

Abstract
Peroxisome proliferator-activated receptor γ (PPARγ) is a nuclear receptor that is important in many physiological and pathological processes, such as lipid metabolism, insulin sensitivity, inflammation, cell proliferation, and carcinogenesis. Several studies have shown that PPARγ plays an important role in gastric mucosal injury due to Helicobacter pylori (H. pylori). As H. pylori infection is the main etiologic factor in chronic gastritis and gastric cancer, understanding of the potential roles of PPARγ in H. pylori infection may lead to the development of a therapeutic target. In this paper, the authors discuss the current knowledge on the role of PPARγ in H. pylori infection and its related gastric carcinogenesis.
1. Introduction
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors and members of the nuclear hormone receptor superfamily. To date, three isoforms of PPARs (PPARα, PPARδ/β, and PPARγ) have been identified in mammals. PPAR forms a heterodimer with its preferential binding partner—retinoid X receptor (RXR). The function of PPAR/RXR heterodimer depends on its interactions with cofactor complexes (coactivators or corepressors). After activation by ligand, the PPAR/RXR heterodimer binds to specific DNA response elements called peroxisome proliferator response elements (PPREs) of the target genes. This results in transcription regulation of these genes (Figure 1) [1]. PPARs play a significant role in regulation of fatty acid oxidation and glucose utilization [2]. PPARγ was originally identified as a differentiation transcription factor for adipose tissue [3]. In addition, PPARγ is involved in the control of inflammation and glucose metabolism and participates in the processes of cellular proliferation, differentiation, and apoptosis [4]. Natural ligands for PPARγ are 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and various polyunsaturated fatty acids [5, 6]. The insulin sensitizing thiazolinediones, which are selective ligands of the nuclear transcription factor PPARγ, were the first drugs used to treat insulin resistance in patients with type II diabetes [7].
Figure 1.

A basic mechanism of PPAR signaling. Following ligand binding, PPAR forms a heterodimer with RXR, which binds to the PPRE of target genes and regulates the transcription of these genes.
Helicobacter pylori (H. pylori) infection is the main etiologic agent in gastric inflammation, and longstanding infection with this organism is linked to gastric cancer [8]. Based on epidemiological studies, the risk of gastric cancer conferred by H. pylori has been estimated to be 75% [9]. Although the mechanism of H. pylori-induced carcinogenesis is still being investigated, inflammation is the strongest risk factor in the carcinogenic process [10], because it affects host responses such as epithelial cell proliferation and apoptosis [9]. PPARγ may be involved in the regulation of gene expression associated with inflammation and cancer. This paper reviews current knowledge of the role of PPARγ in H. pylori infection and its related gastric carcinogenesis.
2. PPARγ Expression in H. pylori Infection
PPARγ is predominantly expressed in adipose tissue, intestinal epithelium, monocytes and macrophages, the retina, skeletal muscle, and lymphoid organs [1]. Braissant et al. demonstrated PPARγ expression in the adult rat gastric mucosa by in situ hybridization and immunohistochemistry [11]. Several studies have found that PPARγ expression increases during H. pylori infection [12–14]. First, Konturek et al. demonstrated that PPARγ gene and protein expression were significantly higher in the gastric mucosa of H. pylori-positive gastric cancer patients than in H. pylori-negative healthy controls [12]. In addition, H. pylori eradication significantly reduced PPARγ expression. We demonstrated previously that PPARγ expression, identified by immunohistochemistry, was mostly detected in the nucleus of the foveolar epithelial cells in gastric mucosa and the intensity of PPARγ expression was significantly higher in the 18 patients with H. pylori-associated chronic gastritis than in the 21 H. pylori-negative patients (Figure 2) [13]. However, there was no correlation between the numbers of neutrophils and PPARγ expression in the two groups. Haruna et al. reported results similar to ours [14]. In this study, cyclooxygenase-2 (COX-2) and PPARγ mRNA expression in the gastric mucosa of children were found to be increased with H. pylori infection. The expression of COX-2, which plays an important role in inflammation, carcinogenesis, and development, is regulated by a negative feedback loop mediated through PPARγ [15]. Overexpression of both PPARγ and COX-2 was detected in the gastric mucosa of Mongolian gerbils infected with H. pylori [16]. Taking these findings together, enhanced PPARγ expression in gastric mucosa infected with H. pylori may have anti-inflammatory and cytoprotective effects.
