

Abstract
In no other field has the function of clusterin (CLU) been more controversial than in cancer genetics. After more than 20 years of research, there is still uncertainty with regard to the role of CLU in human cancers. Some investigators believe CLU to be an oncogene, others - an inhibitor of tumorigenesis. However, owing to the recent efforts of several laboratories, the role of CLU in important cellular processes like proliferation, apoptosis, differentiation and transformation is beginning to emerge. The “enigmatic” CLU is becoming less so. In this chapter we will review the work of research teams interested in understanding how CLU is regulated by oncogenic signalling. We will discuss how and under what circumstances oncogenes and epigenetic factors modify CLU expression, with important consequences for mammalian tumorigenesis.
I. Introduction
The role of CLU in cancer has been the matter of debate for many years. There are many reports, mainly based on studies with cancer cell lines, indicating that CLU is involved in promotion of tumorigenesis and in conferring resistance to chemotherapeutic drugs (Chi et al., 2008; Chung et al., 2004; Miyake et al., 2003; Sallman et al., 2007). However, more recent studies using mouse models of neuroblastoma and prostate cancer have established that an important function of CLU is to restrict tumour development [(Chayka et al., 2009) and Bettuzzi et al, in press.] While some of the contrasting results observed so far could be explained by the use of different types of cell lines, reagents or procedures, we suggest here that CLU, lying at the crossroad of life and death, is at the same time an oncogene and a tumour suppressor gene. This concept will be developed and clarified in the course of this review.
The categorization of genes in strict functional classes clearly does not reflect the complexity of biological systems. The boundaries dividing gene functions are becoming blurred and to classify genes as oncogenes or tumour suppressors is, in the light of the more recent literature, an anachronism. Classical tumour suppressor genes like pRb, PML and p21WAF-Cip are now found to promote human cancer in specific contexts (Cote et al., 1998; Ito et al., 2008; Morris et al., 2008; Viale et al., 2009). Conversely, proto-oncogenes like E2F1 and MYB have been shown to restrict tumour growth or promote the maintenance of normal cell division (Morris et al., 2008; Pierce et al., 1999; Tarasov et al., 2008). Additionally, it is useful to keep in mind that association (“post hoc”) doesn’t imply causation (“propter hoc”). Just because a gene is over-expressed or under-expressed in certain tumours, one cannot conclude that it is driving or inhibiting neoplastic growth. Instead, its deregulation might be a defence mechanism that the host employs to maintain tissue homeostasis. Research on clusterin could well be a case study in cancer gene complexity.
CLU is the prototypical multifunctional gene: it was found to regulate apoptosis, cell - cell interactions, protein stability, cell signalling, proliferation and, finally, transformation. In spite of the multiple functions that have been ascribed to CLU, its genetic inactivation in mice is well tolerated and animals develop and live normally (McLaughlin et al., 2000). Since CLU expression in mammalian cells is highly modulated by certain pathological processes or exposure to physical and chemical agents, it is tempting to speculate that CLU is mainly required to respond to exogenous or endogenous stress signals. Indeed, CLU knockout mice are more susceptible than wild type mice to experimentally-induced autoimmune diseases, and fibroblasts derived from CLU knockout mice are more sensitive to thermal injury (McLaughlin et al., 2000; Santilli et al., 2005). In cancer, expression of CLU has been shown to be either up- or down-modulated, although the data available on the Oncomine web site, which represents a very large and growing collection of cDNA microarray experiments, shows that in the most cancer types CLU is downregulated (Figure 1.) It is still unclear whether the opposing observations published in the literature are caused by technical reasons - i.e. use of different antibodies, cell lines, patients, etc – or they reflect the fact that CLU can be a tumour suppressor and promoter, at the same time, depending on the specific biology of the disease and its phase of progression.
Figure 1. CLU expression in primary tumors.
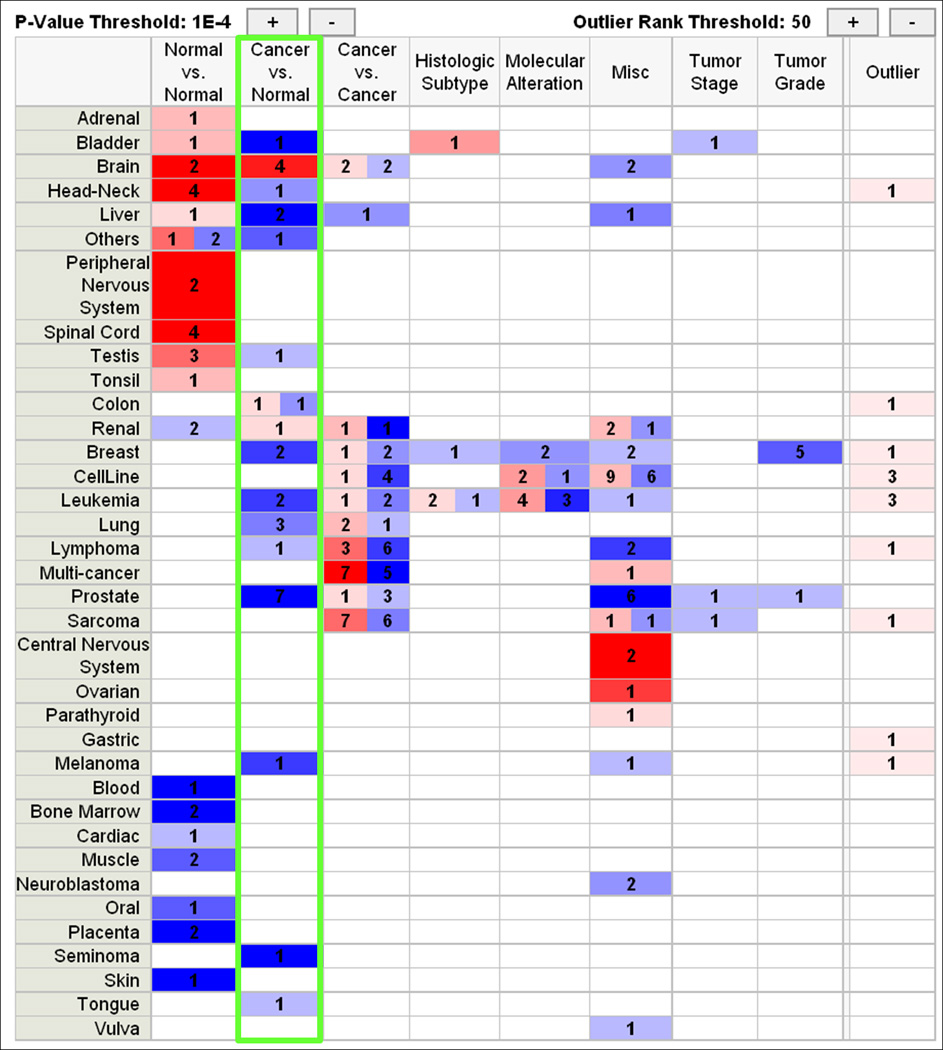
Expression of CLU mRNA in Affymetrix experiments as represented on the Oncomine web site (www.Oncomine.org). Shades of blue color indicate under- expression, whereas shades of red indicate over-expression. The intensity of the color is proportional to the statistical significance of the difference. Numbers indicate how many independent experiments show a significant difference in each tissue. Note that in the «Cancer vs Normal» column (green rectangle) all the transformed tissues, with the exception of brain, show significant downregulation of CLU with respect to the corresponding normal samples.
