

Abstract
A series of 6H-thiopyran-2,3-dicarboxylate derivatives 4a–d were synthesized and evaluated for their cytotoxic effect against HCT-15 colon and MCF-7 breast cancer cell lines using Sulforhodamine B (SRB) assay. The results showed that these compounds could exhibit a good cytotoxicity to both cell lines. In addition, these compounds were found to exhibit significant DNA-binding affinity. Ultraviolet–visible light (UV–Vis) spectroscopy was conducted to determine the ability of the ligand under analysis. The effect of ligand complexation on DNA structure led to overall affinity constants of K4a=3.5×104 M–1, K4b=6.4×104 M–1, K4c=3.2×104 M–1, and K4d=2.4×104 M–1. Our findings could provide new evidence showing the relationship between the chemical structure and biological activity and may be useful for the discovery of new anti-cancer drugs.
Introduction
In spite of the fact that the chemistry of thiopyrans has been less investigated than that those of pyran analogues, currently the interest in the heterocycles containing sulfur has been considerably increased since a wide range of biological activities associated with the scaffold have been identified (Scheller, 1975). Thiopyrans are used as key units in medicinal chemistry and as versatile building blocks in organic synthesis (Vedeje and Krafft, 1982). For example, it has been reported that thiopyrans were widely used in the construction of analogues of natural products with various pharmaceutical activities such as anti-bacteria (Brown et al., 2002), anti-hyperplasia (Quaglia et al., 2002), anti-psychiatric (Van Vliet et al., 2000), and anticancer activities (Wang et al., 2006). Studies on the anticancer activity of thiopyran analogs showed that they are one of active antiproliferative agents against tumor cell lines (Sugita et al., 2001) and possess high lipophilicity and could cause insufficient biomembrane permeability. It has been also reported that the substituted thiopyrans are potent inhibitors of deoxyribonucleic acid-protein kinase (Hollik et al., 2003). In view of their wide range of biological activities, we investigated the antiproliferative and anticlonogenic effects of new analogue of 6H-thiopyran-2,3-dicarboxylates on two cancer cell lines, MCF-7 (breast cancer) and HCT-15 (colon cancer), which also showed effect on cell invasion and adhesion (Rajabi et al., 2011). Therefore, in continuation of our interest in the biological properties of thiopyran on human cancer cell lines, we have prepared the compounds 4a–d in 85–92% yield (Fig. 1). The synthesized thiopyrans could represent of a potential class of new cytotoxic agents, since the antiproliferative activity of these compounds has not been investigated yet. Herein, we have treated HCT-15 colon cancer and MCF-7 breast cancer cell lines with different concentrations of compounds 4a–d and the effect on cell cycle for 96 hours. Interaction study and binding properties of compounds 4a–d with DNA have been investigated since DNA binding molecules represent a valuable portion of the clinically useful antitumor drugs (Wheate et al., 2007; CHAIRES, 2006). Most of the drugs that noncovalently bind to DNA selectively interact with the nucleic acid along the minor groove or by intercalation (Hrley, 2002; Minotti et al., 2004).
FIG. 1.

Chemical structures of 6H-thiopyran-2,3-dicarboxylate derivatives 4a [dimethyl 5-acetyl-4-methyl- 6-(4-methoxyphenylimino)-6H-thiopyran-2,3-dicarboxylate]; 4b [dimethyl 5-acetyl-4-methyl-6-(3-fluorophenylimino)-6H-thiopyran-2,3-dicarboxylate]; 4c [diethyl 5-acetyl-4-methyl-6-(3-bromophenylimino)-6H-thiopyran-2,3-dicarboxylate]; and 4d [dimethyl 5-acetyl-4-methyl-6-(phenylimino)-6Hthiopyran-2,3-dicarboxylate].
Materials and Methods
Materials
Trypsin, trypan blue, antibiotic and antimycotic agent, fetal bovine serum (FBS), sulforhodamine B (SRB), DNA extracted from fish sperm, and dimethyl sulfoxide were purchased from Sigma Chemical Co. (St. Louis, MO). Dulbecco's modified Eagle's medium (DMEM) culture media, non-essential amino acids, glutamine, and fetal bovine serum were purchased from EuroClone Life Science Division (Milan, Italy); penicillin and streptomycin were from Invitrogen (Carlsbad, CA). All reagents were of the commercial quality and were used without purification. 1H and 13C nuclear magnetic resonance (NMR) results were obtained with a Bruker AV-400 spectrometer with chemical shifts (d) reported in parts per million downfield from tetramethylsilane and referenced from solvent references. Infrared (IR) spectra were obtained using a PerkinElmer 2000 Fourier Transform Infrared Spectroscopy instrument. Melting points were determined by an X-6 micro-melting point apparatus and uncorrected. Absorption spectra were determined on PGENERAL TU-1901UV–VIS spectrophotometer. Reactions were followed by thin layer chromatography on Merck aluminum silica gel (60 F254) sheets that were visualized under a UV lamp. Column chromatography was performed using silica gel 200–300 mesh.
