

Abstract
Agrobacterium species are plant-associated relatives of the rhizobia. Several species cause plant diseases such as crown gall and hairy root, although there are also avirulent species. A. tumefaciens is the most intensively studied species and causes crown gall, a neoplastic disease that occurs on a variety of plants. Virulence is specified by large plasmids, and in the case of A. tumefaciens this is called the Ti (tumor-inducing) plasmid. During pathogenesis virulent agrobacteria copy a segment of the Ti plasmid and transfer it to the plant, where it subsequently integrates into the plant genome, and expresses genes that result in the disease symptoms. A. tumefaciens has been used extensively as a plant genetic engineering tool and is also a model microorganism that has been well studied for host-microbe associations, horizontal gene transfer, cell-cell communication, and biofilm formation. This unit describes standard protocols for simple phenotypic characterizations of A. tumefaciens.
Keywords: Agrobacterium, Taxonomy, Opines, Plant association, Virulence, Plasmids, Attachment, Biofilms
Introduction
A. tumefaciens has agricultural implications and is an important tool in genetic engineering. Hence, it has been most intensively studied as a plant pathogen. The protocols outlined below are centered on phenotypes that are undoubtedly important for pathogenesis. However, not all species of A. tumefaciens are pathogenic, and field sampling studies suggest that as few as 10% of Agrobacterium strains carry a Ti plasmid. The ability to attach and form biofilms are fundamental aspects of Agrobacterium biology, for avirulent types as well as pathogens living outside of the tumor environment.
Basic Protocol 1describes a potato tumor assay, a method that allows for qualitative assessment of virulence. Basic Protocol 2describes a β-galactosidase assay, that can be used to quantify promoter activity and levels gene expression. Basic Protocols 3–5 describe association with surfaces, with 3focused on full biofilm formation on abiotic surfaces, 4on evaluating cellular attachment to abiotic surfaces and 5, a method for visualizing A. tumefaciens attachment to plant roots. Basic Protocol 6 describes a straightforward assay for motility, and for staining and visualizing bacterial flagella.
BASIC PROTOCOL 1: POTATO TUMOR ASSAY
Virulence on plants is one of the primary features of interest for A. tumefaciens. There are any number of ways to test virulence on a wide variety of plant and fungal hosts. We use the potato assay as a semi-quantitative baseline virulence determination (Anand, 1977).
Inoculate one set of potato tuber tissue discs (one set = 5 discs) with a range of dilutions for each strain. Literature indicates a linear relationship between number of tumors formed and quantity of bacteria ranging from 107 to 109 cfu/ml (107 cfu/ml is approximately equivalent to an optical density at 600 nm OD600=0.1; so begin by innoculating at OD600=0.1, 0.01, 0.001). For a negative control, use an avirulent A. tumefaciens derivative (eg. Ti-plasmidless NTL4 grows well and is non-tumorigenic).
Materials
Large, red-skinned, organically grown potatoes – purchased no more than 2 days prior to beginning assay. (To calculate number of potatoes required: each large potato yields two cylinders, each cylinder yields 10 discs, 5 discs yield one set, so each potato yields 4 sets of discs. The experiment requires one set per dilution per dilution per strain tested).
Sterile 1.5% agar water plates (100 mm diameter) – require one plate per set of potato discs.
Metal Cork borer, size 6. (Must be resistant to EtOH dipped flame sterilization) Scalpel
Solution of saturated potassium sulfate
Sealable container (e.g. Tupperware, etc).
Prepare inoculum -The night before preparing potatoes, start cultures of all strains to be tested. Include your positive (e.g. A. tumefaciens C58) and negative controls (e.g. A. tumefaciens NTL4).
The day of tissue inoculation, measure the OD600 of cultures. Subculture as necessary to set all cultures at approximately the same mid-log density at the time of inoculation.
Prepare appropriate dilutions of all cultures in either media or buffer.
Surface sterilize potatoes. First, rinse them in sterile freshwater, and then soak them in 1.05% sodium hypochlorite (bleach) solution for 20 min.
-
Prepare potato discs
-
a
With surface-sterilized cork borer, bore 2 cylinders from each potato.
-
b
Transfer cylinders to a sterile surface, such as a dissecting tray.
-
c
With a surface-sterilized scalpel, remove a 2-cm piece from each end of the cylinder and discard.