As we try to avoid the dualistic, Cartesian classification of cancer genes into oncogenes and tumour suppressors, we submit that a gene can inhibit tumour growth under untreated conditions (i.e., be “pro-host”) but at the same time act “anti-patient” by rendering tumours resistant to chemo-, radio-, or biological therapy. Conversely, there are well recognized examples when the oncogene initiates or even promotes tumour growth (or acts “anti-host”) but at the same time confers chemosensitivity (or acts “pro-patient”.) While these distinctions have been largely absent from the clusterin literature, for other cancer genes these complexities are well-appreciated. The c-Myc proto-oncogene is a case in point.
At the cellular level many putative Myc target genes pertain to cell proliferation. Among them are ornithine decarboxylase, cyclins A and D2, cdc25A, cdk4, Id2, and telomerase. In addition, entry into the cell cycle is facilitated by repression of several genes, such as assorted cdk inhibitors, gadd45, etc. [reviewed recently in (Meyer and Penn, 2008)]. Consistent with these observations, activation of Myc forces quiescent fibroblasts to re-enter the cell cycle (Eilers et al., 1989), and rodent fibroblasts with targeted disruption of Myc are severely deficient in cell proliferation (Mateyak et al., 1997). Furthermore, at least in mice decreased expression of Myc results in hypoplasia (Trumpp et al., 2001). Conversely, when overexpressed in many transgenic settings, c-Myc initiates tumour growth with very high penetrance (Morgenbesser and DePinho, 1994). Furthermore, c-Myc is overexpressed in a variety of spontaneous human cancers, making it a classical oncogene in the view of both mouse and human geneticists.
This “anti-host” properties of Myc are balanced to some extent by the potentially “pro-patient” propensity of Myc to induce apoptosis [reviewed in (Evan and Vousden, 2001)], both intrinsic and extrinsic. To accomplish the former, Myc activates p53 via the ARF pathway (Zindy et al., 1998). In addition, Myc appears to potentiate the extrinsic pathway triggered by ligation of a death receptor. Indeed, Myc has been shown to participate in apoptosis induced by Fas/CD95 ligand (Hueber et al., 1997), TNFα (Klefstrom et al., 1994), and TRAIL (Ricci et al., 2004).
To the extent that cytotoxic drugs inflict DNA damage and activate the intrinsic apoptotic pathway, one might predict that deregulation of Myc would be associated with chemosensitivity. Indeed, as exemplified by studies on human colon carcinoma, low-level Myc amplification (combined with wild type p53 expression) increases susceptibility to 5-fluorouracil in vivo (Arango et al., 2001). Similarly, human Burkitt’s lymphomas with Myc overexpression appear to be intrinsically sensitive to TRAIL in the clinical setting [reviewed in (Finnberg and El-Deiry, 2008)]
The clusterin situation has many interesting parallels with the Myc story. Not only is clusterin down-regulated by c-Myc (see below) but it has been reported to mediate TRAIL resistance in prostate cancer cells (Sallman et al., 2007). Thus, it is tempting to extend this parallel and propose that clusterin is to Myc as yin is to yang. According to this framework, clusterin inherently inhibits cell proliferation and neoplastic growth (i.e., acts “pro-host”) but confers resistance to therapy (i.e., acts “anti-patient”) - hence its overexpression under certain conditions. Taking this view helps make sense of some famously contradictory data.
For example, the group lead by Martin Gleave has published a number of studies which suggest that expression of CLU is enhanced in human prostate cancer and antisense oligonucleotides targeting CLU expression inhibit prostate tumorigenesis in vivo and in vitro (Chi et al., 2008; Miyake et al., 2005). These results are contrasted by the work of the Bettuzzi group, which showed reduced expression of CLU during mouse and human prostate cancer progression (Caporali et al., 2004; Scaltriti et al., 2004b). The analysis of several gene expression studies available in the Oncomine database shows that there is a significant downregulation of the CLU mRNA in almost all types of cancer, as compared to matched normal tissue controls, corroborating the hypothesis that CLU expression is generally silenced in primary (i.e., frequently untreated) human cancers (Figure 1.)
As a further indication that CLU expression might be inactivated in mammalian tumorigenesis, CLU KO mice are more prone than wild type counterparts to oncogene-induced tumorigenesis (Chayka et al., 2009; Thomas-Tikhonenko et al., 2004). This introduces the theme of this assay, namely, how CLU is regulated by oncogenic signalling and what role CLU plays in inhibiting mammalian cell transformation, as opposed to potentially promoting chemoresistance.
II. Regulation of CLU expression by transforming oncogenes: early evidence
Early reports demonstrated that expression of CLU is modulated during cell transformation. CLU expression was reported to be increased in gliomas compared to normal brain tissue (Danik et al., 1991). In 1993 the group of Michael Sporn observed that expression of CLU was increased after malignant transformation of the rat prostate caused by chemical carcinogenesis (Kadomatsu et al., 1993). It should be noted that only the mRNA expression was detected in these early studies, leaving open the question of whether CLU protein was also up-regulated. Further evidence that CLU might be important in human tumorigenesis originates from the observation that CLU expression is often modulated during apoptosis. A theory was elaborated suggesting that CLU is secreted during injury as a survival response in the face of apoptosis (Koch-Brandt and Morgans, 1996). Consequently, some research groups started to investigate whether oncogenic transcription factors could induce CLU, facilitating cell survival, transformation and/or resistance to chemotherapeutic drug killing.
The first evidence that CLU expression is modulated by oncogenic activity was published in 1989 when it was first reported that a thermally inducible gene, called T64, was activated in avian cells by retroviral oncogenes with protein kinase activity such as v-src, v-fps and v-mil (Michel et al., 1989). Sequencing of T64 revealed that it was the avian orthologue of rat CLU. Subsequent investigations revealed that induction by the oncogenic kinases was dependent on the AP-1 binding site present in close proximity to the clusterin transcription start site. Similarly, in their 1992 study, Herault et al found that the gene most strongly overexpressed upon Rous sarcoma virus infection in quail neuroretina cells was CLU. Mutation of the TGACTCA motif in the CLU promoter abolished CAT activity of the reporter suggesting that the AP-1 binding site was required for induction by Src (Herault et al., 1992).