General procedure for synthesis of thiopyran
To a stirred solution of dimethyl acetylenedicarboxylate 2, (2 mmol) and arylisothiocyanate 1, (2 mmol) in 10 mL of acetonitrile was added mixture of 1,3-dicarbonyl 3 (2 mmol) and sodium hydride (2 mmol) in acetonitrile at room temperature. The reaction mixture was stirred for 8 hours. The solvent was removed under reduced pressure and the residue was separated by column chromatography using n-hexane-EtOAc (5:1) as eluent to give 4a–d.
Dimethyl 5-acetyl-4-methyl- 6-(4-methoxyphenylimino)-6H-thiopyran-2,3-dicarboxylate (4a)
Oil, yield: (87%). IR (KBr) (νmax/cm–1): 1738 (C=O), 1724 (C=O), 1647 (C=O), 1436, 1294, 1224, 1127 cm−1. 1H-NMR (500 MHz, CDCl3): δ=2.00 (3H, s, Me), 2.46 (3H, s, Me), 3.67 (3H, s, MeO), 3.78 (3H, s, MeO), 3.84 (3H, s, MeO), 6.95 (2H, d, 3J=7.6 Hz, 2CH), 7.83 (2H, d, 3J=8.0 Hz, 2CH). 13C-NMR (125.7 MHz, CDCl3): δ=17.4 (Me), 31.3 (Me), 51.8 (MeO), 52.6 (MeO), 54.5 (MeO), 118.4 (C), 122.6 (2CH), 132.7 (C), 133.4 (2CH), 133.9 (C), 136.2 (C), 141.3 (C), 147.5 (C–N), 154.2 (C=N), 161.4 (C=O), 167.6 (C=O), 212.6 (C=O).
Dimethyl 5-acetyl-4-methyl-6-(3-fluorophenylimino)-6H-thiopyran-2,3-dicarboxylate (4b)
Oil, yield: (89%). IR (KBr) (νmax/cm−1): 1725 (C=O), 1712 (C=O), 1658 (C=O), 1547, 1421, 1358, 1268, 1124 cm−1. 1H-NMR (500 MHz, CDCl3): δ=2.01 (3H, s, Me), 2.49 (3H, s, Me), 3.80 (3H, s, MeO), 3.90 (3H, s, MeO), 6.60–6.85 (3H, m, 3CH), 7.35–7.37 (1H, m, CH). 13CNMR (125.7 MHz, CDCl3): δ=17.5 (Me), 30.8 (Me), 53.2 (MeO), 53.7 (MeO), 106.7 (d, 2JCF=22.9 Hz, CH), 111.7 (d, 2JCF=21.0 Hz, CH), 128.2 (d, 3JCF=7.8 Hz, CH), 129.6 (C), 131.4 (d, 4JCF=3.8 Hz, CH), 134.4 (C), 135.7 (C), 140.3 (C), 150.7 (d, 3JCF=3.4 Hz, C–N), 154.0 (C=N), 161.1 (C=O), 162.4 (d, 1JCF=324.2 Hz, C), 166.2 (C=O), 202.4 (C=O).
