

Abstract
The phase II metabolism sulfation and glucuronidation, mediated by sulfotransferases (SULTs) and UDP-glucuronosyltransferases (UGTs) respectively, are significant metabolic pathways for numerous endo- and xenobiotics. Understanding of SULT/UGT substrate specificity (including regioselectivity (i.e., position preference)) is of great importance in predicting contribution of sulfation/glucuronidation to drug and metabolite disposition in vivo. This review summarizes regioselective sulfation and glucuronidation of phenolic compounds with multiple hydroxyl (OH) groups as the potential conjugation sites. The strict regioselective patterns were highlighted for several SULT and UGT isoforms towards flavonoids, a large class of natural polyphenols. To seek for a molecular-level explanation, the enzyme structures (i.e., SULT crystal structures and homology-based UGT structure models) combined with molecular docking was employed. In particular, the structural bases for regioselective metabolism of flavonoids by SULT1A3 and UGT1A1 were discussed. It was concluded that the regioselective nature of these phase II enzymes was determined by the size and shape of binding pocket. While the molecular structures of the enzymes can be used to explain regioselective metabolism regarding the binding property, predicting the turnover at different positions remains a particularly difficult task.
Keywords: Phenolics, Flavonoids, Phase II metabolism, Sulfation, Glucuronidation, SULTs, UGTs, Homology modeling
1 Introduction
Phenolics refer to wide varieties of structurally-related compounds that possess aromatic hydroxyl group(s), including the chemotherapeutic agents (e.g., SN-38 and flavopiridol), non-chemotherapeutic drugs (e.g., raloxifene and ezetimibe), and bioactive natural polyphenols (e.g., hydroxycinnamic acids and flavonoids). Many phenolic compounds are subjective to extensive sulfation and glucuronidation, resulting in poor bioavailabilities and/or lack of efficacies [1–3]. For example, approximately 59% (47% sulfate and 12% glucuronide) of the parent drug is rapidly conjugated after oral administration of phenylephrine (a decongestant found in many cold remedies) [4–6].
Sulfation and glucuronidation,two most prevalent phase II metabolic pathways for drugs, are mediated by sulfotransferases (SULTs) and UDP-glucuronosyltransferases (UGTs), respectively. Human SULTs are divided into four families: SULT1 (phenol SULTs), SULT 2 (hydroxylsteroid SULTs), SULT4, and SULT6. Sulfation of phenolic xenobiotics is mediated primarily by SULT1 family, and sometimes by SULT2 family [7]. Human SULT1 and SULT2 families presently include 12 members (8 from SULT1 and 4 from SULT2): SULT1A1, 1A2, 1A3, 1B1, 1C1, 1C2, 1C3, 1E1, 2A1, 2A3, 2B1a, and 2B1b. Human liver expresses a panel of SULT isoforms including SULT1A1, 1B1, 1E1 and 2A1 [8,9]. SULT1A1, 1A3 and 1B1 are highly expressed in gastrointestinal tract [8,9]. Human UGTs are classified into four families: UGT1, UGT2, UGT3, and UGT8 [10]. The isoforms that are responsible for glucuronidating phenolics (O-glucuronidation) are mainly from UGT1A subfamily, especially, 1A1, 1A3, and 1A7~1A10 [1]. Substrate specificity of the six main contributors towards the phenolics often exhibits significant overlaps [1]. Among UGT1A isoforms, 1A1 and 1A9 are most abundantly expressed isoforms in the liver. By contrast, UGT1A8 and 1A10 were found predominantly in the intestine and colon. UGT1A7 is expressed only in the proximal tissues of the gastrointestinal tract, such as the esophagus and stomach [11].
The list of SULT/UGT substrates is expanding significantly in recent years [1,12]. This and other laboratories have reported that various SULT/UGT isoforms showed regioselectivity (i.e., the preferential formation of one conjugate isomer over the other) in the metabolism of phenolics with more than one conjugation site, including hydroxycinnamic acids (e.g., caffeic acid) [13], isoflavone (e.g., genistein) [14], and stilbene (e.g., piceatannol) [15]. Characterization of the regioselectivity would provide important insights into enzymatic mechanisms with respect to substrate binding and selectivity. The successfully (in silico) modeling of UGT1A9-mediated glucuronidation on a particular position (3-OH) of flavonols suggests that multiple binding modes from a single substrate exist within the catalytic domain for the regiospecific metabolism [16].