The role of AP-1 (a complex containing the Jun and Fos oncoproteins) in regulating CLU expression was confirmed later on in other contexts. For example, it was shown that TGFβ positively modulates CLU expression via activation of an AP-1 site in the mammalian CLU promoter (Jin and Howe, 1999). In that work, the authors proposed that the mechanism of activation is the removal of the trans-repression effect of c-Fos by TGFβ. In another study, exposure of HaCaT keratinocyte cells to vanadium was shown to induce apoptosis, c-Fos expression, and a switch from secreted to nuclear CLU (Markopoulou et al., 2009). Ectopic expression of c-fos also induced apoptosis and nuclear CLU expression in HaCaT cells. The authors inferred that c-Fos controls the ratio of cytoplasmic vs nuclear fraction of CLU. However, it has not been resolved whether c-fos directly regulates the levels of the different CLU protein isoforms, or apoptosis resulting from c-Fos overexpression is actually causing the isoform switch.
Claudia Koch-Brandt’s group was one of the first to study the role of two classical proto-oncogenes, namely c-MYC and Ha-RAS, in regulation of CLU expression. it was reported that overexpression of Ha-RAS, but not of c-MYC, in the rat embryo fibroblast cell line Rat-1 caused repression of CLU expression at the mRNA level (Klock et al., 1998). There had been no attempt to understand the mechanism by which Ha-RAS was causing the inhibitory effect, but this was clarified in subsequent studies that will be discussed later. This early evidence linking the activity of proto-oncogenes to CLU expression and the emerging role of CLU as a modulator of apoptosis prompted many other groups to study the relationship between oncogenic transcription factors and CLU.
III. Regulation of CLU expression by proto-oncogenic transcription factors
Transcription factors are the essential molecular tools, with which the cell is able to respond to changing environmental conditions, stress, differentiating stimuli or proliferative cues. Transcription factors can be tissue-specific or ubiquitously expressed, and oncogenic versions can be found in both categories. In the following sections we will discuss in detail which oncogenic transcription factors have been found to regulate CLU and the biological consequences of its deregulation.
MYC
MYC is a small family of transcription factors composed of the prototype member, c-MYC, the neuronal specific MYCN and the less studied L-MYC. C-MYC is a major player in human tumorigenesis and its function in human cancer has been discussed in detail in many reviews [see references above and also (Lutz et al., 2002; Vita and Henriksson, 2006; Yaylim-Eraltan et al., 2008)]. Although it was initially thought that c-MYC could not regulate the expression of CLU (Klock et al., 1998) the group lead by Andrei Thomas-Tikhonenko reported that ectopic levels of c-MYC could strongly repress the expression of CLU in murine colonocytes or human keratinocytes. One of the most interesting observations in this paper is that forced overexpression of CLU could inhibit, at least in part, c-MYC-dependent tumorigenesis. Indeed, CLU could attenuated proliferation of colonocytes transformed by c-MYC, and mice with a disrupted CLU gene were more prone to develop papillomas as a consequence of exposure to carcinogens (Thomas-Tikhonenko et al., 2004). The concept that CLU could behave as an inhibitor of cell proliferation was not without precedent. In 2002 the Bettuzzi group showed that forced overexpression of CLU induced cell cycle arrest of human prostate cells in vitro (Bettuzzi et al., 2002).
Neuronal MYC (MYCN) is also a negative regulator of CLU. It has been recently shown (Chayka et al., 2009) that CLU is downregulated in the paediatric cancer neuroblastoma. Neuroblastoma is characterized by the amplification of MYCN, which is necessary and sufficient to induce transformation of embryonal sympathetic cells into malignant neuroblasts. In tumours with amplified MYCN, CLU is strongly downregulated and MYCN appears to induce CLU downregulation at least in part through transcriptional induction of the six-microRNA cluster miR-17-92 (composed of miR-17, -18, -19a/b, -20, and -92) (Dews et al., 2006; O'Donnell et al., 2005). These and other microRNAs are short non-coding RNAs that can specifically decrease protein output by decreasing translation and/or by mRNA destabilization (Mendell, 2008). In Chayka et al it was demonstrated that the MYCN-induced miR-17-92 cluster downregulates CLU expression in neuroblastoma cells. The MYCN-CLU axis is functionally important, since mice with a disrupted CLU gene are more prone to the formation of neuroblastomas induced by transgenic expression of MYCN, thus suggesting that CLU is a repressor of MYCN tumorigenesis (Chayka et al., 2009). A still unpublished study is yielding evidence suggesting that MYCN can also directly repress transcription of CLU through an E-box in the CLU 5’ flanking region which is conserved in different mammalian species.
More careful examination of the connection between miR-17-92 cluster members and clusterin has revealed several surprises. While the Miranda algorithm (John et al., 2004) predicts binding sites for several members of the miR-17-92 cluster within the 3’-UTR of human clusterin, these predictions could not be confirmed experimentally using the luciferase sensor assay or gain-of-function microRNA mimic screens (Dews et al, submitted.) This suggests that clusterin may not be a direct molecular target for miR-17-92 and that instead this cluster targets an upstream activator of clusterin expression.
As mentioned above, in some cell lines clusterin can be induced by the TGFβ signalling pathway (Jin and Howe, 1997; Jin et al., 1999). This idea had been also promulgated by David Boothman and his colleagues (Bey et al., 2006). Thus, it was tempting to propose that perhaps down-regulation of CLU by miR-17-92 is in fact lack of activation by TGFβ. Indeed, very recent work from the Thomas-Tikhonenko laboratory demonstrated that Myc-overexpressing cells contain defects in several key components of the TGFβ signalling pathway, including TGFβ receptor II and activating Smads (Bierie and Moses, 2006; Massague, 2008). As predicted previously (Volinia et al., 2006), miR-17-5p and miR-20 reduce levels of the type II TGFβ receptor (TGFBR2) and miR-18 was found to target Smad4 and in some cell lines - Smad2. Overall, weakened TGFβ signalling in Myc and/or miR-17-92 overexpressing cells resulted in very poor induction of CLU by TGFβ.