Figure 2.
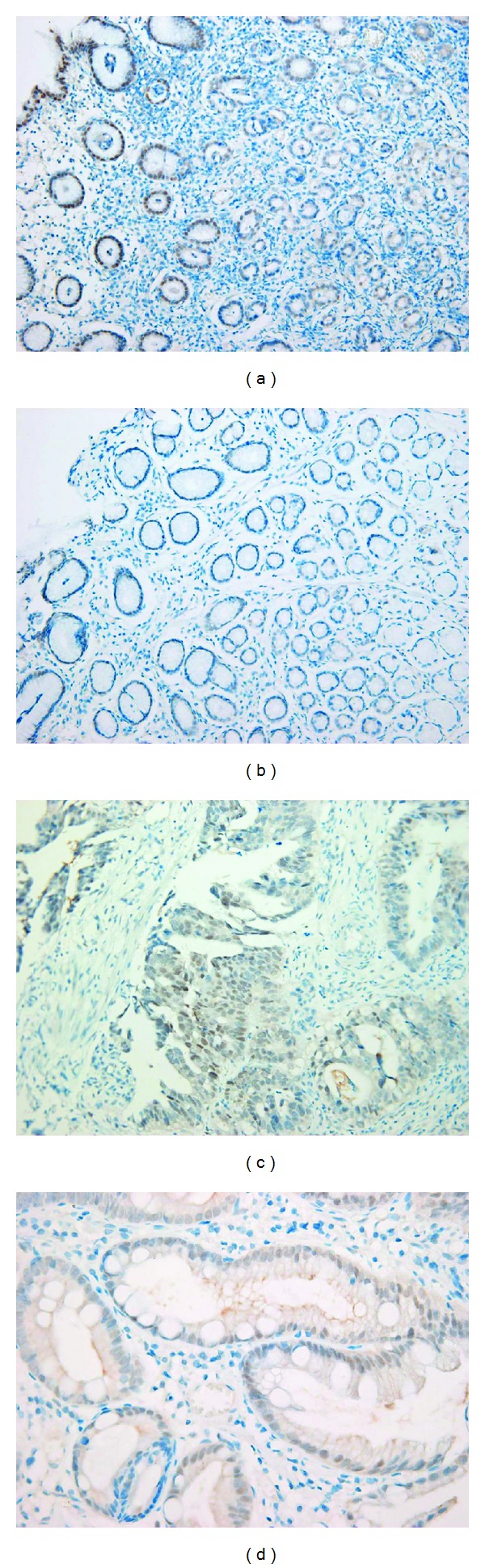
PPARγ protein expression in various tissues. Strong nuclear staining in gastric epithelial cells of H. pylori-infected patients (a) and weak nuclear staining in H. pylori-negative subjects (b). PPARγ protein is also expressed in the nucleus of tumor cells in gastric adenocarcinoma (c) and in noncancerous tissue with intestinal metaplasia adjacent to cancer tissue (d).