To date, crystal structures have been determined for all known SULT1 and SULT2 isoforms (Table 1). The bank of published crystal structures provides a strong basis to understand the substrate specificity differences between various SULT isoforms. For example, SULT1A3 specifically sulfonates dopamine, but shows limited activity towards p-nitrophenol. This is adequately explained by the fact that the amino group of dopamine forms electrostatic interactions with residues Glu146 and Asp86 (so called “substrate selector”), which however disfavor the positioning of the nitro group of p-nitrophenol into the binding site [17]. Unlike human SULTs, which are cytosolic proteins, UGTs are membrane-anchored. Because of the exceeding difficulties in crystallize them in active form(s), there is no complete three-dimensional structure of any human UGTs. Nevertheless, a partial crystal structure (C-terminal or UDPGA binding domain) of human UGT2B7 was determined and agrees well with its counterparts in plant UGT crystal structures [18,19–23]. This fact instills confidence to model human UGT structure using homology-based approach (homology modeling or comparative modeling). Presently, four 2-domain homology models have been published for human UGTs: three for the human UGT1A1 [24–26], and one for the human UGT1A9 [27].
Table 1.
List of available human SULT crystal structures, the structures can be found at http://www.rcsb.org/pdb/home/home.do.
| Human SULT | Crystal name | In complex with | Reference |
|---|---|---|---|
| SULT1A1 | 1LS6 | PAP, 4-Nitrophenol | [39] |
| 1Z28 | PAP | [42] | |
| SULT1A2 | 1Z29 | PAP | [42] |
| SULT1A3 | 1CJM | PAP, Dopamine | [17] |
| SULT1B1 | 3CKL | PAP, Resveratrol | N/A |
| SULT1C1 | 3BFX | PAP | [68] |
| SULT1C2 | 2GWH | PAP, Pentachlorophenol | [7] |
| SULT1C3 | 2H8K | PAP | [7] |
| 2REO | PAP | N/A | |
| SULT1E1 | 1HY3 | PAPS | [40] |
| SULT2A1 | 3F3Y | PAP, Lithocholic acid | [41] |
| SULT2A3 | 1EFH | PAP | [7] |
| SULT2B1 | 1Q20 | PAP, Pregnenolone | [69] |
| 1Q22 | PAP, Dehydroepiandrosterone | [69] |
N/A, paper is yet to published.
Understanding of SULTs and UGTs’ regioselectivity would allow prediction of the site(s) of sulfation/glucuronidation on a phenolic and identification of the metabolites likely to be formed in vivo. Since different metabolites (e.g., morphine-6-glucuronide vs. morphine-3-glucuronide) can have vastly different pharmacological and toxicological effects, in silico prediction of the metabolism, especially with respect to regioselectivity, is vital for identification of potential promises and problems associated with metabolism, which can facilitate the clinical development of a drug [28,29]. One main objective of this review is to summarize those phenolic compounds with regioselective SULT/UGT activities. Several SULT and UGT isoforms that display strict regioselective patterns are highlighted. Molecular basis for such metabolic patterns is summarized together with relevant discussion using crystal structures (for SULTs) or homology-modeled structures combined with molecular docking (for UGTs).
2 Regioselective Sulfation of Phenolics
2.1. Hydroxycinnamic acids
Caffeic (1, Fig 1) and dihydrocaffeic acids (2, Fig 1) both possess a catechol group, and sulfation occurred preferentially at the 3-OH group by human liver S9 fraction, intestinal S9 fraction, and SULT1A1 (Table 2). Phenotyping reaction showed that SULT1A1 was most active in the sulfation of caffeic and dihydrocaffeic acids [13]. This suggested that SULT1A1 was primary responsible for 3-OH sulfation of these two compounds in liver and intestinal S9 fraction, as SULT1A1 is highly expressed in both human liver and intestine [8]. SULT1A3 also preferrentially sulfonated 3-OH group of caffeic acid and dihydrocaffeic acids at 50 µM. By contrast, SULT1E1 only generated 4-O-sulfate from caffeic acid, but not sulfate from dihydrocaffeic acid [13].
