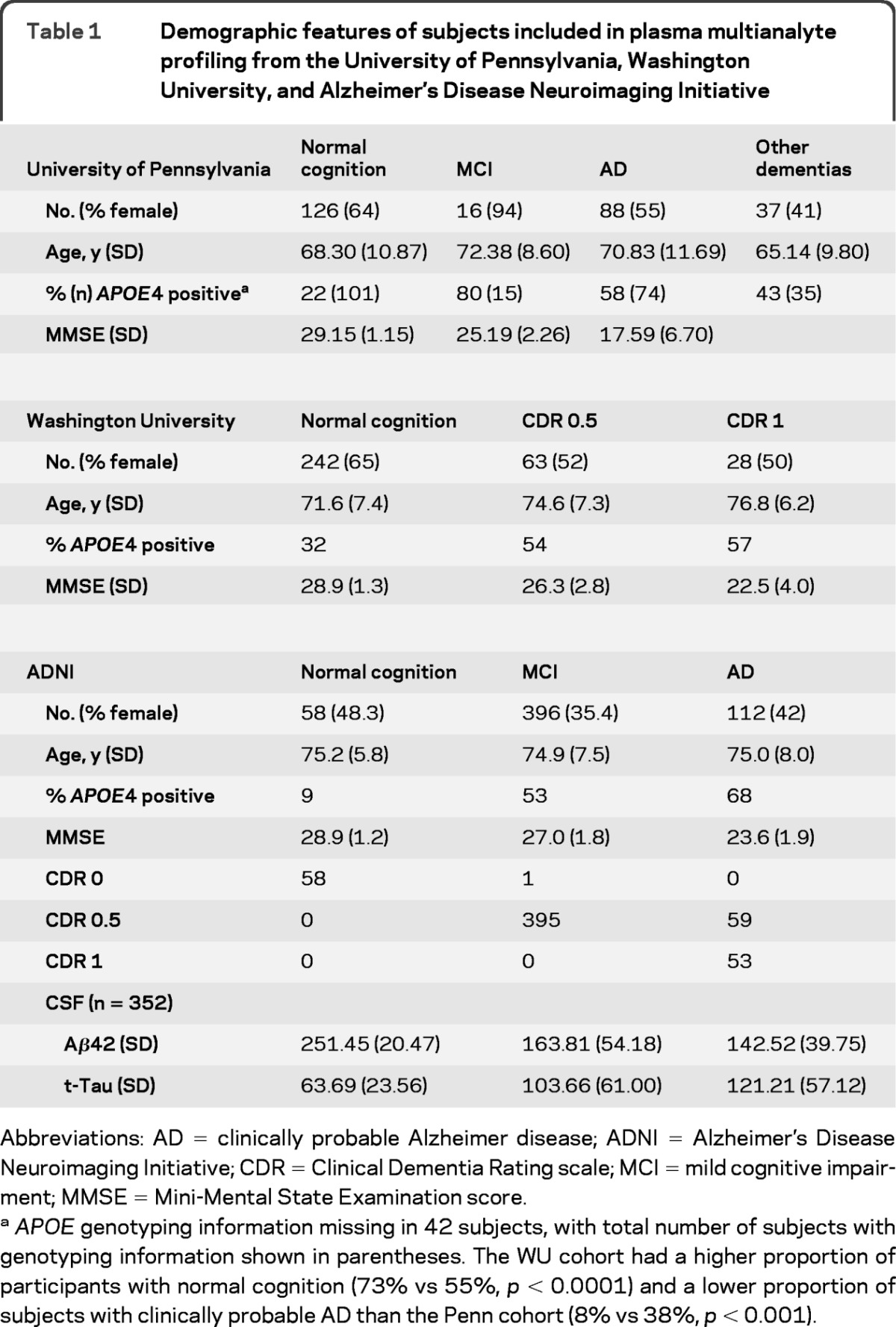
Abbreviations: AD = clinically probable Alzheimer disease; ADNI = Alzheimer's Disease Neuroimaging Initiative; CDR = Clinical Dementia Rating scale; MCI = mild cognitive impairment; MMSE = Mini-Mental State Examination score.
APOE genotyping information missing in 42 subjects, with total number of subjects with genotyping information shown in parentheses. The WU cohort had a higher proportion of participants with normal cognition (73% vs 55%, p < 0.0001) and a lower proportion of subjects with clinically probable AD than the Penn cohort (8% vs 38%, p < 0.001).
At WU, participants (n = 333) were community-dwelling volunteers enrolled in longitudinal studies of healthy aging and dementia at the Knight AD Research Center at Washington University. Clinical diagnosis was evaluated based on criteria from the National Institute of Neurological and Communicative Diseases and Stroke–Alzheimer's Disease and Related Disorders Association.19 Cognitive status was rated with the Clinical Dementia Rating scale (CDR): CDR of 0 indicates no dementia, CDR 0.5 indicates very mild dementia, and CDR 1 indicates mild dementia.23 Some of the CDR 0.5 participants in the study met the criteria for MCI and some were less impaired and were considered “pre-MCI.”18,24APOE genotyping was performed for all subjects enrolled at WU.