3. The Role of PPARγ Activation in H. pylori Infection
Several studies have demonstrated that PPARγ has an overall anti-inflammatory effect [2, 17]. Molecular mechanisms include inhibition of signaling pathways regulating expression of proinflammatory genes (e.g., nuclear factor (NF)-κB) and stress-kinase pathways [17]. Gupta et al. demonstrated that PPARγ activation suppresses H. pylori-induced apoptosis in gastric epithelial cells and that this effect is mediated by direct inhibition of H. pylori-induced NF-κB activation [18]. H. pylori lipopolysaccharide (LPS), a component of the outer membrane, is a potent virulence factor for mucosal inflammatory changes, and its mechanism is mediated by increased proinflammatory cytokine production, excessive nitric oxide (NO) and PG generation, and epithelial cell apoptosis [19, 20]. B. L. Slomiany and A. Slomiany reported that PPARγactivation by ciglitazone inhibits gastric mucosal inflammation and that this effect is mediated by reduced apoptosis, mucosal PGE2 generation, expression of COX-2, and inducible nitric oxide synthase (NOS-2) [20]. In addition, ciglitazone impedes the inhibition of H. pylori LPS on gastric mucin synthesis, an effect likely dependent on the activation of the extracellular signal-related kinase (ERK) pathway by phosphatidylinositol 3-kinase (PI3K) [21]. These findings suggest that PPARγactivation may provide therapeutic benefits in H. pylori-associated gastric inflammation.
The transactivation of epidermal growth factor receptor (EGFR) is strongly linked to H. pylori infection, gastric epithelial hyperplasia, and gastric atrophy [10]. It depends on genes in the cag pathogenicity island, secreted proteins, and host factors such as TLR4 and NOD1 [22]. The biological responses to EGFR transactivation include increased proliferation, reduced apoptosis, altered cell polarity, and enhanced migration [10]. Although the underlying mechanism involved in the differential activation by ciglitazone of the EGFR/Erk mitogen activated protein kinases (MAPK) pathway is not well understood, this effect can be mediated by activation of Erk, an event requiring Src-kinase-dependent EGFR transactivation [23]. Ciglitazone has been shown to suppress H. pylori LPS inhibition of gastric mucin synthesis mediated by Src-kinase-dependent EGFR transactivation [24].
4. The Role of PPARγ in H. pylori-Related Gastric Carcinogenesis
Although the incidence of gastric adenocarcinoma is decreasing, it remains the second most common cause of cancer-related mortality worldwide [25]. There are large variations in the incidence and death rates among racial and ethnic groups, and the gastric cancer incidence and death rate are twice as high as in Asian American/Pacific islanders compared with Caucasians, reflecting an increased prevalence of chronic H. pylori infection [26]. Although gastric cancer treatments are continuously improving, the prognosis for this disease is poor and the survival rate is less than 40% even after curative resection and adjuvant chemotherapy [27].
Although the involvement of PPARγ in the development of cancer in various tissues remains controversial, PPARγ activation has antitumorigenic effects due to its antiproliferative and prodifferentiation activities [1]. Several in vitro studies have found that PPARγ activation results in cell cycle arrest and/or apoptosis of gastric cancer cells [28–32]. First, Takahashi et al. demonstrated that a human gastric cancer cell line, MKN45, expressed PPARγ mRNA and protein, and that PPARγ activation inhibited cell growth and induced apoptosis in gastric cancer cells [28]. Sato et al. used immunohistochemistry to show that the PPARγ protein is expressed in surgically resected specimens from well-, moderately, and poorly differentiated gastric adenocarcinomas as well as in noncancerous gastric mucosa with intestinal metaplasia [29]. The results of our study of PPARγ protein expression in gastric adenocarcinoma and normal mucosa with intestinal metaplasia adjacent to cancer were consistent with Sato's results (Figure 2) [13]. However, recent studies have reported that redistribution of PPARγ expression occurs in human gastric adenocarcinoma [33–35]. The immunohistochemical staining pattern of PPARγ is nuclear in the normal gastric mucosa but primarily cytoplasmic in intestinal metaplasia (IM) [33]. The high cytoplasmic-to-nuclear expression ratio of PPARγ decreases as the differentiation stage changes from IM to adenoma, and to well-, moderately-, and poorly-differentiated cancers. PPARγ is expressed primarily in the nucleus in metastatic gastric cancer [34]. Burgermeister et al. demonstrated that the molecular mechanisms of PPARγ redistribution include interaction with Ras/mitogen activated protein kinases (MAPKs) such as caveolin-1 (Cav1) and docking protein 1 (Dok1) [35].