MYB
MYB, similarly to MYC, is a family of transcription factors which includes the tissue specific c-MYB and A-MYB and the ubiquitous B-MYB, a positive regulator of cell proliferation and survival (Lipsick et al., 2001; Oh and Reddy, 1999; Sala and Watson, 1999). Interestingly, B-MYB is overexpressed or amplified in various types of human cancer suggesting that it too is a proto-oncogene (Nakajima et al., 2008; Raschella et al., 1999; Sala et al., 1999). In the Sala laboratory, it has been shown that B-MYB binds to and positively regulates the CLU promoter through a MYB-consensus sequence. It has also been shown that CLU mediates, at least in part, the antiapoptotic effects of B-MYB. B-MYB-induced CLU can confer resistance to doxorubicin killing of human LAN5 neuroblastoma cells. Furthermore, thermal injury is more pronounced in fibroblasts transfected with a construct expressing dominant negative B-MYB, which also blunts thermal induction of CLU (Cervellera et al., 2000; Santilli et al., 2005). These results are in agreement with evidence correlating decreased expression of secreted CLU and B-MYB with apoptosis induced by all-trans-retinoic acid in smooth muscle cells (Orlandi et al., 2005).
NF-κB
NF-κB is a multifunctional transcription factor that has central importance in immunity and cancer. NF-κB is activated in response to external stimuli - such as engagement of the TNFα receptor by its ligand, and by the IKK kinases alpha, beta and gamma (the latter known as NEMO) which phosphorylate the inhibitors of κB (IκBs), liberating the transcriptionally active NF-κB molecule (Gilmore, 2006; Perkins, 2007). The first evidence that NF-κB regulates CLU expression was provided by Kenneth Marcu and co-workers. In their study, the authors carried out a systematic analysis to isolate all NF-κB target genes in mouse embryo fibroblasts. They used a molecular inhibitor of NF-κB in the presence or absence of TNFα, a classical NF-κB inducer. Among the plethora of genes activated by NF-κB, CLU was one of the most highly regulated (Li et al., 2002). Interestingly, knockout of either one of the three IKKs resulted in lack of activation of CLU, suggesting that its activation is dependent on the whole NF-κB signalsome.
These results were later confirmed by another group that showed that CLU can be induced in glial and astrocyte cells by the bacterial lipopolisaccharide LPS (Saura et al., 2003). LPS is a known activator of NF-κ κB, and the use of aspirin or MG132 as indirect means to inhibit NF-κB resulted in the inhibition of CLU expression. Intriguingly, it was later shown that CLU regulates NF-κB activity in a negative manner by stabilizing IκBs (Devauchelle et al., 2006; Santilli et al., 2003; Savkovic et al., 2007; Takase et al., 2008a; Takase et al., 2008b). This leads to the hypothesis that CLU participates in a negative loop in which transcriptional activation of CLU is evoked to dampen NF-κB activity. This would be especially important when there is a need to control the secretion of potentially harmful cytokines regulated by NF-κB. This hypothesis is corroborated by the study in which it has been shown that abnormally low CLU levels cause excessive NF-κB activation and pathological cytokine secretion in rheumatoid arthritis (Devauchelle et al., 2006).
Egr1
The group lead by David Boothman was the first to show that secreted CLU is induced by ionizing irradiation (Yang et al., 2000). The same group later showed that irradiation leads to the activation of a signalling pathway that emanates from two growth factors receptors: EGFR and IGFR. It was demonstrated that IGFR, but not EGFR, mediates the induction of secreted CLU in response to irradiation (Criswell et al., 2005). Notably, the Src/Map kinase cascade that is triggered downstream of IGFR ultimately signals to the transcription factor Egr1, which, in turn, binds to the CLU promoter and induces up-regulation of CLU mRNA. In this context secreted CLU is induced as a protective response to damaging stress since knockdown of CLU by RNAi accelerates cell death.
Stat1
Stats are a group of transcription factors implicated in transducing survival or apoptotic signalling downstream of a class of receptor-associated molecules called JAKs. In an Affymetrix screen to search for genes involved in conferring resistance to the chemotherapeutic drug docetaxel, Djeu and co-workers identified CLU and Stat-1 as docetaxel-inducible genes that inhibit drug-induced apoptosis. Interestingly, Stat-1 seems to lie upstream of CLU since its depletion by siRNA induces a 50% reduction of CLU expression in prostate cancer cells (Patterson et al., 2006). It is not clear whether Stat-1 can directly regulate CLU gene expression, but the presence of putative Stat-binding sites in the CLU promoter suggests that this could be the case.
GLI and TCF
Recent studies have placed CLU downstream of the two protooncogenic transcription factors activated by the signalling molecules Hedgehog and Wnt, GLI-2 and TCF-1, respectively. Hedgehog and Wnt play important roles in normal development and cancer (Jiang and Hui, 2008; Nusse, 1992; Polakis, 2000). Signaling emanating from these developmental factors is relayed to nuclear transcription factors belonging to the GLI and TCF families. Abnormal activation of GLI is often detected in medulloblastomas with disruption of the Sonic Hedgehog antagonist Patched, which results in constitutive activation of Hedgehog signalling (Villavicencio et al., 2000). TCF family members are usually activated in epithelial tumours in which Wnt signalling is increased by stabilization of beta-catenin, an essential partner in transcriptional regulation (Ilyas, 2005; Rask et al., 2003).
The group led by Marin Gleave has found that knockdown of GLI-2 in prostate cancer cells results in suppression of proliferation and increased apoptosis and concurrent inactivation or activation of several genes. Interestingly, CLU protein expression increased after treatment with GLI-2 antisense oligonucleotides, although CLU mRNA expression did not change (Narita et al., 2008). In another study, it was found that TCF-1 mediates activation of a short mRNA isoform of CLU (Schepeler et al., 2007). In both studies, no attempts were made to understand the functional role of CLU in these signalling pathways and whether CLU is a positive or negative modulator of Wnt and Hedgehog pathways remains to be determined.
IV. Oncogenic signalling and epigenetic regulation of CLU expression
As mentioned in previous paragraphs, c-Myc, N-Myc, and Ras cause silencing of CLU expression, probably facilitating tumorigenesis. The mechanisms of repression by Myc family members appear to be complex and are still a matter of active investigation. Recent studies suggest that RAS-mediated silencing is epigenetic. Analysis of gene expression in rat fibroblasts transformed with activated Ha-RAS revealed that several genes, including CLU, are silenced. Interestingly, RAS first induces deacetylation of the CLU promoter followed by methylation of a CpG island located in proximity of the transcription start site via MEK/ERK signalling (Lund et al., 2006). Curiously, as mentioned in the previous section, the Boothman group has shown that an IGFR-dependent MEK-ERK-EGR signalling pathway mediates activation of CLU by ionizing radiation (Criswell et al., 2005). The apparent contradiction could be explained if one hypothesized that the MEK-ERK pathway could feed into different downstream effectors in a stimulus-dependent manner (i.e. irradiation-induced MEK-ERK activates gene transcription via transcription factors whereas ras-induced MEK-ERK inactivates gene expression by inducing histone deacetylases).
