

Our perception of intracellular protein degradation has changed dramatically during the recent decade. From a scavenger, unregulated, and nonspecific “end point” process, it has become clear that proteolysis of cellular proteins is a highly complex, temporally controlled, and tightly regulated process that plays major roles in a variety of basic pathways during cell life and death. Two major proteolytic cascades have been described. Caspases are involved in programmed cell death (apoptosis), whereas the degradation of most short-lived regulatory cellular proteins is mediated by the ubiquitin-proteasome pathway. Among these are regulators of cell cycle and division such as mitotic and G1 cyclins and cyclin-dependent kinase inhibitors, growth regulators such as c-Fos and c-Jun, tumor suppressors such as p53, surface receptors such as the growth hormone receptor, and ion channels, cystic fibrosis transmembrane conductance regulator (CFTR) for example. The system is also involved in selective proteolysis of abnormal/mutated proteins and in the processing of major histocompatibility complex (MHC) class I-restricted antigens. The discovery that the system is involved in the degradation of c-myc and in the two-step proteolytic activation of NF-κB, for example, signaled the “entry” of ubiquitin-mediated degradation into the area of transcriptional regulation. Via the degradation of short-lived and key regulatory proteins, the system appears to play important roles in a variety of basic cellular processes. Among these are regulation of cell cycle and division, involvement in the cellular response to stress and to extracellular modulators, morphogenesis of neuronal networks, modulation of cell surface receptors, ion channels and the secretory pathway, DNA repair, biogenesis of organelles, and regulation of the immune and inflammatory responses. Recent evidence indicates that the system is involved in apoptosis as well. With such a broad range of substrates and processes, it is not surprising that aberrations in the process recently have been implicated in the pathogenesis of several diseases, both inherited and acquired. Among these are muscle degeneration that follows denervation or prolonged immobilization, certain forms of Alzheimer’s disease, male sterility, and Angelman’s syndrome (for recent reviews of the ubiquitin system, see refs. 1–4).
Degradation of a protein via the ubiquitin pathway proceeds in two discrete and successive steps: (i) covalent attachment of multiple ubiquitin molecules to the protein substrate, and (ii) degradation of the targeted protein by the 26S proteasome complex with the release of free and reusable ubiquitin. To ensure efficient and specific removal of a certain protein at a certain time point, both ubiquitin conjugation and degradation of the tagged substrates must be tightly regulated. In a study published in this issue of the Proceedings (5), Zhang and colleagues report the identification of an activation region in the α subunit of the proteasome activator PA28 (REG). To incorporate this finding into the appropriate biochemical and physiological context, we briefly shall review our current understanding of the ubiquitin proteolytic pathway. The system (depicted in Fig. 1) consists of several components that act in concert. One of these, ubiquitin, an evolutionarily conserved protein of 76 residues, is activated in its C-terminal Gly to a high-energy thiol ester intermediate, a reaction catalyzed by the ubiquitin-activating enzyme, E1. After activation, one of several E2 enzymes (ubiquitin-carrier proteins or ubiquitin-conjugating enzymes, UBCs) transfers the activated ubiquitin moiety from E1 to a member of the ubiquitin-protein ligase family, E3, to which the substrate protein is specifically bound. E3 catalyzes the last step in the conjugation process, covalent attachment of ubiquitin to the substrate. The first ubiquitin moiety is transferred to the ɛ-NH2 group of a Lys residue of the protein substrate to generate an isopeptide bond. In successive reactions, a polyubiquitin chain is synthesized by processive transfer of additional activated moieties to Lys48 of the previously conjugated ubiquitin molecule. The chain serves, most probably, as a recognition marker for the proteasome (see below). Ubiquitin K48R or methylated ubiquitin (in which all the free amino groups were chemically modified) cannot generate polyubiquitin chains and serve as chain terminators. Consequently, when overexpressed in cells or introduced into cell-free systems, they inhibit proteolysis. The binding of the substrate to E3 is specific and implies that E3s play a major role in recognition and selection of proteins for conjugation and subsequent degradation. The structure of the system appears to be hierarchical: a single E1 appears to carry out activation of ubiquitin required for all modifications. Several major species of E2 enzymes were characterized in mammalian cells. It appears that each E2 can act with one or more E3 enzymes. Although only a relatively few E3 enzymes have been described so far, it appears that the ubiquitin ligases belong to a large, still-growing family of enzymes. As for the mode of recognition of the ligases, except for a few cases, it is unlikely that each E3 targets a single substrate. Rather, it is conceivable that several different cellular proteins are recognized by a single ligase via a similar, but clearly not identical, structural motif. A few proteins may be recognized via their free and “destabilizing” N-terminal residue (“N-end rule”; ref. 6). However, the vast majority of cellular proteins are acetylated at their N termini or have “stabilizing” amino termini and are targeted through different signals. Some are recognized via primary sequences that reside downstream from the N-terminal residue. Others are targeted via secondary, posttranslational modification(s) such as phosphorylation, or after association with ancillary proteins such as oncoproteins or molecular chaperones.