The inhibitory effect of PPARγ on gastric cancer may be attributed to various mechanisms. Ligand-induced activation of PPARγ was found to inhibit c-MET (an important protooncogene-encoding receptor for hepatocyte growth factor) [36] and expression of cyclin D1 [37] and COX-2 [31], induce expression of p27 [38], p21 [32], and p53 [39], and suppress gastrin (a potent cancer cell growth promoting factor) [40]. An in vivo animal study determined that heterozygous (PPARγ +/−) knockout mice were more susceptible to N-methyl-N-nitrosourea-induced gastric carcinoma than homozygotes (PPARγ +/+), but troglitazone only reduced the incidence of gastric cancer in homozygotes [41].
Konturek et al. reported that expression of tissue PPARγ, tissue levels of proinflammatory cytokines (IL-1β and IL-8), and plasma gastrin concentrations were significantly higher in H. pylori-positive gastric cancer compared to H. pylori-negative controls, but H. pylori eradication reduced these parameters [12]. These findings suggest that these parameters could be implicated in H. pylori-related gastric carcinogenesis. An in vitro study found that H. pylori infection downregulates the expression of p27 in gastric epithelial cells even in the absence of inflammation [42]. Reduced expression of p27 is found in H. pylori-associated intestinal metaplasia and H. pylori eradication reverses the aberrant expression of p27 [43]. Low p27 protein expression has been reported to be associated with increased expression of p27-specific F-box protein Skp2 and H. pylori eradication reverses the aberrant expression of p27 and Skp2 protein [44]. However, p27 and Skp2 mRNA levels were unaffected by H. pylori eradication, suggesting that H. pylori may influence cell cycle progression and carcinogenesis post-translational effects on specific gene expression. We have shown that rosiglitazone inhibited the growth of H. pylori-infected gastric epithelial cells [45]. These effects of rosiglitazone were associated with decreased Skp2 expression, thereby promoting p27 accumulation in H. pylori-infected gastric epithelial cells.
5. PPARγ Polymorphism in H. pylori-Related Gastric Carcinogenesis
A common PPARγ polymorphism, a C to G substitution in exon B, results in a proline to alanine exchange at codon 12 (Pro12Ala) [46]. Functionally, this Ala variant has been reported to show decreased binding to the response element and a lower capacity for activating target genes [47]. PPARγ polymorphism (Pro12Ala) has been found to be associated with various diseases including type II diabetes, cardiovascular disease, and several types of cancer [48]. Pro12Ala polymorphism lowers the risk of diseases in colorectal cancer and type II diabetes [49]. These results could be partly explained by the etiological link between type II diabetes and colorectal cancer. On the contrary, several studies have demonstrated that the Pro12Ala polymorphism is associated with the high risk of gastric adenocarcinoma [48, 50–53]. Liao et al. reported that the carriage of G phenotype or Ala allele in codon 12 of PPARγ was associated with a 2.5-fold increase in the risk of noncardia gastric cancer in Chinese, and that the risk was higher when this polymorphism was combined with H. pylori infection [50]. A recent meta-analysis suggested that carriers of Pro12Ala have a 2.31-fold (95% CI = 1.59–3.36, P heterogeneity = 0.941) increased gastric cancer risk [48]. Considering that PPARγ activation inhibits the growth of gastric cancer cells, these results suggest that gastric carcinogenesis may have a different genetic background than colorectal carcinogenesis.
The role of Pro12Ala polymorphism in peptic ulcer disease remains controversial. Prasad et al. showed that patients with Pro12Ala polymorphism were susceptible to peptic ulcer disease in the presence of H. pylori infection [52]. Meanwhile, Bazargani et al. found no significant increase in the risk of peptic ulcer formation among patients with Pro12Ala polymorphism [53]. This discrepancy may be due to the role of other bacterial virulence factors and/or host factors.