Other research groups have observed epigenetic silencing of CLU in transformed cells and cancer. For example, Nuutinen et al have shown that CLU is silenced by gene methylation and deacethylation in human neuroblastoma or neuronal cell lines (Nuutinen et al., 2005). In murine and human prostate cancer cell lines CLU expression is silenced by gene methylation and/or histone deacetylation (Rauhala et al., 2008). In line with these results, the Bettuzzi group had previously shown that CLU expression is downregulated during progression of human and murine prostate cancer and that CLU promotes slowdown of prostate cell proliferation (Bettuzzi et al., 2000; Bettuzzi et al., 2002; Caporali et al., 2004; Scaltriti et al., 2004b).
Moreover, CLU is one of the genes most highly induced by histone deacetylase inhibitors and inhibitors of DNA methylation in tumour endothelial cells. Most notably, suppression of CLU expression by shRNA drives increased proliferation, migration and sprouting of tumour endothelial cells (Hellebrekers et al., 2007). Overall, these results invoke a scenario in which oncogenic stimuli provoke chromatin rearrangements that result in suppression of genes, like CLU, that are implicated in restraining tumour proliferation and angiogenesis. Indeed, most recent work from Thomas-Tikhonenko laboratory provides direct evidence that clusterin overexpression severely limit neovascularisation of murine and human colon carcinomas (Dews et al, submitted) potentially affecting tumour metabolism (Chapter 3.)
V. Concluding remarks
These are exciting times for researchers studying CLU and cancer. CLU is emerging as an important player in human cancer, although its role is more complex than anticipated. Regarded initially as a mere extracellular chaperone or a scavenging protein, CLU has been proven to be an important mediator of cell signalling as well. Its documented ability to interfere with NF-κB, PI3 kinase, or MAP kinase signalling could perhaps explain its role as a tumour modifier. Cancer cells often hijack cellular signalling to their advantage and become “addicted” to a specific molecular pathway. By interfering in a negative or a positive manner with such pathways, CLU could either promote or restrict neoplastic disease. In the light of recent evidence gathered using mouse models of human cancer where CLU has been genetically ablated, we hypothesize that CLU is mainly required to restrict the early stages of mammalian tumorigenesis and metastatic spread while assisting established tumours in becoming chemo- and radio-resistant.
While the mechanism by which CLU acts as a tumour suppressor gene is not entirely clear, there is some evidence to suggest that suppression of the NF-kB signalling could be involved. It is tempting to speculate that very aggressive clones of cancer cells arising after chemotherapeutic drug treatments or natural selection could re-activate the expression of CLU. This hypothesis was recently corroborated in experimental models in which initial up-regulation of CLU was found to induce clonogenic toxicity, thus killing the majority of prostate cancer cells, while the rare surviving clones were expressing CLU solely in the cytoplasm. This could lead to the development of anti-apoptotic properties and the ability to survive the mitotic catastrophe, if only at the cost of acquiring an altered phenotype with impaired mitosis, endoreduplication and genetic instability (Scaltriti et al., 2004a; Scaltriti et al., 2004c). Therefore, high expression of secreted or cytoplasmic CLU could be advantageous because it confers increased resistance to killing by anti-cancer drugs or enhances tumour cell survival in specific niches. The opposite roles played by CLU in early versus late stages of tumorigenesis could also explain why epigenetic inactivation of CLU, but not gene rearrangements or mutations, is commonly detected in mammalian cancers.
Acknowledgments
Research in the authors’ laboratories has been supported by: Sporting Aiding Medical Research for Kids (SPARKS) and Neuroblastoma Society (A.S.); FIL 2008 and FIL 2009, University of Parma, Italy, AICR (UK) Grant No. 06-711, and Istituto Nazionale Biostrutture e Biosistemi (INBB), Roma, Italy (S.B.); US National Institutes of Health grant CA 122334 (ATT).
References
- Arango D, Corner GA, Wadler S, Catalano PJ, Augenlicht LH. c-myc/p53 interaction determines sensitivity of human colon carcinoma cells to 5-fluorouracil in vitro and in vivo. Cancer Res. 2001;61:4910–4915. [PubMed] [Google Scholar]
- Bettuzzi S, Davalli P, Astancolle S, Carani C, Madeo B, Tampieri A, Corti A, Saverio B, Pierpaola D, Serenella A, Cesare C, Bruno M, Auro T, Arnaldo C. Tumor progression is accompanied by significant changes in the levels of expression of polyamine metabolism regulatory genes and clusterin (sulfated glycoprotein 2) in human prostate cancer specimens. Cancer Res. 2000;60:28–34. [PubMed] [Google Scholar]
- Bettuzzi S, Scorcioni F, Astancolle S, Davalli P, Scaltriti M, Corti A. Clusterin (SGP-2) transient overexpression decreases proliferation rate of SV40-immortalized human prostate epithelial cells by slowing down cell cycle progression. Oncogene. 2002;21:4328–4334. doi: 10.1038/sj.onc.1205594. [DOI] [PubMed] [Google Scholar]
- Bey EA, Wuerzberger-Davis SM, Pink JJ, Yang CR, Araki S, Reinicke KE, Bentle MS, Dong Y, Cataldo E, Criswell TL, Wagner MW, Li L, Gao J, Boothman DA. Mornings with Art, lessons learned: feedback regulation, restriction threshold biology, and redundancy govern molecular stress responses. J Cell Physiol. 2006;209:604–610. doi: 10.1002/jcp.20783. [DOI] [PubMed] [Google Scholar]
- Bierie B, Moses HL. Tumour microenvironment: TGFβ: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- Caporali A, Davalli P, Astancolle S, D'Arca D, Brausi M, Bettuzzi S, Corti A. The chemopreventive action of catechins in the TRAMP mouse model of prostate carcinogenesis is accompanied by clusterin over-expression. Carcinogenesis. 2004;25:2217–2224. doi: 10.1093/carcin/bgh235. [DOI] [PubMed] [Google Scholar]