Procedures.
Samples were collected from Penn and WU subjects according to strict protocols without protease inhibitors. At sample collection, participants were ≥50 years of age and in good general health (including no evidence of clinically significant liver disease or renal failure), having no other psychiatric or medical diagnoses that could contribute importantly to cognitive impairment or dementia other than the primary neurodegenerative disorder. At Penn, plasma was collected in 10 mL K2 EDTA tubes (BD Vacutainer®) without overnight fasting and refrigerated immediately (4°C) before transporting to a central site on ice for centrifuge (2,000 g × 15 minutes at 4°C) separation into plasma and cellular components within 4 hours of collection. Plasma aliquots (0.5 mL) were prepared, bar-coded, and then stored in polypropylene vials at −80°C until analysis. Quality control samples to determine coefficients of variation (CV) included duplicate plasma samples from 3 control subjects analyzed at the same time as the remaining Penn subjects, and an average intra-assay CV was obtained for each analyte of interest. At WU, plasma was collected in polypropylene tubes after overnight fasting between 7:30 and 8:00 am and centrifuged (2,000 g × 15 minutes at 4°C) for separation into plasma and cellular components. Plasma aliquots (0.5 mL) were stored at −80°C until analyzed.
Plasma aliquots from each center were interrogated consecutively in 2 batches (1 batch per center) in 2009 by Rules-Based Medicine (RBM, Austin, TX) for levels of 190 analytes using the multiplex Human DiscoveryMAPTM panel and a Luminex 100 platform (table e-1 on the Neurology® Web site at www.neurology.org).12,25,26 The 190 analytes were assembled into preformatted assays designed for different diseases including cancer, autoimmune disorders, AD, Parkinson disease, and frontotemporal degeneration.27 Plasma levels of 190 analytes in 566 subjects from the ADNI cohort (table 1) were also measured at RBM in 2010 using the same multiplexed immunoassays. Analytes below threshold of detection (11 for Penn and 21 for WU) were excluded. Dynamic range for each plasma analyte in the RBM panel is provided on the ADNI Web site (http://adni.loni.ucla.edu). A total of 352 subjects (56 normal cognition, 195 MCI, and 101 AD) also had CSF AD biomarker levels provided by the ADNI Biomarker Core.3
Statistical analysis.
Statistical analysis in this study was performed in SPSS 17.0 (Chicago, IL) and significance analysis of microarrays (SAM).28 In each cohort, cognitively impaired individuals (Penn: MCI and AD, WU: CDR 0.5 and 1) were grouped together as we sought to identify plasma analytes altered across various stages of the very mild dementia/MCI/AD spectrum, and because of the differential distribution of subjects within each impaired category. Power calculation was performed in PASS 11 (Kaysville, UT), which showed 89.0% power in the Penn cohort and 93.8% power in the WU cohort for each of 190 analytes to detect a true difference in expression of at least 0.5 with estimated group SD of 1.0 and a false discovery rate of 0.10 using a 2-sided 2-sample t test. All raw levels were log-transformed to achieve normal distribution. For initial identification of individual analytes different between normal cognition and very mild dementia/MCI/AD, logistic regression model was used adjusting for age and gender. We then used a model based on Intersection Union Test (IUT), which involves identification of overlapping results (analytes in the current study, genes in microarray studies) from distinct datasets.29 As this method may be overly conservative and reduce the power in detecting true positives,30 we used a more liberal threshold of significance at the univariate analysis stage of p < 0.10 (after adjusting for age and gender) to reduce type II errors. We further reduced type I errors by applying 2 additional filters by identifying 1) analytes from the modified IUT with common direction of change (vector direction) and 2) analytes from (1) that fulfill strict Bonferroni correction at the validation phase. Analytes with similar associations with very mild dementia/MCI/AD in each discovery cohort were then analyzed in the ADNI cohort (n = 566) for association with the diagnosis of MCI/AD with an α value of 0.0036 (0.05/14) for the 14 analytes that passed first level screening (figure). Univariate analysis was also repeated within each cohort using SAM,28 and analytes found to be significant in more than 2 cohorts were identified.
Figure. Flow diagram of subjects included in the plasma multianalyte profiling study and general analytical strategy.

Subjects from University of Pennsylvania (Penn) and Alzheimer's Disease Neuroimaging Initiative (ADNI) were stratified into normal cognition, mild cognitive impairment (MCI), and clinically probable Alzheimer disease (AD) according to published criteria, along with additional patients from Penn with non-AD dementia. Subjects from Washington University were stratified according to Clinical Dementia Rating scale (CDR) according to published criteria (see Methods). In each cohort, mild impairment likely due to AD (mild cognitive and AD, or CDR of 0.5 and 1) were grouped and compared with subjects with normal cognition or CDR of 0.