6. Conclusions
In this paper, we focused on the role of PPARγ in H. pylori infection and its related gastric carcinogenesis. PPARγ suppresses inflammation in H. pylori infection and tumor growth in gastric cancer. Emerging evidence indicates that mutations in PPARγ may play a crucial role in the development of noncardia gastric cancer in H. pylori-infected patients. Therefore, further studies are needed to investigate modulation of PPARγ as an effective therapy for chemoprevention and treatment of inflammation in H. pylori infection and gastric cancer.
Acknowledgment
This work was supported, in part, by a National Research Foundation of Korea (NRF) grant funded by the Ministry of Education, Science, and Technology (2010-0023295).
References
- 1.Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nature Reviews Cancer. 2004;4(1):61–70. doi: 10.1038/nrc1254. [DOI] [PubMed] [Google Scholar]
- 2.Kersten S, Desvergne B, Wahli W. Roles of PPARS in health and disease. Nature. 2000;405(6785):421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- 3.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell. 1994;79(7):1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 4.Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. Journal of Endocrinology. 2001;169(3):453–459. doi: 10.1677/joe.0.1690453. [DOI] [PubMed] [Google Scholar]
- 5.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-Δ12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ . Cell. 1995;83(5):803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 6.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell. 1995;83(5):813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 7.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ) The Journal of Biological Chemistry. 1995;270(22):12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 8.Peek RM, Jr., Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nature Reviews Cancer. 2002;2(1):28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 9.Herrera V, Parsonnet J. Helicobacter pylori and gastric adenocarcinoma. Clinical Microbiology and Infection. 2009;15(11):971–976. doi: 10.1111/j.1469-0691.2009.03031.x. [DOI] [PubMed] [Google Scholar]
- 10.Polk DB, Peek RM., Jr. Helicobacter pylori: gastric cancer and beyond. Nature Reviews Cancer. 2010;10(6):403–414. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braissant O, Foufelle F, Scotto C, Dauça M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-α, -β, and -γ in the adult rat. Endocrinology. 1996;137(1):354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- 12.Konturek PC, Kania J, Kukharsky V, et al. Implication of peroxisome proliferator-activated receptor γ and proinflammatory cytokines in gastric carcinogenesis: link to Helicobacter pylori-infection. Journal of Pharmacological Sciences. 2004;96(2):134–143. doi: 10.1254/jphs.fpj04016x. [DOI] [PubMed] [Google Scholar]
- 13.Son SH, Kim HK, Ji JS, et al. Expression of peroxisome proliferator-activated receptor (PPAR) gamma in Helicobacter pylori-infected gastric epithelium. The Korean Journal of Gastroenterology. 2007;49(2):72–78. in Korean. [PubMed] [Google Scholar]
- 14.Haruna H, Shimizu T, Ohtsuka Y, et al. Expression of COX-1, COX-2, and PPAR-γ in the gastric mucosa of children with Helicobacter pylori infection. Pediatrics International. 2008;50(1):1–6. doi: 10.1111/j.1442-200X.2007.02504.x. [DOI] [PubMed] [Google Scholar]
- 15.Inoue H, Tanabe T, Umesono K. Feedback control of cyclooxygenase-2 expression through PPARγ . The Journal of Biological Chemistry. 2000;275(36):28028–28032. doi: 10.1074/jbc.M001387200. [DOI] [PubMed] [Google Scholar]