Figure.1. Chemical structures of phenolic compounds with multiple possible conjugation sites (i.e., hydroxyl groups).

The structures are randomly numbered (in parenthesis).
Table 2.
Summary of kinetic constants for regioselective sulfation of phenolics
| Substrates | Position | Enzyme source |
Km(µM) | Vmax(pmol/min/mg) or Kcat |
Vmax/Km (µl/min/mg) or Kcat/Km |
|---|---|---|---|---|---|
| Caffeic acid# [13] | 3-OH | Liver S9 | 13.4 | 204 | 15.2 |
| 4-OH | 11.9 | 52.7 | 4.43 | ||
| 3-OH | Intestine S9 | 34.5 | 682 | 19.8 | |
| 4-OH | 41.3 | 297 | 7.2 | ||
| 3-OH | SULT1A1 | 11.6 | 62.1 | 5.35 | |
| 4-OH | 14.8 | 17.6 | 1.19 | ||
| Dihydrocaffeic acid# [13] | 3-OH | Liver S9 | 106 | 165 | 1.56 |
| 3-OH | Intestine S9 | 15.8 | 592 | 37.4 | |
| 3-OH | SULT1A1 | 34.1 | 108 | 3.17 | |
| Daidzein$ [14] | 7-OH | SULT1A1 | 0.47 | 1.27 | 2702 |
| 4’-OH | 0.54 | 0.33 | 611 | ||
| 7-OH | SULT1A3 | N.D. | N.D. | N.D. | |
| 4’-OH | 407.1 | 0.30 | 0.7 | ||
| 7-OH | SULT1E1 | 2.57 | 5.71 | 2216 | |
| 4’-OH | 3.24 | 6.39 | 1972 | ||
| 7-OH | SULT2A1 | 45.6 | 0.21 | 4.7 | |
| 4’-OH | 51.1 | 0.02 | 0.4 | ||
| Genistein$ [14] | 7-OH | SULT1A1 | 0.51 | 6.71 | 13,157 |
| 4’-OH | 0.52 | 0.78 | 1500 | ||
| 7-OH | SULT1A3 | N.D. | N.D. | N.D. | |
| 4’-OH | 371.8 | 1.46 | 3.9 | ||
| 7-OH | SULT1E1 | 2.79 | 7.25 | 2,682 | |
| 4’-OH | 2.94 | 7.45 | 2,530 | ||
| 7-OH | SULT2A1 | 25.5 | 0.37 | 14 | |
| 4’-OH | 55.9 | 0.09 | 1.6 | ||
| 7-hydroxyflavone* | 7-OH | SULT1A3 | 2.80 | 85,100 | 30,400 |
| 7,4’-dihydroxyflavone* | 7-OH | SULT1A3 | 3.42 | 20,800 | 6,090 |
| 3,7-dihydroxyflavone* | 7-OH | SULT1A3 | 1.85 | 79,700 | 43,000 |
| Chrysin* | 7-OH | SULT1A3 | 2.73 | 87,800 | 32,200 |
| Galangin [31] | 7-OH | SULT1A1 | 0.21 | 3,270 | 15,572 |
| 7-OH | SULT1A3 | 37.1 | 822 | 22.2 | |
| 7-OH | SULT1E1 | 1.13 | 948 | 839 | |
| Hesperetin [33] | 7-OH | SULT1A2 | N.D. | N.D. | N.D. |
| 3’-OH | 0.5 | 454,000 | 881,553 | ||
| 7-OH | SULT1A3 | 12.5 | 89,900 | 7,178 | |
| 3’-OH | 13.2 | 276,000 | 20,964 | ||
| 7-OH | SULT1B1 | 3.9 | 3,540 | 899 | |
| 3’-OH | 4.3 | 21,500 | 5,003 | ||
| 7-OH | SULT1C4 | 0.1 | 87,400 | 1,117,263 | |
| 3’-OH | N.D. | N.D. | N.D. | ||
| 7-OH | SULT1E1 | N.D. | N.D. | N.D. | |
| 3’-OH | 2.5 | 538,000 | 219,242 | ||
| Resveratrol [36] | 3-OH | Liver cytosol | 3.56 | 20.1 | 5.65 |
| 4’-OH | 30.2 | 1.12 | 0.04 | ||
| 3-OH | SULT1A1 | 79.8 | 46,484 | 582.4 | |
| 4’ -OH | 61.5 | 282 | 4.59 | ||
| 3-OH | SULT1E1 | 48.4 | 2,845 | 58.8 | |
| 4’ -OH | 119 | 5,491 | 46.1 | ||
| Dopamine [37] | 3-OH | SULT1A3 | 2.59 | 344,000 | 134,000 |
| 4-OH | 2.21 | 45,400 | 20,800 |
Meng et al. (submitted); N.D., not detectable;
Parameters Kcat (S−1) and Kcat/Km (µM−1•S−1) were calculated;
Parameters Kcat (Min−1) and Kcat/Km (mM−1 •Min−1) were calculated.