Figure 1.
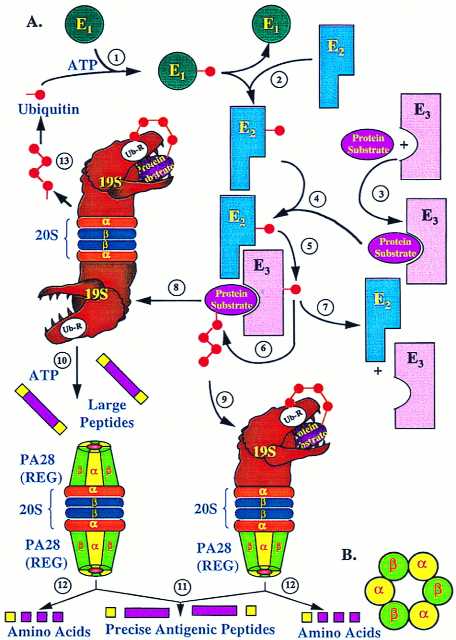
(A) The ubiquitin-proteasome pathway. Conjugation of ubiquitin to the target protein: 1) activation of ubiquitin by E1. 2) Transfer of the activated ubiquitin moiety to a member of the E2 family of enzymes. 3) Formation of a binary complex between E3 and the protein substrate. 4) Formation of a ternary E2-E3-protein substrate complex. 5) Transfer of the activated ubiquitin moiety from E2 to E3. 6) Synthesis of protein substrate-anchored polyubiquitin chain. 7) Recycling of E2 and E3. Degradation of the polyubiquitin-conjugated substrate by the proteasome: 8) Transfer of the protein substrate-ubiquitin adduct to the symmetrical 19S-20S-19S proteasome. 9) Transfer of the protein substrate-ubiquitin adduct to the putative asymmetrical 19S-20S-PA28 proteasome. 10) Generation of large peptides by the symmetrical 19S-20S-19S proteasome. 11) “Trimming” of the large peptides into precise antigenic epitopes in a two-step mechanism by the asymmetrical 19S-20S-PA28 or the symmetrical PA28-20S-PA28 proteasomes. 12) Generation of free amino acids by the symmetrical or asymmetrical PA28-containing proteasomes. 13) Recycling of ubiquitin by ubiquitin C-terminal hydrolases (isopeptidases). Ub-R denotes ubiquitin chain receptor (binding) subunit of the 19S complex. (B) The (αβ)3 structure of the PA28 complex.
After conjugation, the protein moiety of the adduct is degraded by the proteasome complex, and free and reusable ubiquitin is released (for recent reviews on proteasomes, see refs. 7–10). Although the current consensus is that 26S proteasome serves as the main proteolytic arm of the ubiquitin system, the substrate spectrum of the enzyme may be broader and also include nonubiquitinylated proteins. One well-studied case is that of ornithine decarboxylase (ODC). ODC is degraded after a noncovalent association with antizyme that may function in the recognition process as a substrate-specific ubiquitin-like chaperone. Other substrates, mostly endoplasmic reticulum (ER) proteins, also may be degraded by the proteasome without previous ubiquitinylation, although this has not been firmly established. These may include, for example, misfolded MHC heavy chain molecules (HCs), the mammalian 3-hydroxy-3-methyl glutaryl CoA (HMG-R), and the Z variant of the α-1-antitrypsin (α1-ATZ) (reviewed in ref. 11). Also, it is clear now that not all ubiquitinylated proteins are targeted for degradation by the proteasome. This is true mostly for cell surface mature membrane proteins such as the growth hormone receptor. For these proteins, ubiquitin modification occurs after ligand binding and is required for their endocytosis and targeting to the lysosome (reviewed in ref. 12). Degradation of membrane-anchored proteins by the ubiquitin system raise important, yet unresolved, mechanistic problems that are mostly related to the question of how two topologically distinct events such as misfolding in the ER and degradation in the cytosol, or ligand binding in the extracellular domain and ubiquitinylation on a cytosolic tail of a receptor come together. For cell surface membrane proteins an obvious problem relates to the role of ubiquitin modification: is it required for endocytosis of the tagged protein or in a later stage, for its specific targeting and uptake by the lysosome. For ER proteins degraded by the cytosolic proteasome, important questions involve the mechanisms that underlie retrieval of these proteins across the membrane back into the cytosol. The transmembrane domain of membrane-anchored proteins is hydrophobic and its removal from the membrane probably involves a specialized, energy-dependent, channel-based transport. For lumenal ER proteins the question centers on how they are transported back into the cytosol.