- 16.Konturek PC, Kania J, Konturek JW, Nikiforuk A, Konturek SJ, Hahn EG. H. pylori infection, atrophic gastritis, cytokines, gastrin, COX-2, PPARγ and impaired apoptosis in gastric carcinogenesis. Medical Science Monitor. 2003;9(7):SR53–SR66. [PubMed] [Google Scholar]
- 17.Dubuquoy L, Dharancy S, Nutten S, Pettersson S, Auwerx J, Desreumaux P. Role of peroxisome proliferator-activated receptor γ and retinoid X receptor heterodimer in hepatogastroenterological diseases. The Lancet. 2002;360(9343):1410–1418. doi: 10.1016/S0140-6736(02)11395-X. [DOI] [PubMed] [Google Scholar]
- 18.Gupta RA, Polk DB, Krishna U, et al. Activation of peroxisome proliferator-activated receptor γ suppresses nuclear factor κB-mediated apoptosis induced by Helicobacter pylori in gastric epithelial cells. The Journal of Biological Chemistry. 2001;276(33):31059–31066. doi: 10.1074/jbc.M104141200. [DOI] [PubMed] [Google Scholar]
- 19.Piotrowski J, Piotrowski H, Skrodzka D, Slomiany A, Slomiany BL. Induction of acute gastritis and epithelial apoptosis by Helicobacter pylori lipopolysaccharide. Scandinavian Journal of Gastroenterology. 1997;32(3):203–211. doi: 10.3109/00365529709000195. [DOI] [PubMed] [Google Scholar]
- 20.Slomiany BL, Slomiany A. Suppression of gastric mucosal inflammatory responses to Helicobacter pylori lipopolysaccharide by peroxisome proliferator-activated receptor γ activation. IUBMB Life. 2002;53(6):303–308. doi: 10.1080/15216540213459. [DOI] [PubMed] [Google Scholar]
- 21.Slomiany BL, Slomiany A. Impedance of Helicobacter pylori lipopolysaccharide interference with gastric mucin synthesis by peroxisome proliferator-activated receptor γ activation involves phosphatidylinositol 3-kinase/ERK pathway. IUBMB Life. 2003;55(2):97–102. doi: 10.1002/tbmb.718540877. [DOI] [PubMed] [Google Scholar]
- 22.Basu S, Pathak SK, Chatterjee G, Pathak S, Basu J, Kundu M. Helicobacter pylori protein HP0175 transactivates epidermal growth factor receptor through TLR4 in gastric epithelial cells. The Journal of Biological Chemistry. 2008;283(47):32369–32376. doi: 10.1074/jbc.M805053200. [DOI] [PubMed] [Google Scholar]
- 23.Gardner OS, Dewar BJ, Earp HS, Samet JM, Graves LM. Dependence of peroxisome proliferator-activated receptor ligand-induced mitogen-activated protein kinase signaling on epidermal growth factor receptor transactivation. The Journal of Biological Chemistry. 2003;278(47):46261–46269. doi: 10.1074/jbc.M307827200. [DOI] [PubMed] [Google Scholar]
- 24.Slomiany BL, Slomiany A. Role of epidermal growth factor receptor transactivation in PPARγ-dependent suppression of Helicobacter pylori interference with gastric mucin synthesis. Inflammopharmacology. 2004;12(2):177–188. doi: 10.1163/1568560041352248. [DOI] [PubMed] [Google Scholar]
- 25.Crew KD, Neugut AI. Epidemiology of gastric cancer. World Journal of Gastroenterology. 2006;12(3):354–362. doi: 10.3748/wjg.v12.i3.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA: A Cancer Journal for Clinicians. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 27.Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. The New England Journal of Medicine. 2001;345(10):725–730. doi: 10.1056/NEJMoa010187. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi N, Okumura T, Motomura W, Fujimoto Y, Kawabata I, Kohgo Y. Activation of PPARγ inhibits cell growth and induces apoptosis in human gastric cancer cells. FEBS Letters. 1999;455(1-2):135–139. doi: 10.1016/s0014-5793(99)00871-6. [DOI] [PubMed] [Google Scholar]