-
d
Again using the surface-sterilized scalpel, slice the remainder of the cylinder into 0.5 cm thick discs.
-
b
Transfer 5 discs to each 1.5% water agar plates, being certain that no plate receives more than one disc from a given cylinder.
-
a
-
Inoculate Potato Discs
-
Transfer 100 μl of the appropriate bacterial suspension to the top surface of each disc and spread to cover disc. Cover plate.
It is critical that potato discs be inoculated within 1 hour of slicing in order to avoid drying and ensure proper results!
Allow bacterial suspension to penetrate potato tissue.
Carefully wrap plate with parafilm to maintain moisture level in the plate.
Transfer plates to a sealable container containing a saturated solution of potassium sulfate to maintain constant humidity.
Store undisturbed at room temperature. The first tumors may be observed as early as day 10. Tumors appear as rough white to reddish-white bumps covering the surface of the potato tissue (See Figure 1).
-
Figure 1.
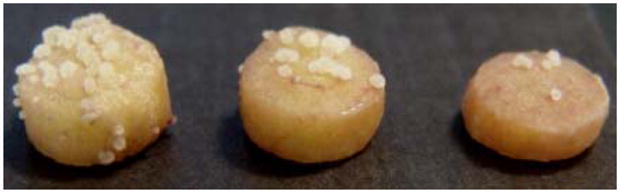
Photo depicting potato disc tumors induced by Agrobacterium tumefaciens C58. These tumors developed after 14 days of incubation.
BASIC PROTOCOL 2: β-GALACTOSIDASE ASSAY FOR AGROBACTERIUM TUMEFACIENS
This is a common assay by which the expression of specific genes, fused to lacZ can be measured under conditions of interest. This protocol is very similar to standard assays, but has been optimized for work on A. tumefaciens.
Materials
28°C incubator with shaking or rotating capablities
5 ml culture tubes
2 ml microfuge tubes
spectrophotometer
microcentrifuge
Z-buffer (see recipe)
Chloroform
0.05% SDS
1M Na2CO3
ONPG (ortho-nitrophenyl-β-galactoside) -Sigma Cat No. N1127
Grow overnight cultures of strain(s) of interest.
Measure the OD600 of the culture at the time of harvesting (0.3–0.6 OD600).
-
To each 2 ml microfuge tube (each test sample)
0.1 mls bacterial cells*
0.9 mls Z-Buffer
2 drops 0.05% SDS
3–4 drops chloroform
* For a negative control, instead of adding culture, add uninoculated media (the same as used in the experiment)
* You can add up 0.5 mls of culture -simply compensate by using less Z-Buffer.
Cap and vortex hard 10 sec.
Add 100 μl of 4 mg/ml ONPG (made up in Z-buffer). Immediately start timer.
Stop reaction with 0.6 mls of 1M Na2CO3 once the solution is visibly medium yellow. Record reaction time in minutes.
Microfuge tubes at full speed for 3 min to remove cell debris.
-
Measure absorbance at 420 nm (A420).
Miller unit equation:
BASIC PROTOCOL 3: STATIC BIOFILM COVERSLIP ASSAY
This assay provides a basic assessment of the ability A. tumefaciens to form biofilms, using polyvinyl chloride as a surface for attachment and crystal violet staining to obtain a quantitative measure of the phenotype. Most wild-type strains will form robust biofilms 24–48 h post-inoculation.
Materials
Solution of saturated potassium sulfate.
Sealable container (e.g. Tupperware, etc)
12-well tissue culture plates: Fisher catalog number 07-200-81
PVC coverslips: Fisher catalog number 12–547
0.1% crystal violet stain
33% acetic acid
UV light source
small plastic weigh boats (large enough to hold a coverslip)
spectrophotometer
5 ml glass culture tubes
28°C incubator
scissors
ATGN or other culture media
Microtiter plates or cuvettes
Day 1: Start overnight cultures of your strains of interest in the appropriate media with any required antibiotics (adjust culture volume according to your experiment).
Day 2:
-
Prepare plates. You will require 4 wells per sample per time-point (3 replicates will be measured for biofilm biomass and 1 will be kept as a visual representation).
The coverslips fit vertically into the wells of the 12-well plate with the narrow edge of the coverslip at the bottom of the well. However, the coverslips must be trimmed in order with to prevent displacement of the lid of the plate. (To trim, cut 2–3 mm off the edge of the coverslip using scissors)
Using an aerosol duster (such as VWR Whoosh-Duster, catalogue #16650-027), remove lint from coverslips and plates.