Recent evidence suggests that the principle of ubiquitin modification is not limited to targeting of proteins for degradation. It appears that ubiquitin is a member of a larger family, and several ubiquitin-like proteins also have been described. One interesting case involves targeting into complex structures. It was found that localization of RanGAP1, a Ran GTPase activating protein, to the nuclear pore complex protein RanBP2, is dependent on a single and stable covalent modification by a 11.5-kDa, 101-residue ubiquitin-like protein, SUMO-1 (13). The activation reaction is similar to that of ubiquitin and involves E1 and a specific E2, UBC9. Thus, posttranslational covalent modification by ubiquitin and ubiquitin-like molecules serves a broad spectrum of functions. It is involved in targeting of proteins for degradation by the proteasome and the lysosome, but also serves nonproteolytic functions.
The best-studied proteasome complex involved in degradation of ubiquitin-tagged proteins is the 26S proteasome complex. It is a “dumbshell-shaped” symmetric structure composed of a core catalytic unit, a barrel-shaped 20S proteasome complex, which is flanked at both sides by regulatory 19S proteasome complexes (19S-20S-19S). The crystal structure of the eukaryotic (yeast) 20S proteasome has been resolved at 2.4 Å (14). The study has corroborated previous predictions on the structure of the complex, but also revealed some unexpected features. The yeast complex is arranged as a stack of four rings, each containing seven distinct subunits, α1–7β1–7β1–7α1–7. The different α and β subunits have molecular masses in the range of 25–30 kDa. The active sites reside in three β subunits, β1, β2, and β5, but not in α subunits. Topological analysis of the location of the different subunits has revealed that for the three distinct proteolytic activities, the trypsin-like, the chymotrypsin-like, and the postglutamyl peptidyl hydrolytic activities, the active sites are generated by adjacent pairs of identical β-type subunits residing in different β rings. Analysis of the active site residues reveals a new kind of proteolytic mechanism. The three β subunits undergo autocatalysis between the last Gly residue of the pro-peptide and Thr1 of the mature subunit that participates in the autocatalytic process and becomes an essential part of the catalytic site. The resolution of the crystal structure also enabled better understanding of the mode of action of the different proteasome inhibitors. Thr1 in β1, β2, and β5 binds the less-specific calpain I inhibitor Acetyl-Leu-Leu-Norleucinal (ALLN). Lactacystin, a more specific inhibitor, can be covalently bound to β5 where it can generate, in addition, a host of hydrogen bonds with other surrounding side chains. Mutational analysis has revealed that the other β subunits, in particular β4 and β7 that reside adjacently to β5 and β1, affect the activity of these subunits. β6 and β7 also are generated from pro-proteins via specific processing. β3 and β4 are not processed. Because of their possible role in the establishment of intersubunit contacts and stabilization of the complex structure, it appears that the propeptides are essential for the biogenesis and stabilization of the proteasomal structure, and processing occurs only after assembly. The structure of the crystal also has shown that the α chains, although catalytically inactive, play an essential role in stabilizing the two-ring structure of the β chains. They also must play a role in the binding of the 19S cap complexes, but the structure of the contacts and mechanisms of binding will be elucidated only when the crystal structure of the 26S complex will be resolved. The crystal structure also has revealed a distance of 28 Å between Thr1 residues of adjacent active β subunits. This distance probably determines the length of the peptides generated during the proteolytic process (≈8-aa residues) and explains the role of the proteasome in generation of antigenic peptides presented on class I MHC molecules. The existence, however, of intermediate products of a variable length suggests a second hydrolytic site downstream to Thr1 (see below).