- 29.Sato H, Ishihara S, Kawashima K, et al. Expression of peroxisome proliferator-activated receptor (PPAR)γ in gastric cancer and inhibitory effects of PPARγ agonists. British Journal of Cancer. 2000;83(10):1394–1400. doi: 10.1054/bjoc.2000.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y-X, Zhong X-Y, Qin Y-F, Bing W, He L-Z. 15d-PGJ2 inhibits cell growth and induces apoptosis of MCG-803 human gastric cancer cell line. World Journal of Gastroenterology. 2003;9(10):2149–2153. doi: 10.3748/wjg.v9.i10.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leung WK, Bai AHC, Chan VYW, et al. Effect of peroxisome proliferator activated receptor γ ligands on growth and gene expression profiles of gastric cancer cells. Gut. 2004;53(3):331–338. doi: 10.1136/gut.2003.021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheon CW, Kim DH, Kim DH, Cho YH, Kim JH. Effects of ciglitazone and troglitazone on the proliferation of human stomach cancer cells. World Journal of Gastroenterology. 2009;15(3):310–320. doi: 10.3748/wjg.15.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nomura S, Nakajima A, Ishimine S, Matsuhashi N, Kadowaki T, Kaminishi M. Differential expression of peroxisome proliferator-activated receptor in histologically different human gastric cancer tissues. Journal of Experimental and Clinical Cancer Research. 2006;25(3):443–448. [PubMed] [Google Scholar]
- 34.He Q, Chen J, Lin H-L, Hu P-J, Chen M-H. Expression of peroxisome proliferator-activated receptor γ, E-cadherin and matrix metalloproteinases-2 in gastric carcinoma and lymph node metastases. Chinese Medical Journal. 2007;120(17):1498–1504. [PubMed] [Google Scholar]
- 35.Burgermeister E, Friedrich T, Hitkova I, et al. The Ras inhibitors caveolin-1 and docking protein 1 activate peroxisome proliferator-activated receptor γ through spatial relocalization at helix 7 of its ligand-binding domain. Molecular and Cellular Biology. 2011;31(16):3497–3510. doi: 10.1128/MCB.01421-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitamura S, Miyazaki Y, Hiraoka S, et al. PPARγ inhibits the expression of c-MET in human gastric cancer cells through the suppression of Ets. Biochemical and Biophysical Research Communications. 1999;265(2):453–456. doi: 10.1006/bbrc.1999.1715. [DOI] [PubMed] [Google Scholar]
- 37.Yoshida K, Tanabe K, Fujii D, Oue N, Yasui W, Toge T. Induction mechanism of apoptosis by troglitazone through peroxisome proliferator-activated receptor-γ in gastric carcinoma cells. Anticancer Research. 2003;23(1):267–273. [PubMed] [Google Scholar]
- 38.Takeuchi S, Okumura T, Motomura W, Nagamine M, Takahashi N, Kohgo Y. Troglitazone induces G1 arrest by p27Kip1 induction that is mediated by inhibition of proteasome in human gastric cancer cells. Japanese Journal of Cancer Research. 2002;93(7):774–782. doi: 10.1111/j.1349-7006.2002.tb01319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagamine M, Okumura T, Tanno S, et al. PPARγ ligand-induced apoptosis through a p53-dependent mechanism in human gastric cancer cells. Cancer Science. 2003;94(4):338–343. doi: 10.1111/j.1349-7006.2003.tb01443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Konturek PC, Kania J, Kukharsky V, Ocker S, Hahn EG, Konturek SJ. Influence of gastrin on the expression of cyclooxygenase-2, hepatocyte growth factor and apoptosis-related proteins in gastric epithelial cells. Journal of Physiology and Pharmacology. 2003;54(1):17–32. [PubMed] [Google Scholar]
- 41.Lu J, Imamura K, Nomura S, et al. Chemopreventive effect of peroxisome proliferator-activated receptor γ on gastric carcinogenesis in mice. Cancer Research. 2005;65(11):4769–4774. doi: 10.1158/0008-5472.CAN-04-2293. [DOI] [PubMed] [Google Scholar]