2.2. Flavonoids
2.2.1. Isoflavones
In the study of Nakano et al., preferential sulfation of daidzein (3, Fig.1) and genistein at the 7-OH position (4, Fig.1) was observed in human liver cytosols. The ratios for 7- to 4’-sulfate formation were 4.5:1 for daidzein and 8.4:1 for genistein [14]. In addition, SULT1A1 exhibited highest catalytic efficiency for 7- and 4’-sulfation of these substrates, followed by SULT1E1, SULT2A1 and SULT1A3 (Table 2). SULT1A1 catalyzed 7-O-sulfation of daidzein and genistein at rates 4.4- and 8.8-fold higher, respectively, than their respective 4’-O-sulfation [14]. However, with SULT1E1, catalytic efficiency was very similar for sulfation at either 7-O or 4’-O position. It is noted that daidzein and genistein essentially are a very poor substrate for SULT1A3 with strikingly high Km values (~400 µM) for 4’-O-sulfation, and no detectable activity for 7-O-sulfation (Table 2).
2.2.2. Flavones
SULT1A1 and 1A3 isoforms use chrysin (5, Fig.1) as a substrate, and the sulfation occurs at 7-OH group only. The apparent Km values for the isoform-mediated 7-O-sulfation of chrysin were 0.05 and 3.1 µm, respectively [30]. The latter is quite similar to the one (2.73 µM) we recently determined (Table 2). Significant amount of 7-O-sulfate was also found when incubating 50 µM chrysin with Caco-2 and HepG2 cells [30]. Likewise, SULT1A3 selectively conjugated 7-OH group of 7,4’-dihydroxyflavone (6, Fig.1) with a Km value of 3.4 µM (Meng et al., submitted).
2.2.3. Flavonols
Sulfation of galangin (7, Fig.1) by the human liver cytosol was mediated mainly by SULT1A1, with some contributions from SULT1A3 and 1E1 [31]. There was only one sulfate peak (i.e., 7-O-sulfate) identified, although the compound has two other potential sulfation sites (i.e., 3-OH a n d 5-OH) (Table 2). Consistently, SULT1A3 only generated 7-O-sulfate from 3,7-dihydroyflavone (8, Fig.1) and kaempferol (9, Fig.1). In the study of Ung and Nagar [32], quercetin (10, Fig.1) was an active substrate for both SULT1E1 and SULT1A1*1. Unfortunately, the authors quantified the metabolism rate by a radiometric method and were unable to provide any regioselective information.
2.2.4. Flavanone
Sulfation is a predominant metabolic pathway (faster than glucuronidation?) for the flavanone hesperetin (11, Fig.1) [33]. SULT1A2 catalyzed preferably and most efficiently the formation of hesperetin 3’-O-sulfate. SULT1A3, 1B1, and 1E1 also generated 3’-O-sulfate much efficiently than 7-O-sulfate from hesperetin [33]. By contrast, SULT1C4 catalyzed preferably and most efficiently the formation of hesperetin 7-O-sulfate [33].
2.2.5. Chalcone
SULT1A1*2, 1A2, and 1E1 were the most active SULT isoforms responsible for sulfation of xanthohumol (12, Fig.1), whereas SULT1A3 and 2A1 were of minor importance for the conjugation of xanthohumol [34]. Three mono-sulfates were identified and designated as MSulf1, MSulf2, and MSulf3 (ordered by elution time). MSulf1 was predominantly formed by SULT1E1 and 2A1, MSulf2 by SULT1A1*2 and 1A3, and MSulf3 by SULT1A2 [34]. The site of sulfation was not identified in the report.