Diethyl 5-acetyl-4-methyl-6-(3-bromophenylimino)-6H-thiopyran-2,3-dicarboxylate (4c)
Oil, yield: (90%). IR (KBr) (νmax/cm−1): 1728 (C=O), 1722 (C=O), 1625 (C=O), 1584, 1412, 1329, 1204, 1100 cm−1. 1H-NMR (500 MHz, CDCl3): δ=1.28 (3H, t, 3J=7.4 Hz, Me), 1.36 (3H, t, 3J=7.2 Hz, Me), 2.02 (3H, s, Me), 2.49 (3H, s, Me), 4.29 (2H, q, 3J=7.3 Hz, OCH2), 4.37 (2H, q, 3J=7.3Hz, OCH2), 6.71 (1H, t, 3J=7.5 Hz, CH), 7.07 (1H, d, 3J=7.4 Hz, CH), 7.27 (1H, d, 3J=7.5 Hz, CH), 7.30 (1H, s, CH). 13C-NMR 25.7 MHz, CDCl3): δ=13.8 (Me), 13.9 (Me), 17.5 (Me), 30.7 (Me), 62.4 (OCH2), 63.3 (OCH2), 117.9 (CH), 122.5 (CH), 123.4 (C), 127.8 (CH), 129.0 (C), 130.1 (C), 131.3 (CH), 134.3 (C), 135.8 (C), 150.5 (C–N), 154.4 (C=N), 160.7 (C=O), 166.3 (C=O), 202.5 (C=O).
Dimethyl 5-acetyl-4-methyl-6-(phenylimino)-6Hthiopyran-2,3-dicarboxylate (4d)
Oil, yield: (85%). IR (KBr) (νmax/cm−1): 1725 (C=O), 1720 (C=O), 1685 (C=O), 1587, 1432, 1129 cm−1. 1H-NMR (500 MHz, CDCl3): δ=2.25 (3H, s, Me), 2.37 (3H, s, Me), 3.65 (3H, s, MeO), 3.82 (3H, s, MeO), 7.53 (2H, t, 3J=7.2 Hz, 2 CH), 7.61 (1H, t, 3J=7.2 Hz, CH), 8.02 (2H, d, 3J=7.3 Hz, 2 CH). 13C-NMR (125.7 MHz, CDCl3): δ=16.8 (Me), 28.4 (Me), 52.5 (MeO), 53.0 (MeO), 122.3 (2 CH), 123.6 (CH), 126.2 (C), 129.1 (2 CH), 133.5 (C), 134.3 (C), 138.5 (C), 148.7 (C–N), 157.4 (C=N), 160.7 (C=O), 161.5 (C=O), 207.1 (C=O).
Cell line and culture
Human breast cancer MCF-7 and colon cancer HCT-15 cell lines were supplied from the American Type Cell Culture Collection (ATCC) and maintained in the standard medium and grown as a monolayer in DMEM containing 10% FBS, 2 mM glutamine, 100 units/mL penicillin, and 100 g/mL streptomycin. Cultures were maintained at 37°C with 5% CO2 in a humidified atmosphere and were passaged weekly using 0.25% trypsin.
Cell proliferation study by SRB assay
Cells were seeded in 96-well tissue culture plates at 2×103 cells per well and were allowed to adhere for 24 hours before treatment. Serial dilutions of individual compounds were added and cellular growth was assessed after 4 days by SRB assay. Briefly, proteins were precipitated with 10% (final concentration) trichloroacetic acid for 1 hour at 4°C and stained for 30 minutes with SRB dye 0.4% w/v in acetic acid 1% v/v. Finally, precipitated proteins were washed and solubilized in Tris buffer 10 mM. Absorbance (optical density, OD) was measured and read on an ELISA plate reader at a wavelength of 540 nm and used as a relative measure of viable cell number. The percentage of growth inhibition was calculated by using the equation: percentage growth inhibition (1 – OD TC tx/OD TC t0 )×100, where OD TC tx is the mean optical density of treated cells at time x and OD TC t0 is the value at time zero, respectively. Half maximal inhibitory concentration (IC50) was determined by interpolation from dose–response curves.
Evaluation of cell morphology
Human breast cancer MCF-7 and colon cancer HCT-15 cell lines plated at about 20,000 cells per well on chamber-slides were treated with 0 and 5 μM of 4a for 96 hours. After rinsing in phosphate-buffered saline (PBS), cells were fixed in methanol 100% and photographed using a Nikon camera attached to the microscope.
Cell cycle analysis by fluorescence-activated cell sorting
Apoptosis and cell cycle profile were assessed by flow cytometry. MCF-7 and HCT-15 cells were plated at a density of 5×105 cells per well on 6-well plates. HCT-15 cells treated with 4a at concentration of 3.5 μM and 4.5 μM for MCF-7 cells. After 96 hours incubation, the cells were harvested, rinsed in PBS, suspended in 600 μL of PBS containing 1% FBS, fixed by 1.4 mL 80% ethanol, and stored at −20°C in fixation buffer. Then the pellets were suspended in 1 mL of fluorochromic solution [0.08 mg/mL propidium iodide (PI) in 1× PBS] at room temperature in the dark for 60 minutes. The DNA content was analyzed by FACScan flow cytometer (Beckman Counter, cytomics FC 500) and CellQuest software (Becton Dickinson). The population of apoptotic nuclei (subdiploid DNA peak in the DNA fluorescence histogram) was expressed as the percentage in the entire population.