In addition, we determined the relationship between novel plasma MAP biomarkers and CSF AD biomarker-drive diagnosis (CSF Aβ42 levels <193 pg/mL and t-tau/Aβ42 >0.39)3 adjusting for age and gender (p < 0.0036), and the correlation between CSF AD biomarker and plasma marker levels using linear regression analysis. In these models, CSF AD biomarker (Aβ42, t-tau/Aβ42) levels were dependent variables, age and gender were entered in the first stage as independent variables, and number of APOE4 alleles and plasma biomarker levels were then entered in a stepwise fashion. Finally, as pancreatic peptide levels are influenced by ChEI therapy,31 we analyzed the correlation between plasma and CSF AD biomarkers among subjects without ChEI (including donepezil, galantamine, and rivastigmine), including 58 subjects with normal cognition, 226 subjects with MCI, and 20 subjects with AD.
RESULTS
Univariate analysis in Penn and WU cohorts.
Among 190 analytes measured, 41 and 51 analytes were associated with mild dementia/MCI/AD in the Penn and WU cohorts (p < 0.10, table e-2). Among 23 analytes identified in both cohorts, 6 (apolipoprotein A1, apolipoprotein H, cystatin C, fibrinogen, myeloperoxidase, and neutrophil gelatinase-associated lipocalin; table 2) demonstrated opposite directions of association with diagnosis and were excluded from further analysis. In the remaining 17 analytes, 5 were reported by 1 previous serum study using the RBM panel to correlate similarly with clinical AD,8 including C-reactive protein (CRP), interleukin (IL)-10, IL-15, pancreatic polypeptide, and resistin. Only 1 (IL-3) from a prior study using plasma and a different multiplex platform was in this list of 17, and showed an opposite direction of association with AD.7 SAM identified analytes associated with mild dementia/MCI/AD in each cohort, with 6 (α1-antitrypsin, apoE, CRP, N-terminal pro B-type natriuretic peptide, osteopontin, serum amyloid P) identified in both cohorts (table e-3).
Table 2.
Analytes associated with abnormal cognition in the Penn and WU datasetsa

Abbreviations: AD = Alzheimer disease; IFN = interferon; IL = interleukin; MCI = mild cognitive impairment; Penn = University of Pennsylvania; WU = Washington University.
Shown are odds ratios associated with diagnosis of very mild dementia/MCI/AD in the logistic regression models, in comparison to published studies of blood-based multianalyte profiling studies identifying analytes predictive of AD diagnosis. Positive values predict increases in log-transformed analyte levels associated with the diagnosis of AD, while negative values predict decreases in levels.
Univariate analysis in ADNI cohort.
We then sought to confirm the association between the 17 candidate AD plasma biomarkers and clinical diagnosis in ADNI. In the ADNI cohort, B-type natriuretic peptide (BNP) levels were measured instead of N-terminal pro B-type natriuretic peptide levels, and IL-10, IL-12p40, and IL-15 levels were not available. Using Bonferroni correction for the 14 analytes (p < 0.0036), we found 6 to be highly associated with the clinical MCI/AD diagnosis, including apoE, BNP, cortisol, CRP, IL-3, and pancreatic polypeptide (table 3). When ADNI participants with CSF were analyzed (n = 352), CSF findings predictive of underlying AD pathologic changes (i.e., CSF Aβ42 levels <193 pg/mL and CSF t-tau/Aβ42 ratio >0.39) also were associated with levels of 4 plasma proteins, including apoE, BNP, CRP, and pancreatic polypeptide (table 3). All analytes had acceptable intra-assay variability (appendix e-1).
Table 3.
Effects of clinical or CSF-based diagnosis on analyte levels in the ADNI cohort (adjusted for age and gender)a

Abbreviations: Aβ42 = β-amyloid 1–42; AD = Alzheimer disease; ADNI = Alzheimer's Disease Neuroimaging Initiative; BNP = brain natriuretic peptide; CRP = C-reactive protein; IGF-BP2 = insulin-like growth factor binding protein 2; IL = interleukin; MCI = mild cognitive impairment; Penn = University of Pennsylvania; PP = pancreatic polypeptide; SAP = serum amyloid protein; SCF = stem cell factor; t-tau = total tau; WU = Washington University.
Levels of IL-10, IL-12p40, and IL-15 not available in the ADNI cohort at time of manuscript preparation.
Analytes identified from both Penn and WU cohorts (table 3) significantly associated with a clinical diagnosis of MCI/AD or CSF biomarker pattern associated with pathologic AD with odds ratios shown.
Applying SAM (false discovery rate of 10%) and then IUT to all 3 datasets yielded 14 analytes associated with mild dementia/MCI/AD (table e-4), including apoE, CRP, and insulin growth factor binding protein 2 (IGF-BP2), altered in all 3 cohorts, and BNP and pancreatic peptide altered in 2 cohorts. Given the larger type II error associated with applying IUT to all 3 datasets, we consider these results to be consistent with our discovery-validation approach using logistic regression.