An important, yet unresolved, problem involves the entry of protein substrates and exit of proteolytic products from the proteasome. The archaebacterial (Thermoplasma acidophilum) proteasome contains two entry pores of ≈13 Å at the two ends of the cylinder surrounded by defined segments of the seven α chains. In striking and rather surprising contrast, these ports of entry do not exist in the eukaryotic 20S proteasome, and entry to the inter-β rings catalytic chamber is not possible from the ends of the complex. The N-terminal domains of α1, α2, α3, α6, and α7 protrude toward each other and fill the space in several layers of tightly interacting side chains. Thus, entry from the ends will be possible only after substantial rearrangement that can potentially occur after association with the 19S complex. Such a rearrangement also may require metabolic energy that can be provided by the ATPase activity of several of the 19S complex subunits. The yeast complex displays some narrow side orifices, particularly at the interface between the α and β rings. These openings lead directly to the Thr1 active sites. They are coated with polar residues that can potentially rearrange to generate ≈10 Å apertures through which unfolded and extended protein substrates may enter.
Regulation of the 20S proteasome activity occurs at several levels. After stimulation of antigen-presenting cells with interferon γ, three constitutive β subunits, 1, 2, and 5, are replaced with new and distinct β subunits, β1i (LMP2), β2i (MECL1), and β5i (LMP7). The new subunits are relatively more efficient in the generation of antigenic peptides recognized by the MHC class I molecules and the appropriate cytotoxic T cells via the T cell receptors. A different type of regulation involves complex formation with regulatory complexes. The 26S proteasome is generated after ATP-dependent association of the 20S complex with two 19S complexes. The 19S complexes are composed of at least 18 distinct proteins with a molecular mass range of 25–110 kDa. This complex serves as the port of entry into the catalytic core and provides the different regulatory functions that are necessary to ensure selective degradation of ubiquitin-tagged substrates. These include, for example, a binding site for ubiquitin chains, ubiquitin recycling activity, and several ATPases, as well as the ability to stimulate the different peptidase activities of the 20S complex. Indeed, subunits that carry out such activities have been identified. Of particular interest is the ubiquitin chain binding subunit that has been described in both mammals (S5a) and plants (MBP1). These subunits bind ubiquitin monomers; however, polyubiquitin chains, and in particular those that contain more then four moieties, bind at a higher affinity. Recently, the yeast gene encoding the homologous chain, Mcb1, has been cloned. Surprisingly, Δmcb1 deletion mutants do not display any growth defect and degrade normally ubiquitinylated proteins, except for the linear fusion model protein ubiquitin-Pro-β-Gal. They do display, however, a slight sensitivity to stress, such as exposure to amino acid analogs (15). A possible explanation for these unexpected results is that ubiquitinylated proteins are recognized by an additional, yet undefined, proteasomal subunit. One such candidate is the deubiquitinylating enzyme Doa4 that functions to remove ubiquitin moieties from proteolytic substrates. A remote possibility is that the proteasome does not recognize ubiquitin moieties, but similar to molecular chaperones, it associates with ill-defined misfolded motifs in the protein substrate. These motifs are generated after “denaturation” of the protein by ubiquitin tagging. Identification of the specificity of recognition of the proteasome and the role ubiquitin plays in this process are essential for understanding the mechanisms of action of the protease in particular and the ubiquitin system in general. Another regulatory function of the 19S proteasome involves polyubiquitin chain editing. The complex contains a 37-kDa isopeptidase that removes single ubiquitin moieties from the distal end of short poly-ubiquitin chains (16). It is assumed that this isopeptidase is involved in editing and rescue of poorly ubiquitinylated or slowly degraded proteins from degradation. It is different in this respect from Doa4 and an ATP-dependent isopeptidase that is associated with the proteasome. The two enzymes are involved in recycling of ubiquitin and maintenance of the free ubiquitin level in the cell.