DNA titration experiments
The absorbance at 260 and 280 nm was recorded, in order to check the protein content of DNA solution. The A260:A280 ratio was 1.81, showing that the DNA was sufficiently free of protein. DNA (5 mg/mL) was dissolved in distilled water (pH=7) at 4°C for 24 hours with occasional stirring to ensure the formation of a homogeneous solution. The final concentration of the DNA solution was determined spectrophotometrically at 260 nm using molar extinction coefficient ɛ260=6600 cm–1 M–1 (expressed as molarity of phosphate groups). The UV absorbance at 260 nm of a diluted solution (1:250) of DNA used in our experiments was 0.661 and the final concentration of the DNA solution was 12.5 mM in DNA phosphate. The appropriate amounts of 4 (0.05–12.5 mM) were prepared in distilled water and added dropwise to DNA solution in order to attain the desired ligand per DNA [DNA(P)] molar ratios (r) of 1/80, 1/40, 1/20, 1/10, 1/5, 1/2 and 1 with a final DNA(P) concentration of 6.25 mM. The pH of the solutions was adjusted at 7.0±0.2 using NaOH solution.
Results and Discussion
Synthesis of substituted thiopyrans
As indicated in Fig. 2, a facile synthesis of substituted iminothiopyrans is described via reaction between 1,3-dicarbonyls, electron deficient acetylenic compounds such as dimethyl acetylenedicarboxylate and arylisothiocyanate in the presence of sodium hydride as a base in acetonitrile at room temperature (Fig. 2). The advantages of this study are that no catalyst is required for this reaction and that the simplicity of the present procedure makes it an attractive alternative to the complex multistep approaches. The reaction mixture was stirred for 8 hours. The solvent was removed under reduced pressure and the residue was separated by silica gel column chromatography to give 4a–d in 85–90% yield (Fig. 2).
FIG. 2.

Synthesis of substituted iminothiopyrans 4a–4d.
Antiproliferative activity of iminothiopyrans
The in vitro cytotoxicity experiments were performed with the synthesized compounds against HCT-15 colon and MCF-7 breast cancer cell lines from the ATCC. The SRB assay was employed for these antiproliferation studies and the IC50 values are summarized in Table 1. The compound concentration causing a 50% cell growth inhibition (IC50) was determined by interpolation from dose–response curves. Cells were maintained in the standard medium and grown as a monolayer in DMEM containing 10% FBS, 2 mM glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin. Cultures were maintained at 37°C with 5% CO2 in a humidified atmosphere. IC50 values (in μM), which is the concentration required to inhibit 50% of cell viability by the test compounds after exposure to cells, have been determined. Surprisingly, as it is shown in Table 1, all thiopyran derivatives exhibited potent cytotoxic ability in dose-dependent manner against two cancer cell lines with IC50 values ranging from 3.5 to 15 μM. It is noteworthy that compound 4a with methoxy group exhibited the most antiproliferative effect, suggesting either steric or electronic property of methoxy group could increase the antiproliferative activity of this compounds. In particular, compound 4a inhibited the cell growth at IC50 values of 4.5 μM for MCF-7 and 3.5 for HCT-15 respectively, which are 2–4 times more active than the other compounds (Table 1). In this case, the methoxy substituent in the 4-position of the aryl ring led to a significant increase in potency compared to the other analogues. On the other hand, for compounds 4b and 4c, introduction of a halogen substituent in the 3-positions of the aryl ring led to reduction in potency with IC50 values in the range of 10–5 μM. This might be due to the disfavored interactions between the hydrogen-bond acceptor of 4b and 4c and its molecular targets that counteract the overall activity. It is also noteworthy that 4d without any functional groups in the aryl ring exhibited the antiproliferative effect and inhibition cell growth at IC50 values of 9 μM for MCF-7 and 10 for HCT-15 respectively. Therefore, we concluded that selective antiproliferation of both cells to the tested compounds could be mainly attributed to both steric and electronic effects of aryle ring of aryleisothiosyanate 1. Figure 3 demonstrates the incubation of the HCT-15 and MCF-7 cells with different concentrations of 4a after 96-hour treatment, with significant morphological changes, including cell rounding and detachment from the substratum (Fig. 3).