Correlation between plasma and CSF AD biomarkers.
As candidate plasma AD biomarker levels were associated with both the clinical diagnosis and CSF biomarker profiles consistent with AD, we analyzed whether CSF AD biomarker levels correlated with plasma AD biomarker levels. Multivariate linear regression modeling showed CSF Aβ42 levels to be strongly correlated with number of APOE4 alleles adjusting for age and gender (p < 0.001, R = 0.559, adjusted R2 of 0.307), as well as levels of BNP and pancreatic polypeptide (table 4, R = 0.596, adjusted R2 of 0.345). A similar association was found with CSF t-tau/Aβ42 ratios (table 4), although a weaker relationship was found between an increased t-Tau/Aβ42 ratio and candidate AD blood biomarkers (number of APOE4 alleles and plasma pancreatic polypeptide levels, R = 0.404, adjusted R2 of 0.154). Plasma apoE levels correlated with APOE4 allele numbers and were not predictive of CSF biomarker levels independently of the latter. The addition of IGF-BP2 levels from SAM analysis or adjustment for ChEI use in either model did not affect the outcome (appendix e-1).
Table 4.
Linear regression models showing associations between CSF AD biomarker levels (Aβ42 level, ratio of t-tau/Aβ42) and plasma AD biomarkers in all ADNI subjects with CSF and plasma analytes (n = 566)a

Abbreviations: Aβ42 = β-amyloid 1–42; AD = Alzheimer disease; ADNI = Alzheimer's Disease Neuroimaging Initiative; BNP = B-type natriuretic peptide; t-tau = total tau.
Similar correlations were observed in ADNI subjects with CSF and plasma analytes not treated with cholinesterase inhibitors (n = 304; see appendix e-1).
DISCUSSION
The search for novel biochemical predictors of underlying neurodegenerative pathology has been greatly aided by high throughput multiplex platforms.7,8,12 Using 2 large cohorts of well-characterized participants with normal cognition and cognitive impairment, we identified 17 plasma analytes associated with the clinical diagnosis of very mild dementia/MCI/AD in both cohorts. Among these, changes in apoE, BNP, CRP, and pancreatic polypeptide levels were also associated with MCI/AD diagnosis and CSF AD biomarker profiles in ADNI. Importantly, along with the known association between APOE genotyping and CSF AD biomarker levels, these AD plasma biomarkers also correlated with CSF Aβ42 levels and t-tau/Aβ42 ratios. These plasma AD biomarkers may thus help predict underlying AD pathology through their relationships to established CSF biomarkers of AD, and could serve as the basis of a plasma-based screening battery for AD.
A major roadblock in the identification of candidate biomarkers through multi-analyte profiling has been the successful replication of 1 study's “hits” in other studies. There are many reasons for this, including preanalytical variables, different analytical platforms (such as 2D gel electrophoresis and Luminex multiplexing, among others), and lack of platform cross-validation, body fluid types (plasma, serum), subject selection, disease endophenotypes, and analytical algorithms. In the current study, all 3 cohorts (Penn, WU, ADNI) had the same platform, body fluid type, and analytical approaches, with similar methods for subject selection and clinical characterization. Some analytes were previously identified in the multicenter Texas Alzheimer's Research Consortium (TARC) study using the same platform but a different biofluid (serum).8 The difference in results between a plasma-based study and a serum-based study may be due to protein–protein interactions between analytes of interest and clotting factors, or differential interaction between analytes of interest, additives (e.g., EDTA, serum separation substrate), and potentially different plastic used in the construction of plasma (“purple top”) and serum (“gold top”) tubes. We collected plasma in tubes containing EDTA in part due to the indeterminate interaction between other additives (e.g., heparin, clot activator) and our analytes of interest. Beyond fluid type, the analytical platform likely contributes significantly to the lack of overlap between the current study and one prior study using the same biofluid (plasma) but a different platform.7 This discrepancy can potentially be attributed to different antibody affinity, analyte- and platform-specific dynamic range, platform precision, and handling of noise and outliers by each platform and analysis. Finally, the use of IUT may generate conservative estimates of overlap,29 but we focused on analytes with the highest likelihood of replication for further technical refinement. Future analytical strategies may use modified approaches such as Relaxed IUT to generate common lists, but any algorithm that biases discovery will increase the likelihood of type I error. However, the relatively higher degree of “biomarker concordance” between studies using the same platform would support that future discovery-type studies should be mindful of analytical platform selection as well as analyte identity, and analyte identity and levels should be additionally confirmed by independent means.
Among plasma AD biomarkers reported here, BNP, CRP, and pancreatic polypeptide levels were associated with CSF Aβ42 levels and t-tau/Aβ42 ratios regardless of ChEI use. BNP is a well-established marker of left ventricular dysfunction, and is elevated in acute strokes32 and vascular dementia.33 Elevated BNP levels are also associated with cognitive decline in vascular disease34 and the development of AD and vascular dementia (independent of heart failure).35 Elevated BNP may thus reflect shared risk factors between heart failure and AD or an unknown step in AD pathogenesis, and its effects on CSF Aβ42 levels with magnitudes similar to APOE4 genotype warrant further investigation in future prospective studies.