Another complex that associates with the 20S proteasome and enhances dramatically its activity is PA28 (REG or the 11S regulator; refs. 17 and 18). Unlike the association with 19S, complex formation with PA28 is ATP-independent. After association, PA28 increases the Vmax and decreases the Km of the 20S complex toward a whole array of different peptides. The PA28-20S-PA28 complex is inactive, however, toward intact native or ubiquitin-conjugated proteins. The pure activator is a complex of two ≈28-kDa subunits, PA28α and PA28β, which are ≈50% identical. Immunoprecipitation with subunit-specific antibodies and chemical crosslinking experiments revealed that PA28 is a ring-shaped hexamer that is composed of alternating α and β subunits with a stoichiometry of (αβ)3 (19). Interestingly, these subunits are also 30–40% identical to a nuclear protein of hitherto unknown function, the Ki antigen, that reacts with sera from patients with the autoimmune disease systemic lupus erythematosus. The hexameric complex has a ring-shape structure (Fig. 1) and it caps the 20S proteasome complex on either one or both endplates. The cap structure is traversed by a central channel with a 20 Å opening at the extreme end and a 30 Å opening at the proteasome binding surface (20, 21). The openings and the channel serve, most probably, as part of the translocation machinery of peptide substrates en route to the 20S complex catalytic chamber. PA28α can generate hexa- or hepta-homomultimers that associate with the 20S complex and activate it, although, less efficiently then the (αβ)3 heteromultimer. In contrast, PA28β does not associate with the 20S complex and does not have any stimulating activity. The role of the β subunits probably is to modulate the activity of PA28 activity by indirectly influencing the activity of the α subunits or by modifying the affinity of the PA28 particle to the 20S proteasome.
PA28α contains in its central part a unique sequence, the KEKE motif that is a hydrophilic domain composed of alternating positively charged Lys residues and negatively charged Glu residues. It was postulated that this motif, which does not exist in PA28β, promotes protein–protein interaction between the α subunits of the PA28 particle and the α subunits of the 20S proteasome (22). Mutational analysis revealed, however, that ΔKEKE PA28α retains its stimulatory activity (23). Binding to the 20S proteasome, however, requires an intact C terminus of the PA28α and may involve the C2 α subunit of the 20S proteasome (8). It was suggested that phosphorylation activates the stimulatory activity of PA28α, however, this mode of regulation has not been firmly established.
Mutational analysis of PA28α by Zhang and colleagues (5) revealed several inactivating mutations in a defined loop at the base of the molecule. Some of the mutant proteins bind tightly to the 20S complex, but cannot activate it (5). Thus, binding to the proteasome can be clearly separated from the stimulatory activity of PA28. Interestingly, there is a large gap in the distribution of the inactivating mutations spanning amino acid residues 51–122. This mutation-free zone represents a region that is unique to each subunit in the PA28 complex. It is probably not involved in the interaction of PA28 with the 20S complex or in stimulating its proteolytic activity. Rather it may play a role in the function of the particle in the intact cell such as in its intracellular localization or association with other components that determine its specific activity and interactions.
An important problem involves the physiological roles of PA28. Both subunits are markedly induced by interferon γ, suggesting a role for the particle in the antigen processing function of the proteasome. Overexpression of PA28α in a mouse fibroblast line that expresses the cytomegalovirus pp89 protein results in a marked enhancement of recognition by pp89-specific cytotoxic T cells. Similarly, the presentation of an influenza nucleoprotein also was enhanced (24). Studies in a cell-free system revealed that using a coordinated double-cleavage mechanism, the PA28-20S-PA28 complex, can efficiently trim large peptides (that have flanking sequences on both the N and C termini) to the precise antigenic epitopes recognized by the MHC complex and the appropriate cytotoxic T cell (25). Thus, it appears that PA28 plays an important role in processing of antigens for presentation on class I MHC molecules. Because PA28-20S-PA28 proteasome cannot digest intact native or ubiquitinylated proteins, it must act downstream to the 19S-20S-19S proteasome that degrades ubiquitin-tagged proteins or large peptides. It is also possible, although it has not been demonstrated, that a single asymmetrical 19S-20S-PA28 proteasome exists that can carry out this two-step proteolytic process, initial proteolysis to large peptides, and final trimming. The symmetrical or putative asymmetrical PA28-containing proteasomes also may be involved in terminal degradation of peptides to free amino acids, an activity that cannot be catalyzed by the 19S-20S-19S proteasome (Fig. 1). Thus, it appears that elucidation of the cellular roles of the PA28 complex will have to await further experimentation.
Acknowledgments
Work in our laboratories is supported by grants from the Israel Science Foundation founded by the Israeli Academy of Sciences and Humanities—Centers of Excellence Program, the Israeli Ministry of Science, the German-Israeli Foundation for Scientific Research and Development (GIF), and the United Kingdom-Israel Science and Technology Research Fund (to A.C.), and the National Institutes of Health (to A.L.S.).