Table 1.
In Vitro Anticancer Activity Data for Seri of Compounds 4a–4d
| Entry | HCT-15 IC50 (μM) | MCF-7 IC50 (μM) |
|---|---|---|
| 4a | 3.5 | 4.5 |
| 4b | 13 | 15 |
| 4c | 10 | 12 |
| 4d | 10 | 9 |
Dose of the compound required to inhibit cell growth by 50% compared to untreated cell controls; values are derived from half maximal inhibitory concentration (IC50) graphs. All experiments were done in triplicate wells and each experiment was repeated thrice.
4a, dimethyl 5-acetyl-4-methyl- 6-(4-methoxyphenylimino)-6H-thiopyran-2,3-dicarboxylate; 4b, dimethyl 5-acetyl-4-methyl-6-(3-fluorophenylimino)-6H-thiopyran-2,3-dicarboxylate; 4c, diethyl 5-acetyl-4-methyl-6-(3-bromophenylimino)-6H-thiopyran-2,3-dicarboxylate; and 4d, dimethyl 5-acetyl-4-methyl-6-(phenylimino)-6Hthiopyran-2,3-dicarboxylate].
FIG. 3.

(A) Inhibition growth of 4a on HCT-15 colon cancer cell line and morphological analysis for the effects of 4a on HCT-15 after 96-hour incubation. (B) Inhibition growth of 4a on MCF-7 MCF-7 breast cancer cell line and morphological analysis for the effects of 4a on MCF-7 after 96-hour incubation.
Cell cycle analysis
In order to study the mechanism of the antiproliferative activity by 4a in more detail, we analyzed the effects of 4a treatment on cell cycle distributions of MCF-7 and HCT-15 cells. HCT-15 cells treated with 4a at concentration of 3.5 μM and 4.5 μM for MCF-7 cells for 96 hours were subjected to fluorescence-activated cell sorting (FACS) analysis after PI staining of the chromosomal DNA (Fig. 4). In histograms of FACS analysis, untreated proliferative HCT-15 cells showed cell cycle distributions of 70% in G0/G1, 9% in S, 18% in G2/M, and 3% in sub G0/G1 phase, and untreated proliferative MCF-7 cells showed cell cycle distributions of 57% in G0/G1, 12% in S, 27% in G2/M, and 4% in sub G0/G1 phase. In both HCT-15 and MCF-7 cell lines, 4a leads to decrease in the percentage of cells in G0/G1 phase and also to increase percentage of cells in G2/M phase. For instance at 3.5 μM of 4a in treatment with HCT-15, populations reached 56 % for G0/G1, 24% for G2/M phase and 8% for sub G0/G1 phase (Fig. 4A). Also at 3.5 μM of 4a in treatment with MCF-7, populations reached 42 % for G0/G1, 38% for G2/M phase and 6% for sub G0/G1 phase (Fig. 4B). These data indicates that 4a could arrest HCT-15 and MCF-7 cell growth by decreasing the percentage of cells in G0/G1 phase and increasing the percentage of cells in G2/M phase.
FIG. 4.

Flow cytometric analysis of 4a on HCT-15 (A) and 4a on MCF-7 (B) cells. Values are expressed as percentage of the cell population in the G0/G1, S, and G2/M phase of cell cycle.