- 42.Shirin H, Sordillo EM, Kolevska TK, et al. Chronic Helicobacter pylori infection induces an apoptosis-resistant phenotype associated with decreased expression of p27kip1 . Infection and Immunity. 2000;68(9):5321–5328. doi: 10.1128/iai.68.9.5321-5328.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu J, Leung WK, Ng EKW, et al. Effect of Helicobacter pylori eradication on expression of cyclin D2 and p27 in gastric intestinal metaplasia. Alimentary Pharmacology and Therapeutics. 2001;15(9):1505–1511. doi: 10.1046/j.1365-2036.2001.01038.x. [DOI] [PubMed] [Google Scholar]
- 44.Kim SS, Meitner P, Konkin TA, Cho YS, Resnick MB, Moss SF. Altered expression of Skp2, c-Myc and p27 proteins but not mRNA after H. pylori eradication in chronic gastritis. Modern Pathology. 2006;19(1):49–58. doi: 10.1038/modpathol.3800476. [DOI] [PubMed] [Google Scholar]
- 45.Kim SS, Cho YS, Kim HK, et al. The effect of rosiglitazone on the cell proliferation and the expressions of p27 and skp2 in Helicobacter pylori infected human gastric epithelial cells. The Korean Journal of Gastroenterology. 2010;55(4):225–231. doi: 10.4166/kjg.2010.55.4.225. in Korean. [DOI] [PubMed] [Google Scholar]
- 46.Chung-Jen Y, Beamer BA, Negri C, et al. Molecular scanning of the human peroxisome proliferator activated receptor γ (hPPARγ) gene in diabetic Caucasians: identification of a Pro12Ala PPARγ2 missense mutation. Biochemical and Biophysical Research Communications. 1997;241(2):270–274. doi: 10.1006/bbrc.1997.7798. [DOI] [PubMed] [Google Scholar]
- 47.Deeb SS, Fajas L, Nemoto M, et al. A Pro12Ala substitution in PPARγ2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nature Genetics. 1998;20(3):284–287. doi: 10.1038/3099. [DOI] [PubMed] [Google Scholar]
- 48.Xu W, Li Y, Wang X, et al. PPARγ polymorphisms and cancer risk: a meta-analysis involving 32,138 subjects. Oncology Reports. 2010;24(2):579–585. [PubMed] [Google Scholar]
- 49.Koh W-P, Yuan JM, Van Den Berg D, Ingles SA, Yu MC. Peroxisome proliferator-activated receptor (PPAR) γ gene polymorphisms and colorectal cancer risk among Chinese in Singapore. Carcinogenesis. 2006;27(9):1797–1802. doi: 10.1093/carcin/bgl001. [DOI] [PubMed] [Google Scholar]
- 50.Liao S-Y, Zeng Z-R, Leung WK, et al. Peroxisome proliferator-activated receptor-gamma Pro12Ala polymorphism, Helicobacter pylori infection and non-cardia gastric carcinoma in Chinese. Alimentary Pharmacology and Therapeutics. 2006;23(2):289–294. doi: 10.1111/j.1365-2036.2006.02739.x. [DOI] [PubMed] [Google Scholar]
- 51.Tahara T, Arisawa T, Shibata T, et al. Influence of peroxisome proliferator-activated receptor (PPAR)γ Plo12Ala polymorphism as a shared risk marker for both gastric cancer and impaired fasting glucose (IFG) in Japanese. Digestive Diseases and Sciences. 2008;53(3):614–621. doi: 10.1007/s10620-007-9944-8. [DOI] [PubMed] [Google Scholar]
- 52.Prasad KN, Saxena A, Ghoshal UC, Bhagat MR, Krishnani N. Analysis of Pro12Ala PPAR gamma polymorphism and Helicobacter pylori infection in gastric adenocarcinoma and peptic ulcer disease. Annals of Oncology. 2008;19(7):1299–1303. doi: 10.1093/annonc/mdn055. [DOI] [PubMed] [Google Scholar]
- 53.Bazargani A, Khoramrooz SS, Kamali-Sarvestani E, Taghavi SA, Saberifiroozi M. Association between peroxisome proliferator-activated receptor-γ gene polymorphism (Pro12Ala) and Helicobacter pylori infection in gastric carcinogenesis. Scandinavian Journal of Gastroenterology. 2010;45(10):1162–1167. doi: 10.3109/00365521.2010.499959. [DOI] [PubMed] [Google Scholar]