References
- 1.Hershko A. Trends Biochem Sci. 1996;21:445–449. doi: 10.1016/s0968-0004(96)10054-2. [DOI] [PubMed] [Google Scholar]
- 2.Hochstrasser M. Annu Rev Genet. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- 3.Jentsch S, Schlenker S. Cell. 1995;82:881–884. doi: 10.1016/0092-8674(95)90021-7. [DOI] [PubMed] [Google Scholar]
- 4.Ciechanover A. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, Clawson A, Realini C, Jensen C, Knowlton J R, Hill C P, Rechsteiner M. Proc Natl Acad Sci USA. 1998;95:2807–2811. doi: 10.1073/pnas.95.6.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Varshavsky A. Cell. 1992;69:725–735. doi: 10.1016/0092-8674(92)90285-k. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt M, Kloetzel P-M. FASEB J. 1997;11:1235–1243. doi: 10.1096/fasebj.11.14.9409542. [DOI] [PubMed] [Google Scholar]
- 8.Coux O, Tanaka K, Goldberg A L. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 9.Hilt W, Wolf D H. Trends Biochem Sci. 1996;21:96–102. [PubMed] [Google Scholar]
- 10.Stock D, Nederlof P M, Seemüller E, Baumeister W, Huber R, Löwe J. Curr Opin Biotechnol. 1996;7:376–385. doi: 10.1016/s0958-1669(96)80111-x. [DOI] [PubMed] [Google Scholar]
- 11.Sommer, T. & Wolf, D. H. FASEB J. 11, 1227–1233. [DOI] [PubMed]
- 12.Hicke L. FASEB J. 1997;11:1215–1226. doi: 10.1096/fasebj.11.14.9409540. [DOI] [PubMed] [Google Scholar]
- 13.Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. Cell. 1997;88:97–107. doi: 10.1016/s0092-8674(00)81862-0. [DOI] [PubMed] [Google Scholar]
- 14.Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik H D, Huber R. Nature (London) 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 15.van Nocker S, Sadis S, Rubin D M, Glickman M, Fu H, Coux O, Wefes I, Finley D, Vierstra R D. Mol Cell Biol. 1996;16:6020–6028. doi: 10.1128/mcb.16.11.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lam Y A, Xu W, DeMartino G N, Cohen R E. Nature (London) 1997;385:737–740. doi: 10.1038/385737a0. [DOI] [PubMed] [Google Scholar]
- 17.Chu-Ping M, Slaughter C, DeMartino G N. J Biol Chem. 1992;267:10515–10523. [PubMed] [Google Scholar]
- 18.Dubiel W, Pratt G, Ferrell K, Rechsteiner M. J Biol Chem. 1992;267:22369–22377. [PubMed] [Google Scholar]
- 19.Song X, Mott J D, von Kampen J, Pramanik B, Tanaka K, Slaughter C A, DeMartino G N. J Biol Chem. 1996;271:26410–26417. doi: 10.1074/jbc.271.42.26410. [DOI] [PubMed] [Google Scholar]
- 20.Gray C W, Slaughter C A, DeMartino G N. J Mol Biol. 1994;236:7–15. doi: 10.1006/jmbi.1994.1113. [DOI] [PubMed] [Google Scholar]
- 21.Knowlton J R, Johnston S T, Whitby F G, Realini C, Zhang Z, Rechsteiner M, Hill C P. Nature (London) 1997;390:639–643. doi: 10.1038/37670. [DOI] [PubMed] [Google Scholar]
- 22.Realini C, Rogers S W, Rechsteiner M. FEBS Lett. 1994;348:109–113. doi: 10.1016/0014-5793(94)00569-9. [DOI] [PubMed] [Google Scholar]
- 23.Song X, von Kampen J, Slaughter C A, DeMartino G N. J Biol Chem. 1997;272:27994–28000. doi: 10.1074/jbc.272.44.27994. [DOI] [PubMed] [Google Scholar]
- 24.Groettrup M, Soza A, Eggers M, Kuehn L, Dick T P, Schild H, Rammensee H G, Koszinowski U H, Kloetzel P-M. Nature (London) 1996;381:166–168. doi: 10.1038/381166a0. [DOI] [PubMed] [Google Scholar]
- 25.Dick T P, Ruppert T, Groettrup M, Kloetzel P-M, Kuehn L, Koszinowski U H, Stevanovic S, Schild H, Rammensee H G. Cell. 1996;86:253–262. doi: 10.1016/s0092-8674(00)80097-5. [DOI] [PubMed] [Google Scholar]