Binding of iminothiopyrans to DNA
Studying the interaction of small molecules with DNA which can modulate transcription, repair and replication both in vitro and in vivo, is one of the most important aspects of biological investigations aimed at discovering and developing new types of anti-proliferative agents (Nafisi et al., 2008; Rajabi et al., 2010; Milanese et al., 2011). DNA can provide 3 distinctive binding sites for ligands-complexes (groove binding, electrostatic binding to phosphate group, and intercalation). Additionally, DNA-binding molecules represent a valuable portion of clinically useful antitumor drugs (Palchaudhuri and Hergenrother, 2007) and most of the drugs than bind non-covalently to DNA interact selectively with the nucleic acid along the minor groove or by intercalation. The binding mode depends on structural features of these molecules and on the DNA sequences they recognize (Cortesi and Nastruzzi, 2001; Kuwabara et al., 2003). The UV spectra have been recorded for a constant DNA concentration in different [DNA]/[compound] mixing ratios (r). UV spectra of DNA in the presence of a complex derived for diverse r values are shown representatively for 4a–4d in Fig. 5. The intrinsic binding constants, K, of the compounds with DNA have been determined using the UV spectra of the compounds recorded for a constant concentration in the absence or presence of DNA for diverse r-values. Changes in drug absorption properties as a function of DNA concentration were used for evaluation of the overall binding constants. Therefore, to improve the action of drugs on DNA within the cell as a complementary function related to anti-proliferative activity, the interaction of thiopyran derivatives with DNA has been studied by analyzing absorbance changes in the UV–Vis frequency range. The goal is to obtain structural information regarding the thiopyran derivatives binding mode, apparent binding constant, and the effects of conformational changes of native DNA after complexation with thiopyran derivatives. In order to explore the structural changes of DNA on addition of 4a–4d, UV–Vis absorbance spectra of DNA was measured at different concentrations of 4a–4d. The increase in intensity of the characteristic UV–Vis band of the ligand is due to major drug–DNA interaction at the DNA surface. The calculation of the overall binding constants was carried out on the basis of UV absorption as reported. The equilibrium for 4a–4f 4e and 4f have not defined. Please add a definition for these two compounds or revise as needed. and DNA complex can be described as follows:
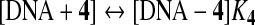 |
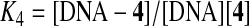 |
FIG. 5.

Plot of 1/[Ligand complexed] versus 1/ligand for DNA and ligand complexes at different ligand concentrations.
The double reciprocal plot of 1/[Ligand complexed] versus 1/[ligand] is linear and the association binding constant (K) is calculated from the ratio of the intercept on the vertical coordinate axis to the slope. The concentration of the complexed ligand was determined by subtracting the absorbance of free DNA at 260 nm from those of the complexed DNA. The concentration of the free ligand was determined by subtraction of the complexed ligand from the total ligand used for the experiment. Our data showed that 1/[complexed ligand] almost proportionally increases as a function of 1/[free ligand] (Fig. 5).
UV–Vis spectroscopic analysis was conducted to determine the binding mode and the binding constant of the ligand, as well as the effects of ligand complexation on DNA structure with overall affinity constants of K4a=3.5×104 M−1, K4b=6.4×104 M–1, K4c=3.2×104 M–1 and K4d=2.4×104 M–1.
Acknowledgments
Dr. Mehdi Rajabi greatly appreciate the financial support from the Molecular Medicine Ph.D. Program at University of Milan and also thank professor Riccardo Ghidoni for allowing him to use his lab facilities.
Author Disclosure Statement
No competing financial interests exist.
References
- BROWN M.J. CARTER P.S. FENWICK A.E. FOSBERRY A.P. HAMPRECHT D.W. HABBS M.J. ARVEST R.L. MENSAH L. MILNER P.H. O'HANLON. P.J., et al. Bioorg. Med. Chem. Lett. 2002;12:3171–3173. doi: 10.1016/s0960-894x(02)00604-2. [DOI] [PubMed] [Google Scholar]