2.2.6. Catechin
Human liver cytosol efficiently sulfated (−)-epicatechin (EC) (13, Fig.1) mainly through the SULT1A1 isoform. In the intestine, SULT1A3 also contributed to the sulfation. Other SULT isoforms contributed little to sulfation of (−)-EC [35]. Interestingly, sulfation of this compound primarily occurred at one particular position, although the exact position was unknown [35].
2.3. Stilbenes
Sulfation of resveratrol (14, Fig.1) and piceatannol (15, Fig.1) was studied by Miksits et al. [15,36], using human liver cytosol and recombinant SULT isoforms. Resveratrol-3-O-sulfate was almost exclusively catalyzed by SULT1A1 and only to a minor extent by SULT1A2, 1A3 and 1E1, whereas resveratrol-4'-O-sulfate was selectively formed by SULT1E1 (Table 2). For piceatannol, two mono-sulfates (i.e., M2 and M3) were identified, M2 was preferentially catalyzed by SULT1A1*2, 1A3 and 1E1. The formation of M3, however, was mainly catalyzed by SULT1A2*1 and SULT1A3 [15]. The exact position of the sulfate was not reported.
2.4. Endogenous phenolics
2.4.1. Dopamine
Dopamine (16, Fig.1) is sulfated at both 3-OH and 4-OH positions by SULT1A3. The apparent Km values for sulfation at both hydroxyl groups were similar (2.21 and 2.59 µM for dopamine -4-O-sulfate and dopamine -3-O-sulfate, respectively), but the Vmax value was approximately six times higher for the formation of the 3-O-sulfate (344 nmol/min/mg protein) than that of 4-O-sulfate (45.4 nmol/min/mg protein) [37]. The preferential sulfation of dopamine at 3-OH position is in accordance with the crystal structure of SULT1A3 with dopamine bound to the active site, and 3-hydroxyl group appropriately aligned to form hydrogen bonds with the catalytic residues (His108 and Lys106) of the enzyme [17].
2.4.2. D-dopa
SULT1A3 has a unique dopa (3,4-dihydroxyphenylalanine; 17, Fig.1)-sulfating activity that was stereo-selective for their D-form enantiomers [38]. Although there is a lack of experimentally derived differentiation between the formation rates of different sulfated metabolites, molecular docking of D-dopa to the SULT1A3 crystal structure rendered the 3-OH group of D-dopa at a closer position to the catalytic residues than 4-OH, suggesting that 3-O-sulfate should be preferentially generated by SULT1A3 [17].
3 3D Structures of four SULT1 isoforms
The 3D structures of SULT1 are highly conserved. Overlay of the coordinates of the crystal structure (Cα’s) of SULT1E1 with SULT1A1, SULT1A3 and SULT1B1 gives RMSD (root mean square deviation) values of 1.4 Å, 1.3 Å and 1.7 Å, respectively. These crystal structures adopt α/β fold and are comprised of a central 4-stranded parallel β-sheet (numbered β1, β2, β3, and β4) surrounded by helices (numbered α1, α2, α3, α4, and α5) on either side (Fig.2). Helices α3 and α4 are divided into 4 and 3 short helices, respectively (Fig.2). The three loops that are less conserved and involved in the substrate binding have been numbered. This numbering is used throughout this review (Fig.2).
Figure.2. Crystal structures of SULT1 isoforms reveal their highly conserved 3D structure.

Panel A: Structure-based sequence alignment of four crystallized SULT1 isoforms, the α-helices are in cyan and β-strands in magenta. The alignment was performed by overlay of the 3D structures. The conserved catalytic residue histidine and lysine are marked with an asterix. The residues determining the regioselectivity of SULT1A3 towards dopamine and D-dopa [15] are marked with a pound sign. Residues reported to form part of the acceptor pocket are highlighted in grey. Panel B: Ribbon diagram of the SULT1A3 crystal structure [15] showing the 3D folding of elements of secondary structure in a spectrum-colored mode. Panel C: Ribbon diagram of the SULT1A3 crystal structure [15]. The regions that form the substrate-binding pocket are shown in red. The PAPS and substrate (dopamine) are shown as black stick models.