Sterilize coverslips and plates by exposing to a UV light source for 20–30 min.
Prepare inocula. You will require 3 ml of culture per well to be inoculated. If your strains have grown at a fairly even pace to mid-log phase, simply prepare a dilution in fresh antibiotic-containing media to OD600=0.05. If strains have not grown evenly, subculture and grow to mid-log phase prior to preparing dilution at OD600=0.05.
Inoculate plates by adding 3 ml of the appropriate strain to each well.
Place plates inside of a sealable container with an open container (e.g., 200 ml beaker) filled with saturated potassium sulfate solution. Store at room temperature undisturbed (without shaking) until time of harvest.
Day 3:
Working with one strain at a time, remove coverslips from wells with forceps and rinse thoroughly with deionized water. Use a squirt bottle to spray water directly onto both sides of the coverslip. This amount of force should not disrupt any biofilm that has formed on the coverslips.
Immerse coverslips in 0.1% crystal violet stain, and incubate for 5–10 min.
-
Remove coverslips from crystal violet, and rinse again as indicated in step 1. Blot coverslip on paper towel. Continue rinsing until the rinse water blotted on the paper towel contains no trace of purple.
Set one coverslip aside to scan and retain as a qualitative image. Immediately process the 3 remaining coverslips to collect quantitative data.
-
Solubilize the crystal violet from the three remaining coverslips:
Place each coverslip into an individual pool of 1 ml of 33% acetic acid. Small plastic weigh boats work well for this task.
Some agitation may help to solubilize the crystal violet.
Transfer 300 μl of the solubilized crystal violet/acetic acid to a microtiter plate or the entire 1 ml to a cuvette.
Measure the absorbance at λ600 (A600).
Finally, measure the OD600 of the resuspended cultures in each well from which a coverslip has been removed. This will allow normalization of biofilm development to culture growth. It is often useful to calculate a ratio of A600/OD600 which normalizes differences in culture growth between strains.
As a starting point, begin by collecting samples at 24, 48, and 72 h time points, which therefore requires 12 wells, or 1 plate, per strain tested.
BASIC PROTOCOL 4: SHORT-TERM BINDING ASSAY
The initial step for biofilm formation and infection of plant tissue is attachment. Wild-type A. tumefaciens cells attach to surfaces at their pole, a process mediated by the extrusion of a unipolar polysaccharide (UPP) comprised at least in part by N-acetyl glucosamine (Tomlinson, 2009). This assay provides a simple method for assessing the ability of cells to attach, and to visualize this polar adhesin.
Materials
28 °C incubator with shaking capabilities
Fluorescence microscope (with appropriate excitation and emission filter sets; Alexafluor 488, excitation 496, emission 519; Alexafluor 594, excitation 590, emission 617)
5 ml glass culture tubes
glass coverslips
glass microscope slides
1x AT Buffer
Alexafluor-594 and Alexafluor-488labeled wheat-germ agglutinin (Sigma-Aldrich Cat. No. W11262and W11261, respectively)
Grow cells in minimal medium (e.g. ATGN) to an OD600 of 0.4–0.6. Prepare enough culture to test each sample in triplicate.
Normalize cultures to have equal OD600 by adding the appropriate volume of media to higher density cultures.
Place coverslips at bottom of 6-well dish (or 33mm petri dishes) and overlay with 2 mL of inoculum. Allow cells to bind for 2 h.
Aspirate unattached cells and rinse several times with 1x AT buffer (pour buffer gently into well and shake)
Lay coverslip over 100 μl of a solution of fluorescently-labelled wheat-germ agglutinin (Alexafluor 594 = redor Alexafluor 488 = green) diluted 1:100 in AT buffer. Allow to stain 20–30 min.
Rinse coverslips (cell-side up) once with 0.05% Tween-20 in 1x AT buffer (rinse by gently pouring buffer into dish and shaking, again, gently) and 3x with 1X AT buffer (no Tween).
Lay coverslip on slide over 1–5 μl of buffer and visualize under microscope. Attached and lectin stained cells per field of view may be counted.
BASIC PROTOCOL 5: PLANT ATTACHMENT ASSAY
It is often useful to evaluate the attachment of A. tumefaciens on plant tissues. Quantitative assays are very difficult and prone to error (Fuqua, 2001), but we find that the following protocol yields repeatable and informative qualitative results.