Pancreatic polypeptide levels were previously identified by TARC.8 Pancreatic polypeptide is a small signaling peptide associated with postprandial appetite suppression36 present in multiple brain regions, including those affected by AD such as the hippocampus37 and locus ceruleus.38 Increased plasma pancreatic polypeptide levels could reflect impaired transport across the blood–brain/CSF barrier through yet unclear mechanisms, but it is also elevated in the CSF of patients with AD.12 Similar changes in patients with non-AD dementia further suggest that elevated pancreatic polypeptide levels may reflect neuronal loss irrespective of etiology, although such elevated levels can still serve as a potential plasma marker of neuronal injury. Similarly, CRP was not specifically associated with CSF Aβ42 levels or t-tau/Aβ42 ratios, although it complemented BNP and pancreatic polypeptides in predicting CSF AD biomarkers. The association between decreased CRP and AD diagnosis is perhaps perplexing, as CRP has been found to be decreased, increased, or unchanged in AD.39 Alterations in CRP levels may again reflect neuronal injury, although its levels may be more susceptible to patient selection and endophenotypes than other plasma biomarkers.
This study included 3 large cohorts but there still exist a number of limitations. We sought to identify plasma biomarkers common to MCI and AD, and different proportion of patients with MCI in each cohort makes it challenging to identify MCI- or AD-specific changes. As few subjects recruited at the 2 centers or within ADNI have neuropathologic analysis, we did not examine which analytes provided the best pathologic prediction. This type of comparative analysis across centers can also be skewed by differential analyte stability, classification algorithm, and the choice of surrogate gold standard (clinical vs CSF-based diagnosis), although in-depth analyses are underway to address these. The differences in collection techniques between Penn and WU (fasting vs nonfasting, immediate centrifugation/freezing vs cold storage at 4°C before processing) and batch-to-batch variability may account for failed replication for some analytes, and the significance of analytes identified in only one cohort but validated in ADNI needs further interrogation. Finally, plasma biomarkers identified here accounted for some, but not all, changes in CSF AD biomarkers in the exploratory portion of the current study, although only analytes that passed our stringent selection criteria were tested in the ADNI cohort. These analytes, along with plasma amyloid peptides and nonamyloid analytes previously identified,8,40 can be jointly analyzed for their correlation to CSF AD biomarkers. Nevertheless, we were successful in identifying plasma analytes whose levels were similarly altered in 3 independent cohorts using highly strict criteria. With 2 of these biomarkers (BNP, CRP) assayed routinely in clinical laboratories, the utility of plasma AD biomarkers in screening patients at risk for AD should be readily assessed in prospectively recruited subjects.
Supplementary Material
ACKNOWLEDGMENT
Data used in preparation of this article were obtained from ADNI database (adni.loni.ucla.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at www.neurology.org.
GLOSSARY
- Aβ42
βamyloid 1–42
- AD
Alzheimer disease
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- BNP
B-type natriuretic peptide
- CDR
Clinical Dementia Rating
- CRP
C-reactive protein
- CV
coefficient of variation
- IGF-BP2
insulin growth factor binding protein 2
- IL
interleukin
- IUT
Intersection Union Test
- MCI
mild cognitive impairment
- p-tau181
tau phosphorylated at threonine 181
- Penn
University of Pennsylvania
- RBM
Rules-Based Medicine
- SAM
significance analysis of microarrays
- t-tau
total tau
- TARC
Texas Alzheimer's Research Consortium
- WU
Washington University
Footnotes
Editorial, page 846
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Drafting/revising the manuscript for content, including medical writing for content: Drs. Hu, Holtzman, Fagan, Shaw, Arnold, Grossman, Xiong, Morris, Trojanowski, Soares. Study concept or design: Drs. Hu, Holtzman, Fagan, Shaw, Perrin, Arnold, Grossman, Xiong, Craig-Schapiro, Clark, McCluskey, Elman, Karlawish, Chen-Plotkin, Hurtig, Siderowf, Lee, Morris, Trojanowski, Soares. Analysis or interpretation of data: Drs. Hu, Holtzman, Shaw, Pickering, Kuhn, Chen, Van Deerlin, Swenson, Trojanowski, Soares. Contribution of vital reagents/tools/patents: Dr. Van Deerlin. Acquisition of data: Drs. Hu, Holtzman, Fagan, Shaw, Perrin, Arnold, Grossman, Craig-Schapiro, Clark, McCluskey, Elman, Karlawish, Chen-Plotkin, Hurtig, Siderowf, Lee, Morris, Trojanowski, Soares. Statistical analysis: Drs. Hu, Pickering. Study supervision or coordination: Drs. Holtzman, Shaw, Arnold, Xiong, Lee, Morris, Trojanowski, Soares. Obtain funding: Drs. Holtzman, Shaw, Arnold, Grossman, Swenson, Lee, Morris, Trojanowski, Soares.