3.1. Ligand binding involves structural changes
Molecular packing in the SULT1 crystals in presence of both cofactor (PAPS) and substrate is very tight, resulting in the formation of two separate binding pockets (Fig.3). The narrow access (especially for big substrates such as troglitazone [39]) to the binding pockets suggests that ligand accommodation occurs following the pocket opening. Compared to SULT1A1 [39] and SUTL1A3 [17], the substrate-free SULT1E1 structure [40] shows a bigger substrate access space (in the middle of loop1 and loop3). This might indicate the flexibility of the loop1 and loop 3 regions involving in the substrate binding. Structural comparisons across a family of SULT structures with or without disordered segments suggest that the presence of ligands (cofactor and/or substrate) is coupled with an increased order, namely the formation of helices α2-2, α2-3, α7, and α8 [7]. The PAP(S)-induced ordering of α7 and α8 may restrict the conformations available for the intervening substrate-binding loop 3 [7]. Therefore, PAPS binding is suggested to prime the loop1 and loop3 for binding to the substrate [7]. In addition, as seen in the SULT2A1 structure [41], binding of two molecules of a ligand might induces a non-productive conformation which is incompatible with PAPS binding. This “adapt interaction” between substrate binding site and PAPS binding site was proposed by Allali-Hassani et al. as an alternative mechanism for substrate inhibition kinetics frequently displayed by SULTs [7].
Figure.3. Surface view of the SULT1A1 crystal [39] showing the buried state of PAPS (red stick) and substrate (blue stick).

3.2. PAPS binding
The binding site for PAPS is nearly identical among SULTs, with highly conserved residues contributing to the binding pocket [7]. The interaction between SULT and PAPS is depicted in Fig.4A, and residues (in red, Fig.4B) involved in PAPS binding are indicated in the primary sequence with secondary structures annotated (Fig.4B). In brief, the short loop following the first strand (β1) of the central β-sheet forms the so-called PSB-loop (residues 45TYPKSGTT52). The last two residues (Thr51 and Thr52) from the PSB-loop form hydrogen bond interaction with 5’-phosphate of PAPS, whose positioning is thought to be important for orienting the cofactor for in-line sulfonyl transfer to the substrate. The PAPS 3’-phosphate interacts with three conserved residues, namely residues 257RKG259 of the GXXGXXK motif (in loop 3) and residues Arg130 and Ser138. The adenine ring of PAPS forms stacking and T-shaped interactions with two conserved residues Trp53 and Phe229, which is stabilized by hydrogen bond interactions between N-6 and Thr227 as well as N-3 and Tyr193. The hydrogen bond interaction between adenine ring (N-3) and Tyr193 is not shown in Fig.4 for the clarity purpose (i.e., to reduce clutter).
Figure.4. Conserved interaction of PAPS with SULT residues.

Panel A: The conserved PAPS interacting residues from the four SULT crystals, showing their interactions with the PAPS. Dashed lines indicate hydrogen bonds. Panel B: Alignment of four SULT1 sequences showing the PAPS-interacting residues (in red) in secondary structures. Residue numbers in SULT1A1 are labeled,
3.3. Substrate binding
The substrate-binding pocket, described as “L-shaped” or “V-shaped” pockets previously [39,42], is formed by less conserved regions. In the structural alignment (Fig.2A), residues forming part of the acceptor pockets in the four SULT crystals are highlighted in grey. The pocket is mainly formed by residues from loop1, loop2 and loop3 (Fig. 2B/C). The catalytic residue histidine and a neighboring residue lysine are situated at the bottom of the pocket. Some residues from α1 and α4-1 also contribute to the formation of the pocket. The binding pocket is very hydrophobic. Take SULT1A3 for example, the pocket incorporates predominately aromatic residues (Phe24, Phe81, Y139, Phe142, Y169, and Y240) and aliphatic residues (Ile21, Pro47, Val84, Ala148, and Leu247). This suggests that hydrophobic molecules would have better affinity for SULTs than hydrophilic ones.