Materials
1/2x Murashige Skoog Salts 1% agar plates with 1% sucrose
1 mM CaCl2, 0.4% sucrose buffer (filter sterilized)
A. thaliana seed stocks
A. tumefaciens derivatives expressing autofluorescent proteins such as GFP parafilm
4°C incubator
28°C incubator
35 mm dishes
spectrophotometer
sterile fine-tipped tweezers
microscope slides
Fluorescence microscope (with appropriate excitation and emission filter sets)
-
Prepare root segments
-
Surface sterilize A. thaliana seed stocks:
5–10 min wash in 70% ethanol with 0.1% Tween-20
5–10 min wash in 70% ethanol
5–10 min wash in 95% ethanol
Plant surface-sterilized A. thaliana on 1/2 MS salts plates with 1% agar and 1% sucrose. Seal edges of plates with parafilm to prevent dehydration of plates and seeds.
Incubate seeded plates at 4°C, in the dark, 18–48 h.
Transfer seeded plates to a 24-h photoperiod, room temperature environment for germination and growth.
Allow plants to grow until roots are approximately 2.5–3 cm long. At this point, plates may be stored at 4°C in the dark until ready to use.
-
-
Prepare of Agrobacterium tumefaciens strains
Inoculate 2 ml cultures of desired strains in ATGN broth with appropriate antibiotics for overnight aerobic growth at 28°C.
After overnight growth of cultures, check OD600 of strains. Calculate dilution necessary to achieve a bacterial suspension at OD600=0.1.
Wash bacteria by centrifuging at 9,000 x g for 3 min at room temperature and resuspending in 1 mM CaCl2, 0.4% sucrose buffer to achieve OD600=0.1.
-
Inoculate root segments with bacteria of interest
Set out and label the appropriate number of 35 mm dishes for your experiment. For each bacterial strain/root type combination to be tested, you will need two dishes per timepoint (one for root tips, one for root midsections). Additionally, you will need two additional plates per root type per timepoint to be tested, for a no bacteria (sterility) control. Into each plate, transfer 1.8 ml 1 mM CaCl2, 0.4% sucrose buffer.
Select healthy plants with a consistent root appearance. Transfer roots from media to a sterile surface (the lid of the plate in which the plants were grown is a useful sterile surface).
Cut roots into two sections, each approx. 1 cm long using a sterile scalpel or razor blade. Transfer segments to appropriate dishes (tip or mid-section, as described in step C1) using sterile forceps. Place 4 root segments into each dish.
After all dishes contain 4 segments of root, inoculate the buffer with 200 μl of the appropriate bacterial suspension (washed and at OD600=0.1, as described in step B3 above).
Carefully wrap dishes with parafilm, place in the dark (i.e., put the dishes in a box), and incubate at room temperature on a rocker for the appropriate time.
-
Prepare slides
At selected timepoint, prepare slides to receive root segments by cleaning with ethanol and labeling.
Set up 100 mm petri dishes with puddles of 1 mM CaCl2, 0.4% sucrose buffer. There should be one puddle per root segment, allowing enough space between them that they do not come in contact with one another.
Using surface-sterilized, very fine-tipped forceps, remove root segments from 35 mm dish containing bacterial suspension. Pass the root segment through a puddle of buffer, and lay on appropriate slide. This helps to rinse any bacteria from the root that are not truly attached.
Mount coverslips with additional 1 mM CaCl2, 0.4% sucrose buffer. Seal the edge of the slide with nail polish.
View slides with the appropriate microscope. 60X magnification is recommended.
BASIC PROTOCOL 6: SWIMMING MOTILITY
This simple assay allows you to evaluate the swimming motility of A. tumefaciens or other bacteria. Swimming bacteria can swim through the pores of low percentage (semi-solid) agar media, and resulting growth of the bacteria results in a visible swim ring on the agar plate. The presence of cells in the agar make it appear cloudy, so that the size of the ring correlates with the swimming velocity, the ability of the bacteria to chemotax along a nutrient gradient, and the dynamics of flagellar propulsion.
Materials
28°C incubator with shaking capabilities
0.3% Bacto Agar ATGN plates
5 ml glass culture tubes
Sealable container (that can accommodate all plates)
Start overnight cultures of test strains in ATGN media so that they are in mid-log phase at the time of inoculation the subsequent day.