Study Funding
This study has been supported by NIH grants AG10124, AG17585, and the Penn-Pfizer Research Alliance at Penn; a grant to Washington University from Pfizer, NIH grants P50 AG05681, P01 AG039919, P01 AG026276, P30 NS057105, and the Charles F. and Joanne Knight Alzheimer Research Initiative at Washington University; and the Viretta Brady Discovery Fund and Emory University Center for Neurodegenerative Diseases at Emory University. Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through contributions from the following: Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson & Johnson, Eli Lilly and Co., Medpace, Inc., Merck and Co., Inc., Novartis AG, Pfizer Inc, F. Hoffman-La Roche, Schering-Plough, Synarc, Inc., as well as non-profit partners the Alzheimer's Association and Alzheimer's Drug Discovery Foundation, with participation from the US Food and Drug Administration. Private sector contributions to ADNI are facilitated by the Foundation for the NIH (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129, K01 AG030514, and the Dana Foundation. This publication was also made possible by Grant Number UL1 RR024992 from the National Center for Research Resources (NCRR), a component of the NIH, and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH.
DISCLOSURE
W. Hu receives research support from the Viretta Brady Discovery Fund. D. Holtzman serves on scientific advisory boards for Satori Pharmaceuticals and EnVivo Pharmaceuticals; may accrue revenue on pending patents re: Methods for measuring the metabolism of neurally derived biomolecules in vivo; Use of anti-AB antibody to treat traumatic brain injury; Methods to treat Alzheimer's disease or other amyloid beta accumulation associated disorders; Humanized antibodies that sequester abeta peptide; Diagnostic for early stage Alzheimer's disease; and Predictive diagnostic for Alzheimer's disease; serves as a consultant to Merck Serono, Eli Lilly and Company, Takeda Pharmaceutical Company Limited, Abbott, Comentis, Inc., Eisai Inc., and AstraZeneca; is cofounder of and receives board of directors compensation from C2N Diagnostics LLC; receives research support from AstraZeneca, Pfizer Inc., Eli Lilly and Company, Elan Corporation, Forest Laboratories, Inc., the NIH, Cure Alzheimer's Fund, and Fidelity Foundation; has received compensation from Washington University from license revenue received for licensing of patent applications to C2N Diagnostics LLC; and may receive future royalty payments for Washington University licensing patents to C2N Diagnostics, LLC, and Eli Lilly and Company. A. Fagan serves on the speakers' bureau for the Alzheimer's Association. L. Shaw and R. Perrin report no disclosures. S. Arnold is a consultant to NIMH Geriatrics Branch and a member of the advisory board to Rush University Medical Center, was a compensated guest lecturer at Vanderbilt University, and was an expert witness to Cozen O'Connor and Reed-Smith. M. Grossman is a consultant to Allon Therapeutics, Forest Labs, and Pfizer Pharmaceuticals; a study section member at the NIH; a member of the editorial board for Wolters Kluwer Health; and a compensated lecturer at Sacred Heart Hospital. C. Xiong and R. Craig-Schapiro report no disclosures. C. Clark is deceased; disclosures are not included for this author. E. Pickering is an employee of Pfizer Global Research and Development. M. Kuhn is an employee of Pfizer Global Research and Development. Y. Chen is an employee of Pfizer Global Research and Development. V. Van Deerlin reports no disclosures. L. McCluskey was an expert witness to Gallagher & Rowan and Kilcoyne & Nesbitt. L. Elman, J.H.T. Karlawish, and A. Chen-Plotkin report no disclosures. H. Hurtig is a consultant to UpToDate. A. Siderowf is supported by a Morris K. Udall Parkinson's Disease Research Center of Excellence grant from NINDS (NS-053488), and has been supported by SAP4100027296, a health research grant awarded by the Department of Health of the Commonwealth of Pennsylvania from the Tobacco Master Settlement Agreement under Act 2001-77. He has received consulting fees from Teva Neuroscience, Supernus Pharmaceuticals, Schering-Plough, and Merck Serono. He has received speaking honorarium from Teva Neuroscience. F. Swenson is an employee of Pfizer Global Research and Development. V.M.