As revealed by the crystal structures [17,39,41,42], a crucial point for SULT activity is the position of the acceptor -OH group(s) amenable to sulfation. The accepting group needs to be positioned close to the sulfur group of PAPS and the amino acid acting as the general base facilitating deprotonation of the acceptor. In the majority of SULT structures, this amino acid is a histidine residue. The correct orientation of the -OH group determines whether an enzymatic reaction can take place or not. Also, correct positioning in the active site of the enzyme should be compatible with accommodation of the rest of the acceptor molecule for activity [17,39,41,42].
3.4. Structural basis for regioselectivity of SULT1 isoforms towards flavonoids
SULT1A3 efficiently metabolizes flavones and flavonols with a preference for the 7-OH position (Table 2), whereas this SULT isoform lacks of the ability to sulfonate the isoflavones such as daidzein and genistein (no detectable and very poor activities for 7-O-sulfation and 4’-sulfation, respectively) [14]. Molecular docking was employed to explain this apparent regioselectivity (and substrate selectivity) of SULT1A3. The automated docking program GOLD (CCDC, Cambridge, UK) was used to dock the flavonoid substrates (sulfate acceptors) into the SULT1A3 active sites (PDB entry code: 2A3R). Default genetic algorithm parameters for controlling the operation of the docking process were used. The co-crystallized substrate dopamine was used as a reference ligand. All docking calculations were restricted to the binding pocket by defining the active site with residues His108 and Lys106. Distance constraints were used to define each potential site for calculation (Meng et al., submitted). GOLDscore was used to identify the lowest energy docking results. There are four potential O-sulfation sites (i.e., 3-OH, 5-OH, 7-OH, and 4’-OH) on the kaempferol molecule; 7-OH group of kaempferol can be easily docked into the active site of the enzyme with the highest fitness score than the other three. Hydrogen bonds analysis showed that only 7-OH orientation allows the hydrogen bond interactions between this sulfation site and the catalytic residue His108 and lys106 (Fig.5D & Table 3). The identification of this catalytic-competent binding mode helps us to understand why 7-O-sulfate is the predominant sulfate generated by SULT1A3 from flavones and flavonols (Meng et al., submitted).
Figure.5. Structural basis of regioselective sulfation of phenolics.

Surface view of the substrate-binding pockets of SULT1A3 (panel A), 1A1 (panel B), and 1E1 (panel C). The size and shape of substrate-binding sites are divergent. SULT1E1 has a wider and larger pocket, compared to SULT1A1 and 1A3. SULT1A3 has a unique hump (circled area) in the pocket. Black arrow indicates a narrow corner in the binding pocket of SULT1A3. Panel D: Surface view of the interaction between SULT1A3 and kaempferol (7-OH). Panel D1: An expanded view of residues potentially involved in interactions with kaempferol in the SULT1A3 crystal. Dashed lines indicate potential hydrogen bonds. Panel E: Surface view of the substrate-binding pocket of SULT1A3 with genistein (7-OH) docked, the B-ring was fitted to the narrow corner.
Table 3.
Summary of docking results of SULT or UGT enzymes with selected substrates
| Enzyme | Substrates | Conjugation site | GoldScore | H-Bond with catalytic His* |
| SULT1A3 | Kaempferol | 3-OH | 27.7 | No |
| 5-OH | 35.4 | No | ||
| 7-OH | 41.2 | Yes | ||
| 4’-OH | 31.0 | No | ||
| Genistein | 5-OH | 45.7 | No | |
| 7-OH | 24.6 | Yes | ||
| 4’-OH | 36.8 | No | ||
| SULT1A1 | Caffeic acid | 3-OH | 39.8 | Yes |
| 4-OH | 36.6 | Yes | ||
| Genistein | 5-OH | 42.6 | Yes | |
| 7-OH | 33.0 | Yes | ||
| 4’-OH | 25.8 | Yes | ||
| SULT1E1 | Caffeic acid | 3-OH | 33.5 | Yes |
| 4-OH | 34.1 | Yes | ||
| Genistein | 5-OH | 39.9 | Yes | |
| 7-OH | 37.5 | Yes | ||
| 4’-OH | 40.2 | Yes | ||
| UGT1A1 | Quercetin | 3-OH | 35.2 | Yes |
| 5-OH | 35.7 | Yes | ||
| 7-OH | 37.6 | Yes | ||
| 3’-OH | 33.6 | Yes | ||
| 4’-OH | 22.1 | No |
SULT1A1 and 1A3, His108; SULT1E1, His107; UGT1A1, His39
The backbone structure of isoflavone differs from that of flavone in that it has the B-ring connected to C3 instead of C2. This structural difference results in a disfavored positioning of 7-OH (genistein), whereby the B-ring of genistein is restricted to a narrow space or corner (indicated by black arrow in Fig.5A/E). This binding corner is evident due to the presence of a hump in the middle of the pocket. It is contributed by GLU146 and ASP48, and is presumably thought to create a steric hindrance that affect the binding affinity locally. Unsurprisingly, docking of 7-OH group (of genistein) gives a poor fitting score (Table 3). As for 5-OH and 4’-OH, their orientations preclude the formation of the hydrogen bond with His108, thus their high fitting scores are considered non-productive.