Prepare water agar using Bacto Agar. Calculate volume of water agar such that the final agar concentration will be 0.3% AFTER addition of 20X AT salts, 20X AT buffer, and 50% glucose. Pour plates approximately 1–2 h prior to inoculation – this allows adequate time for plates to solidify, without concern over evaporation changing the agar concentration.
Normalize all cultures of test strains to the same density (OD600 ~ 0.6)
Inoculate plates by dispensing 5 μl of the appropriate OD600=0.6 suspension to the center of each plate. Cover plates and set on the bench for 1–3 hours, thus allowing inoculum to soak into the media without disturbance.
Carefully transfer inoculated plates to a sealable container (eg. tupperware). Add a small, uncovered beaker of saturated potassium sulfate to this to maintain humidity. Cover and incubate at room temperature without disturbance for 2–4 days.
BASIC PROTOCOL 7: PHENOL FLAGELLAR STAINING
This protocol is highly qualitative, but allows you to visualize the bacterial flagella microscopically. Not all non-motile strains are aflagellate, and the flagella of some A. tumefaciens mutants can deviate from the wild-type lophotrichous (ie. single polar tuft) arrangment. This protocol provides a crude means for visualizing flagella and flagellar arrangement.
Materials
Flagellum Stain (see recipe)
Liquid cultures or colonies of test strain(s)
Glass slides
Glass coverslips
Microscope with 1000X oil immersion
Vortex and centrifuge Staining Solution at high speed for 1 min or filter through a 0.22 micron filter to remove crystals.
Spot 1–3 μl of culture (generally concentrated) on a microscope slide and overlay with a coverslip.
Prop up the slide and apply 10 μl of stain to the junction between the slide and the coverslip and allow capillary action to wick the stain into the sample.
-
View the flagella with a light microscope under oil immersion with 1000X magnification.
Note: Flagella may appear swept to one side due to staining by capillary action.
REAGENTS AND SOLUTIONS
| Z-buffer (weights for 1 L) | |
|---|---|
| Na2HPO4-7H20 | 0.06 M (16.1 g) |
| NaH2PO4-H20 | 0.04 M (5.5 g) |
| KCl | 0.01 M (0.75 g) |
| MgSO4-4H20 | 0.001 M (0.25 g – use MgSO4-7H20) |
| β-mercaptoethanol | 0.05 M (3.5 mL) |
| The pH of this solution should be 7.0. Raise pH by adding NaOH and lower it by adding HCl. Store at room temperature for up to 1 year. | |
| Flagellum Stain Recipe: | |
|---|---|
| Solution 1: | |
| 5% carbolic acid (phenol) solution in H2O | 10 ml |
| Saturated aluminum potassium sulfate: 12 hydrate solution | 10 ml |
| Tannic acid | 2 g |
| Solution 2: | |
| Ethanol | 100 ml |
| Crystal violet | 12 g |
| Stain: | |
| 10 parts Solution 1 | |
| 1 part Solution 2 | |
COMMENTARY
Background Information
Potato Tumor Assay
Infection by pathogenic strains of A. tumefaciens typically occurs at wound sites on plant roots or soil-bound stem tissue, characterized by limited phosphorus, low pH, the presence of certain sugars, and most importantly, the release of specific phenolic compounds. These conditions induce expression of the vir genes, which are required for virulence on plants. Subsequently, a short segment of DNA from the Ti plasmid (the T-DNA) is conjugatively transferred into a plant cell via the activity of the vir gene products, where it is incorporated into the plant genome. Expression of T-DNA genes by the plant results in excess production of growth hormones such as auxins and cytokinins that stimulate rapid cell division in the transformed cells, leading to the formation of tumors.
A. tumefaciens is studied as a model system for plant pathogenesis, and as such, a strategy for assessing virulence has been highly valued. Although this assay has many limitations and yields a limited quantitative measure of virulence, it is a very useful qualitative measure. Larger and more abundant tumors are associated with increased virulence, although interpretations should be made with caution. It should be noted that this assay does not require the bacteria to be motile or to chemotax, which in the natural environment are undoubtedly important traits contributing to pathogenicity.