- Cervellera M, Raschella G, Santilli G, Tanno B, Ventura A, Mancini C, Sevignani C, Calabretta B, Sala A. Direct transactivation of the anti-apoptotic gene apolipoprotein J (clusterin) by B-MYB. J Biol Chem. 2000;275:21055–21060. doi: 10.1074/jbc.M002055200. [DOI] [PubMed] [Google Scholar]
- Chayka O, Corvetta D, Dews M, Caccamo AE, Piotrowska I, Santilli G, Gibson S, Sebire NJ, Himoudi N, Hogarty MD, Anderson J, Bettuzzi S, Thomas-Tikhonenko A, Sala A. Clusterin, a haploinsufficient tumour suppressor gene in neuroblastomas. J Natl Cancer Inst. 2009;101:663–677. doi: 10.1093/jnci/djp063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi KN, Zoubeidi A, Gleave ME. Custirsen (OGX-011): a second-generation antisense inhibitor of clusterin for the treatment of cancer. Expert Opin Investig Drugs. 2008;17:1955–1962. doi: 10.1517/13543780802528609. [DOI] [PubMed] [Google Scholar]
- Chung J, Kwak C, Jin RJ, Lee CH, Lee KH, Lee SE. Enhanced chemosensitivity of bladder cancer cells to cisplatin by suppression of clusterin in vitro. Cancer Lett. 2004;203:155–161. doi: 10.1016/j.canlet.2003.07.008. [DOI] [PubMed] [Google Scholar]
- Cote R, Dunn M, Chatterjee S, Stein J, Shi S, Tran Q, Hu S, Xu H, Groshen S, Taylor C, Skinner D, Benedict W. Elevated and absent pRb expression is associated with bladder cancer progression and has cooperative effects with p53. Cancer Res. 1998;58:1090–1094. [PubMed] [Google Scholar]
- Criswell T, Beman M, Araki S, Leskov K, Cataldo E, Mayo LD, Boothman DA. Delayed activation of insulin-like growth factor-1 receptor/Src/MAPK/Egr-1 signaling regulates clusterin expression, a pro-survival factor. J Biol Chem. 2005;280:14212–14221. doi: 10.1074/jbc.M412569200. [DOI] [PubMed] [Google Scholar]
- Danik M, Chabot JG, Mercier C, Benabid AL, Chauvin C, Quirion R, Suh M. Human gliomas and epileptic foci express high levels of a mRNA related to rat testicular sulfated glycoprotein 2, a purported marker of cell death. Proc Natl Acad Sci USA. 1991;88:8577–8581. doi: 10.1073/pnas.88.19.8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devauchelle V, Essabbani A, De Pinieux G, Germain S, Tourneur L, Mistou S, Margottin-Goguet F, Anract P, Migaud H, Le Nen D, Lequerre T, Saraux A, Dougados M, Breban M, Fournier C, Chiocchia G. Characterization and functional consequences of underexpression of clusterin in rheumatoid arthritis. J Immunol. 2006;177:6471–6479. doi: 10.4049/jimmunol.177.9.6471. [DOI] [PubMed] [Google Scholar]
- Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38:1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers M, Picard D, Yamamoto KR, Bishop JM. Chimaeras of Myc oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature. 1989;340:66–68. doi: 10.1038/340066a0. [DOI] [PubMed] [Google Scholar]
- Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- Finnberg N, El-Deiry WS. TRAIL death receptors as tumor suppressors and drug targets. Cell Cycle. 2008;7:1525–1528. doi: 10.4161/cc.7.11.5975. [DOI] [PubMed] [Google Scholar]
- Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- Hellebrekers DM, Melotte V, Vire E, Langenkamp E, Molema G, Fuks F, Herman JG, Van CW, Griffioen AW, van EM. Identification of epigenetically silenced genes in tumor endothelial cells. Cancer Res. 2007;67:4138–4148. doi: 10.1158/0008-5472.CAN-06-3032. [DOI] [PubMed] [Google Scholar]
- Herault Y, Chatelain G, Brun G, Michel D. V-src-induced-transcription of the avian clusterin gene. Nucl Acids Res. 1992;20:6377–6383. doi: 10.1093/nar/20.23.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hueber AO, Zornig M, Lyon D, Suda T, Nagata S, Evan GI. Requirement for the CD95 receptor-ligand pathway in c-Myc-induced apoptosis. Science. 1997;278:1305–1309. doi: 10.1126/science.278.5341.1305. [DOI] [PubMed] [Google Scholar]
- Ilyas M. Wnt signalling and the mechanistic basis of tumour development. J Pathol. 2005;205:130–144. doi: 10.1002/path.1692. [DOI] [PubMed] [Google Scholar]
- Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, Rosenblatt J, Avigan DE, Teruya-Feldstein J, Pandolfi PP. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453:1072–1078. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15:801–812. doi: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G, Howe PH. Regulation of clusterin gene expression by transforming growth factor β. J Biol Chem. 1997;272:26620–26626. doi: 10.1074/jbc.272.42.26620. [DOI] [PubMed] [Google Scholar]
- Jin G, Howe PH. Transforming growth factor β regulates clusterin gene expression via modulation of transcription factor c-Fos. Eur J Biochem. 1999;263:534–542. doi: 10.1046/j.1432-1327.1999.00533.x. [DOI] [PubMed] [Google Scholar]
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human microRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadomatsu K, Anzano MA, Slayter MV, Winokur TS, Smith JM, Sporn MB. Expression of sulfated glycoprotein 2 is associated with carcinogenesis induced by N-nitroso-N-methylurea in rat prostate and seminal vesicle. Cancer Res. 1993;53:1480–1483. [PubMed] [Google Scholar]
- Klefstrom J, Vastrik I, Saksela E, Valle J, Eilers M, Alitalo K. c-Myc induces cellular susceptibility to the cytotoxic action of TNF-alpha. EMBO J. 1994;13:5442–5450. doi: 10.1002/j.1460-2075.1994.tb06879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klock G, Storch S, Rickert J, Gutacker C, Koch-Brandt C. Differential regulation of the clusterin gene by Ha-ras and c-myc oncogenes and during apoptosis. J Cell Physiol. 1998;177:593–605. doi: 10.1002/(SICI)1097-4652(199812)177:4<593::AID-JCP10>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Koch-Brandt C, Morgans C. Clusterin: a role in cell survival in the face of apoptosis? Prog Mol Subcell Biol. 1996;16:130–149. doi: 10.1007/978-3-642-79850-4_8. [DOI] [PubMed] [Google Scholar]