- CHAIRES J.B. A thermodynamic signature for drug-DNA binding mode. Arch. Biochem. Biophys. 2006;453:26–31. doi: 10.1016/j.abb.2006.03.027. [DOI] [PubMed] [Google Scholar]
- CORTESI R. NASTRUZZI C. Delivery systems for DNA-binding drugs as gene expression modulators. Drug. Dis. 2001;6:893–904. doi: 10.1016/s1359-6446(01)01893-1. [DOI] [PubMed] [Google Scholar]
- HOLLICK J.J. GOLDING B.T. HARDCASTELE I.R. MARTIN N. RICHARDSON C. RIGOREAU L.J. SMITH G.C. GRIFFIN R.J. 2,6-Disubstituted pyran-4-one and thiopyran-4-one inhibitors of DNA-Dependent protein kinase (DNA-PK) Bioorg. Med. Chem. Lett. 2003;13:3083–3086. doi: 10.1016/s0960-894x(03)00652-8. [DOI] [PubMed] [Google Scholar]
- HURLEY L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer. 2002;2:188–200. doi: 10.1038/nrc749. [DOI] [PubMed] [Google Scholar]
- KUWABARA T. NODA T. OHTAKE H. OHTAKE T. YOYAMA S. IKARIYAMA Y. Classification of DNA-binding mode of antitumor and antiviral agents by the electrochemiluminescence of ruthenium complex. Anal. Biochem. 2003;314:30–37. doi: 10.1016/s0003-2697(02)00651-6. [DOI] [PubMed] [Google Scholar]
- MILANESE A. GORINCIOI A. RAJABI M. VISTOLI G. SANTANIELLO E. New synthesis of 6[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid and evaluation of the influence of adamantyl group on the DNA binding of a naphthoic retinoid. Bioorg. Chem. 2011;39:151–158. doi: 10.1016/j.bioorg.2011.07.003. [DOI] [PubMed] [Google Scholar]
- MINOTTI G. MENNA P. SALVATORELLI E. CAIRO G. GIANNI L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- NAFISI S. HASHEMI M. RAJABI M. TAJMIR-RIAHI H. A. DNA adducts with antioxidant flavonoids: morin, apigenin, and naringin. DNA Cell Biol. 2008;27:433–442. doi: 10.1089/dna.2008.0735. [DOI] [PubMed] [Google Scholar]
- PALCHAUDHURI P. HERGENROTHER P. DNA as a target for anticancer compounds: methods to determine the mode of binding and the mechanism of action. Cur. Opin. Biotech. 2007;18:497–503. doi: 10.1016/j.copbio.2007.09.006. [DOI] [PubMed] [Google Scholar]
- QUAGLIA W. PIGINI M. PIERGENTILI A. GIANNELLA M. GENTILI F. MARUCCI G. CARRIERI A. POGGESI E. LEONARDI A. MELCHIORRE C.J. Structure–activity relationships in 1,4-benzodioxan-related compounds: selectivity of 4-phenylchroman analogues for α1–adrenoreceptor subtypes. J. Med. Chem. 2002;45:1633–1638. doi: 10.1021/jm011066n. [DOI] [PubMed] [Google Scholar]
- RAJABI M. KHALILZADEH M.A. MEHRZAD J. Antiproliferative activity of novel derivative of thiopyran on breast and colon cancer lines and DNA binding. DNA Cell Biol. 2011;31:128–134. doi: 10.1089/dna.2011.1291. [DOI] [PubMed] [Google Scholar]
- RAJABI M. SIGNORELLI P. GORINCIOI E. GHIDONI R. SANTANIELLO E. Antiproliferative activity of N6-isopentenyladenosine on MCF-7 breast cancer cells: cell cycle analysis and DNA-binding study. DNA Cell Biol. 2010;29:687–691. doi: 10.1089/dna.2010.1073. [DOI] [PubMed] [Google Scholar]
- SCHNELLER W. Thiochromanones and related compounds. Adv. Heterocycl. Chem. 1975;18:59–97. [Google Scholar]
- SUGITA Y. HOSOYA H. TERASAWA K. YOKOE I. FIJISAWA S. SAKAGAMI H. Cytotoxic activity of benzothiepins against human oral tumor cell lines. Anticancer Res. 2001;21:2629–2635. [PubMed] [Google Scholar]
- VAN VLIET L.A. RODENHUIS N. DIJKSTRA D. WIKSTROM H. PUGSLEY T.A. SERPA K.A. MELTZER L.T. HEFFNER T.G. WISE L.D. LAJINESS M.E., et al. Synthesis and pharmacological evaluation of thiopyran analogues of the dopamine D3 receptor-selective agonist (4aR,10bR)-(+)-trans-3,4,4a,10b- tetrahydro-4-n-propyl-2H,5H-[1]benzopyrano[4,3-b]-1,4-oxazin-9-ol (PD 128907) J. Med. Chem. 2000;43:2871–2876. doi: 10.1021/jm0000113. [DOI] [PubMed] [Google Scholar]
- VEDEJE E. KRAFFT G.A. Cyclic sulfides in organic synthesis. Tetrahedron. 1982;38:2857–2881. [Google Scholar]
- WANG W. LI H. WANG J. ZU L.S. Enantioselective organocatalytic tandem michael–aldol reactions: one-pot synthesis of chiral thiochromenes. J. Am. Chem. Soc. 2006;128:10354–10355. doi: 10.1021/ja063328m. [DOI] [PubMed] [Google Scholar]
- WHEATE N.J. BRODIE C.R. COLLINS J.G. KEMP S. ALDRICH-WRIGHT J.R. DNA intercalators in cancer therapy: organic and inorganic drugs and their spectroscopic tools of analysis. Mini. Rev. Med. Chem. 2007;7:627–48. doi: 10.2174/138955707780859413. [DOI] [PubMed] [Google Scholar]