β-galactosidase Assay
The enzyme β-galactosidase is encoded by the bacterial gene lacZ, and can hydrolyze the chromogenic substrate, o-nitrophenyl-β-D-galactoside (ONPG) to form the products galactose and o-nitrophenol, which is yellow in color. This assay takes advantage of the fact that A. tumefaciens lacks β-galactosidase. The lacZ gene is fused to specific promoters (transcriptional or operon fusions) or coding sequences (translational or protein fusions) of interest. Significant β-galactosidase acitivity results in generation of a yellow color that develops in a reaction of cell lysate (expressing the promoter-lacZ fusion) and ONPG. The amount of yellow color directly reflects the expression of that particular gene.
Static Biofilm Assay
Natural populations of bacteria often exist in surface-associated microbial communities called biofilms. The ability to attach to surfaces allows for stable residence in a favorable environment, and the structure of the biofilm can provide cells with increased resistance to external factors that would otherwise be detrimental to an individual (O’Toole et al., 2000). In addition to this, biofilms also often provide optimal conditions for horizontal genetic exchange of plasmids carrying potentially beneficial genes, and cell-to-cell signaling such as quorum sensing. For example, biofilm formation and dispersal genes are often regulated by AHL signaling molecules (An et al., 2006). Within the rhizosphere, biofilms are a critical component of plant-microbe interactions (Danhorn, 2007; Rudrappa et al., 2008) and are undoubtedly an important factor in establishing infection. A. tumefaciens is capable of forming biofilms on a variety of surfaces in multiple environments (Danhorn,2004). Thus A. tumefaciens is an excellent model organism for studying the genetics and functional significance of the process. The ability of A. tumefaciens to form biofilms on the surface of PVC coverslips has been demonstrated to be representative of biofilms formed on other more biologically relevant surfaces (e.g. plant roots or soil particulates). However, because quantification of biofilm biomass on these other surfaces is difficult, this assay provides an adequate and quantitative means for assessing this phenotype.
Short-term Binding
One of the primary steps for biofilm formation is attachment, and the ability for A. tumefaciens cells to perform this function depends on many genes and can vary dramatically in different conditions. This is a quick assay that allows for visualization of attached bacteria and the production of a polar adhesin, the unipolar polysaccharide (UPP). The adhesin contains the sugar N-acetyl glucosamine, which readily binds to wheat germ agglutinin (WGA). Incubation of attached or attaching cells with fluorescently-labeled WGA allows for a quick and easy way to visualize this structure and identify cells that have irreversibly attached to the surface.
Plant Attachment Assay
Unlike the static biofilm protocol and short-term binding assay, this method allows for assessment of the bacteria’s ability to attach to living root tissue. In the format provided, the bacteria rely on root segments to provide nitrogen, phosphorus and other growth-limiting nutrients (carbon is provided as exogenous sucrose in the suspension buffer).
Analyses of Bacterial Motility and Flagellum Staining
The ability to move is a very important phenotype for bacteria. This is true not only for planktonic bacteria living in a liquid environment, but also for biofilm-associated bacteria where motility allows bacteria to colonize new environments and establish structured surface-associated communities. There are several different types of motility including twitching, gliding, swarming, and swimming. The latter two types require the use of extracellular appendages called flagella. These can be positioned around the cell in a monotrichous (one singular flagellum on one pole of the cell), lophotrichous (multiple flagella on one pole of the cell), amphitrichous (one flagellum on each pole of the cell), or peritrichous (multiple flagella distributed around the entire cell) arrangement. There are also less common examples of bacteria such as spirochetes, which contain periplamsic flagella. Species of Agrobacterium are lophotrichous, and motility has been demonstrated to be important for full virulence, attachment, chemotaxis, and biofilm formation (Merritt et al., 2007)
Critical Parameters and Troubleshooting
Potato Tumor Assay
Fungal growth can be quite a problem for this assay. Bleaching the potatoes, and working in a hood with sterile equipment can help to alleviate most of these issues. Bleach can inhibit growth of the bacteria, so it is important to thoroughly rinse them in sterile water. An alternative method for sterilization, is to UV irradiate the surfaces (15 minutes on each side) prior to extracting cores.
β-galactosidase Assay
For reactions with little or no activity that never reach a medium yellow color, the longer the incubation time, the more accurate the calculated values (up to 36 h). For reactions with high activity, use smaller volumes of cell suspension. Aim for reaction times of 30–60 min. Reaction times that are too short amplify the error. For the inexperienced user it is often helpful to have a color swath with a variety of yellow hues (as provided in paint stores) as a reference, and to designate a specific level of yellow color considered to be readable.