-Y. Lee has received funding for travel and honoraria from Takeda Pharmaceutical Company Ltd.; has received speaker honoraria from Pfizer Inc., BMS, and Merck; may accrue revenue on patents re: Modified avidin-biotin technique, Method of stabilizing microtubules to treat Alzheimer's disease, Method of detecting abnormally phosphorylated tau, Method of screening for Alzheimer's disease or disease associated with the accumulation of paired helical filaments, Compositions and methods for producing and using homogeneous neuronal cell transplants, Rat comprising straight filaments in its brain, Compositions and methods for producing and using homogeneous neuronal cell transplants to treat neurodegenerative disorders and brain and spinal cord injuries, Diagnostic methods for Alzheimer's disease by detection of multiple MRNAs, Methods and compositions for determining lipid peroxidation levels in oxidant stress syndromes and diseases, Compositions and methods for producing and using homogenousneuronal cell transplants, Method of identifying, diagnosing and treating alpha-synuclein positive neurodegenerative disorders, Mutation-specific functional impairments in distinct tau isoforms of hereditary frontotemporal dementia and parkinsonism linked to chromosome-17: genotype predicts phenotype, Microtubule stabilizing therapies for neurodegenerative disorders, and Treatment of Alzheimer's and related diseases with an antibody; and receives research support from the NIH NIA PO1 AG 17586-10, PO1 AG-032953, NINDS P50 NS053488-02, NIA UO1 AG029213-01; and from the Marian S. Ware Alzheimer Program. J. Morris serves on scientific advisory boards for AstraZeneca, Bristol-Myers Squibb, Genentech, Inc., Merck Serono, Novartis, Pfizer Inc, Schering-Plough Corp., Eli Lilly and Company, Wyeth, and Elan Corporation; and receives research support from Elan Corporation, Wyeth, Eli Lilly and Company, Novartis, Pfizer Inc, Avid Radiopharmaceuticals, the NIH, and the Dana Foundation. J. Trojanowski has received funding for travel and honoraria from Takeda Pharmaceutical Company Ltd.; has received speaker honoraria from Pfizer Inc.; may accrue revenue on patents re: Modified avidin-biotin technique, Method of stabilizing microtubules to treat Alzheimer's disease, Method of detecting abnormally phosphorylated tau, Method of screening for Alzheimer's disease or disease associated with the accumulation of paired helical filaments, Compositions and methods for producing and using homogeneous neuronal cell transplants, Rat comprising straight filaments in its brain, Compositions and methods for producing and using homogeneous neuronal cell transplants to treat neurodegenerative disorders and brain and spinal cord injuries, Diagnostic methods for Alzheimer's disease by detection of multiple MRNAs, Methods and compositions for determining lipid peroxidation levels in oxidant stress syndromes and diseases, Compositions and methods for producing and using homogenous neuronal cell transplants, Method of identifying, diagnosing and treating alpha-synuclein positive neurodegenerative disorders, Mutation-specific functional impairments in distinct tau isoforms of hereditary frontotemporal dementia and parkinsonism linked to chromosome-17: genotype predicts phenotype, Microtubule stabilizing therapies for neurodegenerative disorders, and Treatment of Alzheimer's and related diseases with an antibody; and receives research support from the NIH (NIA P01 AG 09215-20 [PI], NIA P30 AG 10124-18 [PI], NIA PO1 AG 17586-10 [Project 4 Leader], NIA 1PO1 AG-19724-07 [Core C Leader], NIA 1 U01 AG 024904-05 [Co-PI Biomarker Core Laboratory], NINDS P50 NS053488-02 [PI], NIA UO1 AG029213-01 [Co-I]; RC2NS069368 [PI], RC1AG035427 [PI], and NIA P30AG036468 [PI]), and the Marian S. Ware Alzheimer Program. H. Soares is an employee of Bristol-Myers Squibb. Go to Neurology.org for full disclosures.
REFERENCES
- 1. Jack CR, Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010;9:119–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature 2009;461:916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jack CR, Jr, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain 2009;132:1355–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Apostolova LG, Hwang KS, Andrawis JP, et al. 3D PIB and CSF biomarker associations with hippocampal atrophy in ADNI subjects. Neurobiol Aging 2010;31:1284–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gerardin E, Chetelat G, Chupin M, et al. Multidimensional classification of hippocampal shape features discriminates Alzheimer's disease and mild cognitive impairment from normal aging. NeuroImage 2009;47:1476–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ray S, Britschgi M, Herbert C, et al. Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nat Med 2007;13:1359–1362 [DOI] [PubMed] [Google Scholar]
- 8. O'Bryant SE, Xiao G, Barber R, et al. A blood-based screening tool for Alzheimer's disease that spans serum and plasma: findings from TARC and ADNI. PloS one 2011;6:e28092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reddy MM, Wilson R, Wilson J, et al. Identification of candidate IgG biomarkers for Alzheimer's disease via combinatorial library screening. Cell 2011;144:132–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Poste G. Bring on the biomarkers. Nature 2011;469:156–157 [DOI] [PubMed] [Google Scholar]
- 11. Clark CM, Xie S, Chittams J, et al. Cerebrospinal fluid tau and beta-amyloid: how well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch Neurol 2003;60:1696–1702 [DOI] [PubMed] [Google Scholar]
- 12. Hu WT, Chen-Plotkin A, Arnold SE, et al. Novel CSF biomarkers for Alzheimer's disease and mild cognitive impairment. Acta Neuropathol 2010;119:669–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 2007;64:343–349 [DOI] [PubMed] [Google Scholar]