- Li X, Massa PE, Hanidu A, Peet GW, Aro P, Savitt A, Mische S, Li J, Marcu KB. IKKα, IKKβ, and NEMO/IKKγ are each required for the NF-κB-mediated inflammatory response program. J Biol Chem. 2002;277:45129–45140. doi: 10.1074/jbc.M205165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsick JS, Manak J, Mitiku N, Chen CK, Fogarty P, Guthrie E. Functional evolution of the Myb oncogene family. Blood Cells Mol Dis. 2001;27:456–458. doi: 10.1006/bcmd.2001.0404. [DOI] [PubMed] [Google Scholar]
- Lund P, Weisshaupt K, Mikeska T, Jammas D, Chen X, Kuban RJ, Ungethum U, Krapfenbauer U, Herzel HP, Schafer R, Walter J, Sers C. Oncogenic HRAS suppresses clusterin expression through promoter hypermethylation. Oncogene. 2006;25:4890–4903. doi: 10.1038/sj.onc.1209502. [DOI] [PubMed] [Google Scholar]
- Lutz W, Leon J, Eilers M. Contributions of Myc to tumorigenesis. Biochim Biophys Acta. 2002;1602:61–71. doi: 10.1016/s0304-419x(02)00036-7. [DOI] [PubMed] [Google Scholar]
- Markopoulou S, Kontargiris E, Batsi C, Tzavaras T, Trougakos I, Boothman DA, Gonos ES, Kolettas E. Vanadium-induced apoptosis of HaCaT cells is mediated by c-fos and involves nuclear accumulation of clusterin. FEBS J. 2009 doi: 10.1111/j.1742-4658.2009.07093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGFβ in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateyak MK, Obaya AJ, Adachi S, Sedivy JM. Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth & Differentiation. 1997;8:1039–1048. [PubMed] [Google Scholar]
- McLaughlin L, Mistry M, Zhu G, Ley-Ebert C, Stuart WD, Florio CJ, Groen PA, Witt SA, Kimball TR, Witte DP, Harmony JA, Aronow BJ. Apolipoprotein J/Clusterin limits the severity of murine autoimmune myocarditis. J Clin Invest. 2000;106:1105–1113. doi: 10.1172/JCI9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133:217–222. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- Michel D, Gillet G, Volovitch M, Pessac B, Calothy G, Brun G. Expression of a novel gene encoding a 51.5 kD precursor protein is induced by different retroviral oncogenes in quail neuroretinal cells. Oncogene Res. 1989;4:127–136. [PubMed] [Google Scholar]
- Miyake H, Hara I, Gleave ME. Antisense oligodeoxynucleotide therapy targeting clusterin gene for prostate cancer: Vancouver experience from discovery to clinic. Int J Urol. 2005;12:785–794. doi: 10.1111/j.1442-2042.2005.01173.x. [DOI] [PubMed] [Google Scholar]
- Miyake H, Hara I, Kamidono S, Gleave ME, Eto H. Resistance to cytotoxic chemotherapy-induced apoptosis in human prostate cancer cells is associated with intracellular clusterin expression. Oncol Rep. 2003;10:469–473. [PubMed] [Google Scholar]
- Morgenbesser SD, DePinho RA. Use of transgenic mice to study myc family gene function in normal mammalian development and in cancer. Sem Cancer Biol. 1994;5:21–36. [PubMed] [Google Scholar]
- Morris EJ, Ji JY, Yang F, Di Stefano L, Herr A, Moon NS, Kwon EJ, Haigis KM, Naar AM, Dyson NJ. E2F1 represses beta-catenin transcription and is antagonized by both pRB and CDK8. Nature. 2008;455:552–556. doi: 10.1038/nature07310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T, Yasui K, Zen K, Inagaki Y, Fujii H, Minami M, Tanaka S, Taniwaki M, Itoh Y, Arii S, Inazawa J, Okanoue T. Activation of B-Myb by E2F1 in hepatocellular carcinoma. Hepatol Res. 2008;38:886–895. doi: 10.1111/j.1872-034X.2008.00324.x. [DOI] [PubMed] [Google Scholar]
- Narita S, So A, Ettinger S, Hayashi N, Muramaki M, Fazli L, Kim Y, Gleave ME. GLI2 knockdown using an antisense oligonucleotide induces apoptosis and chemosensitizes cells to paclitaxel in androgen-independent prostate cancer. Clin Cancer Res. 2008;14:5769–5777. doi: 10.1158/1078-0432.CCR-07-4282. [DOI] [PubMed] [Google Scholar]
- Nusse R. The Wnt gene family in tumorigenesis and in normal development. J Steroid Biochem Mol Biol. 1992;43:9–12. doi: 10.1016/0960-0760(92)90181-h. [DOI] [PubMed] [Google Scholar]
- Nuutinen T, Suuronen T, Kyrylenko S, Huuskonen J, Salminen A. Induction of clusterin/apoJ expression by histone deacetylase inhibitors in neural cells. Neurochem Int. 2005;47:528–538. doi: 10.1016/j.neuint.2005.07.007. [DOI] [PubMed] [Google Scholar]
- O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- Oh IH, Reddy EP. The myb gene family in cell growth, differentiation and apoptosis. Oncogene. 1999;18:3017–3033. doi: 10.1038/sj.onc.1202839. [DOI] [PubMed] [Google Scholar]
- Orlandi A, Pucci S, Ciucci A, Pichiorri F, Ferlosio A, Spagnoli LG. Modulation of clusterin isoforms is associated with all-trans retinoic acid-induced proliferative arrest and apoptosis of intimal smooth muscle cells. Arterioscler Thromb Vasc Biol. 2005;25:348–353. doi: 10.1161/01.ATV.0000152609.28569.e1. [DOI] [PubMed] [Google Scholar]
- Patterson SG, Wei S, Chen X, Sallman DA, Gilvary DL, Zhong B, Pow-Sang J, Yeatman T, Djeu JY. Novel role of Stat1 in the development of docetaxel resistance in prostate tumor cells. Oncogene. 2006;25:6113–6122. doi: 10.1038/sj.onc.1209632. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Pierce AM, Schneider-Broussard R, Gimenez-Conti IB, Russell JL, Conti CJ, Johnson DG. E2F1 has both oncogenic and tumor-suppressive properties in a transgenic model. Mol Cell Biol. 1999;19:6408–6414. doi: 10.1128/mcb.19.9.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- Raschella G, Cesi V, Amendola R, Negroni A, Tanno B, Altavista P, Tonini GP, De Bernardi B, Calabretta B. Expression of B-myb in neuroblastoma tumors is a poor prognostic factor independent from MYCN amplification. Cancer Res. 1999;59:3365–3368. [PubMed] [Google Scholar]
- Rask K, Nilsson A, Brannstrom M, Carlsson P, Hellberg P, Janson PO, Hedin L, Sundfeldt K. Wnt-signalling pathway in ovarian epithelial tumours: increased expression of beta-catenin and GSK3beta. Br J Cancer. 2003;89:1298–1304. doi: 10.1038/sj.bjc.6601265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauhala HE, Porkka KP, Saramaki OR, Tammela TL, Visakorpi T. Clusterin is epigenetically regulated in prostate cancer. Int J Cancer. 2008;123:1601–1609. doi: 10.1002/ijc.23658. [DOI] [PubMed] [Google Scholar]