Static Biofilm Assay
The timing for this assay can vary greatly, so to obtain quantitative differences it is best to measure biofilm formation at a range of time points. In addition to this, a wide range of variables can have a dramatic effect on biolfilm formation, so it is important to repeat the experiment multiple times, on different days. Small changes in iron concentration for example can have dramatic effects on A. tumefaciens biofilm biomass, that can vary between strains. Temperature also can have large effects. If there is crystal violet staining above the meniscus, this is likely do to inadequate rinsing of the coverslips prior to staining them.
Short-term Binding
The most common problem with this assay is having a large amount of background in staining the unipolar polysaccharide. This effect can be minimized by manipulating the amount of Tween-20 detergent that is added to the wash buffer. Too much Tween-20 however, will cause the cells to lyse or detach from the surface.
Plant Attachment Assay
It is easy to damage the roots if this is done on a dry surface (as the roots will stick, rip and tear). The easiest way to transfer the roots without damaging them is to use the following tricks: i. use extremely fine-tipped forceps, ii. make sure that the surface to which you transfer them has a fine mist of liquid on it – this can either be condensation on the lid of the plate the plants were grown on, or a lid that you have sprayed a fine mist of sterile water onto, and iii. use the water droplets to get under the root and lift it.
Swimming Motility
For this assay it is very important to use purified agar since impurities in the media can dramatically influence the results. If testing non-motile mutants, selection for swimming is very high and often flares of increased motility or growth will be observable from the perimeter of the swim ring. These are often suppressor mutants that have regained the ability to swim.
Phenol Flagellar Stain
Because flagella are small and fragile, the results of this assay can vary considerably. It is important to keep in mind that this is not meant to be quantitative, but will allow for the visualization of flagella. Many of the flagella will be sheared from the cells, and this can vary from one slide preparation to the next. It is recommended that multiple slides are prepared, from replicate cultures, experimenting with different stages of growth. Fresh crystal violet works the best since older stain can become clumpy and affect the staining results.
Anticipated Results
Potato Tumor Assay
At around day 10, small, irregular, reddish tumors will begin to form around the potato discs. Avirulent control strains should yield no tumors, although the potato slices will turn more notably reddish in color with time.
β-galactosidase Assay
Depending on the strength of the promoter, tubes will turn a bright yellow color with time. For low expression promotors, such as are common for transcription factors, samples may only reach a light yellow color. Chemiluminescent β-galactosidase substrates are available that provide more sensitivity and a greater dynamic range.
Static Biofilm Assay
Strains of A. tumefaciens that are able to form biofilms will do so at the air water interface. Upon staining with crystal violet, a purple ‘smile’ of bacteria that have attached just below the meniscus to form a biofilm should be readily visible. Some strains will also form what is called a ‘pellicle,’ a biofilm that is floating on the surface of the liquid in the well. When staining for coverslip-associated bacteria, this can simply be removed by washing (but should be considered when calculating the cell density of the liquid).
Short-term Binding
Under phase-contrast microscopy and in standard AT medium, A. tumefaciens cells attach primarily by a single pole. Thus, cells will appear as single dots on the surface of the coverslip. Often, cells will form aggregates that will attach to the coverslip as clumps. These are difficult to count and are often filled with UPP. If staining for visualization of the UPP, single wild-type cells will appear to have a single spot at one pole of the cell. Production of this adhesin is surface-induced and thus, any unattached cells in the background are unlikely to be stained (Li et al. 2012).
Plant Attachment Assay
A. tumefaciens cells attach polarly to surfaces, so in this assay there will likely be a large number of cells lined up parallel to one another along the surface of the root.
Swimming Motility
This assay results in a region of the plate that is notably cloudy in appearance, due the growth of the bacteria swimming through the semi-solid agar. The site of inoculation will be off-white and opaque, and depending on the motility of the strain, there will be a swim ring of a certain size. A highly motile strain can fill the plate if given sufficient time. Ideally, if obtaining quantitative results a time point when all strains have a measurable ring should be chosen for comparison, or measurements should be made over multiple days.
Phenol Flagellar Stain
The nature of this assay will often leave the slide with a gradient of stain, so that one side is completely stained, and the other colorless. The location at which the flagella can be visualized is somewhere within this gradient. As stated in the protocol, the flagella will often appear swept to one side. This assay will cause many flagella to be sheared from the cells, so unattached flagella will likely be visible as well.