- 14. Snider BJ, Fagan AM, Roe C, et al. Cerebrospinal fluid biomarkers and rate of cognitive decline in very mild dementia of the Alzheimer type. Arch Neurol 2009;66:638–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aisen PS, Petersen RC, Donohue MC, et al. Clinical Core of the Alzheimer's Disease Neuroimaging Initiative: progress and plans. Alzheimers Dement 2010;6:239–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trojanowski JQ, Vandeerstichele H, Korecka M, et al. Update on the biomarker core of the Alzheimer's Disease Neuroimaging Initiative subjects. Alzheimers Dement 2010;6:230–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Petersen RC, Aisen PS, Beckett LA, et al. Alzheimer's Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology 2010;74:201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol 1999;56:303–308 [DOI] [PubMed] [Google Scholar]
- 19. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944 [DOI] [PubMed] [Google Scholar]
- 20. Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurol 2007;6:734–746 [DOI] [PubMed] [Google Scholar]
- 21. Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–1554 [DOI] [PubMed] [Google Scholar]
- 22. McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–1872 [DOI] [PubMed] [Google Scholar]
- 23. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414 [DOI] [PubMed] [Google Scholar]
- 24. Storandt M, Grant EA, Miller JP, Morris JC. Longitudinal course and neuropathologic outcomes in original vs revised MCI and in pre-MCI. Neurology 2006;67:467–473 [DOI] [PubMed] [Google Scholar]
- 25. Hu WT, Chen-Plotkin A, Grossman M, et al. Novel CSF biomarkers for frontotemporal lobar degenerations. Neurology 2010;75:2079–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen-Plotkin AS, Hu WT, Siderowf A, et al. Plasma epidermal growth factor levels predict cognitive decline in Parkinson disease. Ann Neurol Epub 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hu WT, Chen-Plotkin A, Arnold SE, et al. Biomarker discovery for Alzheimer's disease, frontotemporal lobar degeneration, and Parkinson's disease. Acta Neuropathol 2010;120:385–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci USA 2002;99:6567–6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Quan H, Bolognese J, Yuan W. Assessment of equivalence on multiple endpoints. Stat Med 2001;20:3159–3173 [DOI] [PubMed] [Google Scholar]
- 30. Deng X, Xu J, Wang C. Improving the power for detecting overlapping genes from multiple DNA microarray-derived gene lists. BMC Bioinformatics 2008;9 suppl 6: S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Damholt MB, Arlien-Soeborg P, Hilsted L, Hilsted J. Is pancreatic polypeptide response to food ingestion a reliable index of vagal function in type 1 diabetes? Scand J Clin Lab Invest 2006;66:279–286 [DOI] [PubMed] [Google Scholar]
- 32. Shibazaki K, Kimura K, Iguchi Y, Okada Y, Inoue T. Plasma brain natriuretic peptide can be a biological marker to distinguish cardioembolic stroke from other stroke types in acute ischemic stroke. Intern Med 2009;48:259–264 [DOI] [PubMed] [Google Scholar]
- 33. Kondziella D, Gothlin M, Fu M, Zetterberg H, Wallin A. B-type natriuretic peptide plasma levels are elevated in subcortical vascular dementia. Neuroreport 2009;20:825–827 [DOI] [PubMed] [Google Scholar]
- 34. Gunstad J, Poppas A, Smeal S, et al. Relation of brain natriuretic peptide levels to cognitive dysfunction in adults >55 years of age with cardiovascular disease. Am J Cardiol 2006;98:538–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kerola T, Nieminen T, Hartikainen S, Sulkava R, Vuolteenaho O, Kettunen R. B-type natriuretic peptide as a predictor of declining cognitive function and dementia: a cohort study of an elderly general population with a 5-year follow-up. Ann Med 2010;42:207–215 [DOI] [PubMed] [Google Scholar]
- 36. Adrian TE, Besterman HS, Cooke TJ, Bloom SR, Barnes AJ, Russell RC. Mechanism of pancreatic polypeptide release in man. Lancet 1977;1:161–163 [DOI] [PubMed] [Google Scholar]
- 37. Inui A, Mizuno N, Ooya M, et al. Cross-reactivities of neuropeptide Y and peptide YY with pancreatic polypeptide antisera: evidence for the existence of pancreatic polypeptide in the brain. Brain Res 1985;330:386–389 [DOI] [PubMed] [Google Scholar]
- 38. Hunt SP, Emson PC, Gilbert R, Goldstein M, Kimmell JR. Presence of avian pancreatic polypeptide-like immunoreactivity in catecholamine and methionine-enkephalin-containing neurones within the central nervous system. Neurosci Lett 1981;21:125–130 [DOI] [PubMed] [Google Scholar]
- 39. Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response?. Nat Med 2006;12:1005–1015 [DOI] [PubMed] [Google Scholar]
- 40. Ellis KA, Bush AI, Darby D, et al. The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer's disease. Int Psychogeriatr 2009;21:672–687 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.