- Ricci S, Jin Z, Dews M, Yu D, Thomas-Tikhonenko A, Dicker DT, El-Deiry WS. Direct repression of FLIP expression by c-myc is a major determinant of TRAIL sensitivity. Mol Cell Biol. 2004;24:8541–8555. doi: 10.1128/MCB.24.19.8541-8555.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala A, Watson R. B-Myb protein in cellular proliferation, transcription control, and cancer: latest developments. J Cell Physiol. 1999;179:245–250. doi: 10.1002/(SICI)1097-4652(199906)179:3<245::AID-JCP1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Sallman DA, Chen X, Zhong B, Gilvary DL, Zhou J, Wei S, Djeu JY. Clusterin mediates TRAIL resistance in prostate tumor cells. Mol Cancer Ther. 2007;6:2938–2947. doi: 10.1158/1535-7163.MCT-07-0345. [DOI] [PubMed] [Google Scholar]
- Santilli G, Aronow BJ, Sala A. Essential requirement of apolipoprotein J (clusterin) signaling for IkappaB expression and regulation of NF-kappaB activity. J Biol Chem. 2003;278:38214–38219. doi: 10.1074/jbc.C300252200. [DOI] [PubMed] [Google Scholar]
- Santilli G, Schwab R, Watson R, Ebert C, Aronow BJ, Sala A. Temperature-dependent modification and activation of B-MYB: implications for cell survival. The Journal of biological chemistry. 2005;280:15628–15634. doi: 10.1074/jbc.M411747200. [DOI] [PubMed] [Google Scholar]
- Saura J, Petegnief V, Wu X, Liang Y, Paul SM. Microglial apolipoprotein E and astroglial apolipoprotein J expression in vitro: opposite effects of lipopolysaccharide. J Neurochem. 2003;85:1455–1467. doi: 10.1046/j.1471-4159.2003.01788.x. [DOI] [PubMed] [Google Scholar]
- Savkovic V, Gantzer H, Reiser U, Selig L, Gaiser S, Sack U, Kloppel G, Mossner J, Keim V, Horn F, Bodeker H. Clusterin is protective in pancreatitis through anti-apoptotic and anti-inflammatory properties. Biochem Biophys Res Commun. 2007;356:431–437. doi: 10.1016/j.bbrc.2007.02.148. [DOI] [PubMed] [Google Scholar]
- Scaltriti M, Bettuzzi S, Sharrard RM, Caporali A, Caccamo AE, Maitland NJ. Clusterin overexpression in both malignant and nonmalignant prostate epithelial cells induces cell cycle arrest and apoptosis. Brit J Cancer. 2004a;91:1842–1850. doi: 10.1038/sj.bjc.6602193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaltriti M, Brausi M, Amorosi A, Caporali A, D'Arca D, Astancolle S, Corti A, Bettuzzi S. Clusterin (SGP-2, ApoJ) expression is downregulated in low- and high-grade human prostate cancer. Int J Cancer. 2004b;108:23–30. doi: 10.1002/ijc.11496. [DOI] [PubMed] [Google Scholar]
- Scaltriti M, Santamaria A, Paciucci R, Bettuzzi S. Intracellular clusterin induces G(2)-M phase arrest and cell death in PC-3 prostate cancer cells. Cancer Res. 2004c;64:6174–6182. doi: 10.1158/0008-5472.CAN-04-0920. [DOI] [PubMed] [Google Scholar]
- Schepeler T, Mansilla F, Christensen LL, Orntoft TF, Andersen CL. Clusterin expression can be modulated by changes in TCF1-mediated Wnt signaling. J Mol Signal. 2007;2:6. doi: 10.1186/1750-2187-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takase O, Marumo T, Hishikawa K, Fujita T, Quigg RJ, Hayashi M. NF-kappaB-dependent genes induced by proteinuria and identified using DNA microarrays. Clin Exp Nephrol. 2008a;12:181–188. doi: 10.1007/s10157-008-0038-5. [DOI] [PubMed] [Google Scholar]
- Takase O, Minto AW, Puri TS, Cunningham PN, Jacob A, Hayashi M, Quigg RJ. Inhibition of NF-kappaB-dependent Bcl-xL expression by clusterin promotes albumin-induced tubular cell apoptosis. Kidney Int. 2008b;73:567–577. doi: 10.1038/sj.ki.5002563. [DOI] [PubMed] [Google Scholar]
- Tarasov KV, Tarasova YS, Tam WL, Riordon DR, Elliott ST, Kania G, Li J, Yamanaka S, Crider DG, Testa G, Li RA, Lim B, Stewart CL, Liu Y, Van Eyk JE, Wersto RP, Wobus AM, Boheler KR. B-MYB is essential for normal cell cycle progression and chromosomal stability of embryonic stem cells. PloS one. 2008;3:e2478. doi: 10.1371/journal.pone.0002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Tikhonenko A, Viard-Leveugle I, Dews M, Wehrli P, Sevignani C, Yu D, Ricci S, El-Deiry WS, Aronow B, Kaya G, Saurat J-H, French LE. Myc-transformed epithelial cells down-regulate clusterin which inhibits their growth in vitro and carcinogenesis in vivo. Cancer Res. 2004;64:3126–3136. doi: 10.1158/0008-5472.can-03-1953. [DOI] [PubMed] [Google Scholar]
- Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, Martin GR, Bishop JM. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature. 2001;414:768–773. doi: 10.1038/414768a. [DOI] [PubMed] [Google Scholar]
- Viale A, De Franco F, Orleth A, Cambiaghi V, Giuliani V, Bossi D, Ronchini C, Ronzoni S, Muradore I, Monestiroli S, Gobbi A, Alcalay M, Minucci S, Pelicci PG. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature. 2009;457:51–56. doi: 10.1038/nature07618. [DOI] [PubMed] [Google Scholar]
- Villavicencio EH, Walterhouse DO, Iannaccone PM. The sonic hedgehog-patched-gli pathway in human development and disease. Am J Human Genet. 2000;67:1047–1054. doi: 10.1016/s0002-9297(07)62934-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Sem Cancer Biol. 2006;16:318–330. doi: 10.1016/j.semcancer.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CR, Leskov K, Hosley-Eberlein K, Criswell T, Pink JJ, Kinsella TJ, Boothman DA. Nuclear clusterin/XIP8, an x-ray-induced Ku70-binding protein that signals cell death. Proc Natl Acad Sci USA. 2000;97:5907–5912. doi: 10.1073/pnas.97.11.5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaylim-Eraltan I, Bozkurt N, Ergen A, Zeybek U, Ozturk O, Arikan S, Erbil Y, Uslu I, Camlica H, Isbir T. L-myc gene polymorphism and risk of thyroid cancer. Exp Oncol. 2008;30:117–120. [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]