Time Considerations
Potato Tumor Assay
Set up for this experiment takes approximately 2 days, allowing for inoculum growth and setting up the potato discs. Formation of tumors can vary from 8–20 days, at which point they usually reach their maximum size.
β-galactosidase Assay
Once cultures to be tested have grown, this assay can range from several hours (for highly expressive promoters) to 24 h. (for weakly expressing promoters).
Static Biofilm Assay
Most strains of A. tumefaciens will form visible biofilms between 24 and 72 h. The time required for set-up and processing them is also minimal and depends on the number of strains and replicates. If biofilms are left for longer than 72 h., it is possible that even biofilm deficient strains will begin to catch up to wild-type.
Short-term Binding
When allowing cells to attach, it is important not to incubate for much longer than 2 h, at which point the attached cells that are doubling can skew the results. Insufficient time (less than 30 min) will make visualization of the UPP more difficult (since there will be less of it). This type of optimization is strain and condition-dependent (e.g. cells grown in phosphorus-limiting conditions attach more quickly and produce more UPP).
Plant Attachment Assay
This assay requires several days to 2 weeks to grow the Arabidopsis seedlings to a sufficient age, followed by 2 days to perform the attachment assay. Extended incubation times will allow too much bacterial growth, obscuring attached cells and those that are passively associated with the root. The roots also begin to degrade with longer incubation times.
Swimming Motility
Cultures are started the day before and are allowed to incubate for 1–3 days.
Phenol Flagellar Stain
This is a very quick assay and can be completed in less than 1 h. Visualizing cells under the microscope takes longer, but slides can also be stored until a later time.
Acknowledgments
Studies of Agrobacterium in the Fuqua Lab are supported by the National Institutes of Health (GM092660 and GM080546).
LITERATURE CITED
- An DD, et al. Quorum sensing and motility mediate interactions between Pseudomonas aeruginosa and Agrobacterium tumefaciens in biofilm cocultures. Proc Natl Acad Sci, USA. 2006;103(10):3828–3833. doi: 10.1073/pnas.0511323103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand VK, Heberlein GT. Crown gall tumorigenesis in potato tuber tissue. Am J Bot. 1977;64:153–158. [Google Scholar]
- Danhorn T, Hentzer M, et al. Phosphorous limitation enhances biofilm formation of the plant pathogen Agrobacterium tumefaciens through the PhoR-PhoB regulatory system. J Bacteriol. 2004;186:4492–4501. doi: 10.1128/JB.186.14.4492-4501.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danhorn T, Fuqua C. Biofilm formation by plant-associated bacteria. Annu Rev Microbiol. 2007;61:401–22. doi: 10.1146/annurev.micro.61.080706.093316. [DOI] [PubMed] [Google Scholar]
- Fuqua C, Matthysse AG. Methods for studying bacterial biofilms associated with plants. Meth Enzymol. 2001;337:3–1. doi: 10.1016/s0076-6879(01)37003-9. [DOI] [PubMed] [Google Scholar]
- Li G, Brown PJB, Tang JX, Xu J, Quardokus EM, Fuqua C, Brun YV. Surface contact stimulates the just-in-time deployment of bacterial adhesins. Mol Microbiol. 2012;83:41–51. doi: 10.1111/j.1365-2958.2011.07909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt PM, Danhorn T, et al. Motility and chemotaxis in Agrobacterium tumefaciens surface attachment and biofilm formation. J Bacteriol. 2007;189(22):8005–14. doi: 10.1128/JB.00566-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murashige T, Skoog F. A revised medium for rapid growth and bio-assays with tobacco tissue cultures. Physiol Plantarum. 1962;15(3):473–497. [Google Scholar]
- O’Toole G, Kaplan HB, Kolter R. Biofilm formation as microbial development. Annu Rev Microbiol. 2000;54:49–79. doi: 10.1146/annurev.micro.54.1.49. [DOI] [PubMed] [Google Scholar]
- Rudrappa T, Biedrzycki ML, Bais HP. Causes and consequences of plant-associated biofilms. Fems Microbiology Ecology. 2008;64(2):153–166. doi: 10.1111/j.1574-6941.2008.00465.x. [DOI] [PubMed] [Google Scholar]