It is noteworthy that SULT1A1 and 1E1 displayed distinct position preferences towards the isoflavones (e.g., daidzein and genistein) based on the determined intrinsic clearance (CLint) values (Table 2). SULT1A1 predominately generated 7-O-sulfate, whereas SULT1E1 equally produced 7-O-sulfate and 4’-O-sulfate. Molecular docking showed that all hydroxyl groups of genistein could be docked close to catalytic histidine in both crystalline structures (Table 3). The pockets of these two SULT isoforms (indicated by red arrows) are much wider (compared to that of SULT1A3, Fig.5A–C) and larger than genistein, and genistein can fit in easily, with any of its hydroxyl groups located ~ 3 Å from the NE2 atom of the catalytic histidine (SULT1A1, H108; SULT1E1, His107). For SULT1A1, 7-OH of genistein gives fitness score slightly higher than that of 4’-OH (33.0 vs. 25.8). The use of docking results did not produce a clear explanation as to why the 7-OH group is the favorable site for sulfation by SULT1A1, because they suggested that the binding affinities (kinetic Km values) for 7-O-sulfation and 4’-O-sulfation are almost identical (Table 2). Also, docking of 3-OH and 4-OH of caffeic acid to SULT1A1 gives similar fitting scores, which cannot explain the enzyme’s preference on 3-OH (Table 2 & 3). However, it is in support of the fact that experimentally determined Km values for 3-O-sulfation and 4-O-sulfation are quite close (11.6 vs. 14.8 µM) [13]. Similar docking observations were made with SULT1E1 (4-OH-prefered enzyme) towards caffeic acid [13]. Taken together, these analyses suggest that while substrate binding can sometimes be predicted from the crystal structures, prediction of the turnover rate at different positions remains a particularly difficult task. Unexpectedly, 5-OH of genistein can also be properly positioned in a catalytic mode, which however is not supported by experimental data in that no 5-O-sulfate was ever detected. The exact reason for this inconsistency is unknown. It is hypothesized that this is in part due to the formation of intermolecular hydrogen bond between the 5-OH and 4-carbonyl groups, preventing its deprotonation by the catalytic histidine, thus disrupting the catalytic completion at this position [52].
4 Regioselective Glucuronidation
4.1. Ester-acylglucuronidation vs. ether-O-glucuronidation
Ester-acylglucuronidation occurs at the carboxylic acid group (e.g., bilirubin), resulting in an acyl-linked glucuronide, whereas ether-O-glucuronidation occurs at a hydroxyl group (e.g., phenolics). Selectivity between ester-acylglucuronidation and ether-O-glucuronidation appears to be substrate- and isoform-dependent.
Mouse ugt1a1 predominantly generated ether-O-glucuronide from mycophenolic acid (18, Fig.6) [ 43]. By contrast, glucuronidation of (−)-11-nor-9-carboxy-tetrahydrocannabinol (THC-COOH; 19, Fig.6) only occurs at the carboxylic acid group [44]; the highest activity towards THC-COOH was observed with UGT1A3, but UGT1A1 and human liver microsomes (HLMs) also showed a significant amount of activity towards this substrate. An examination of kinetic parameters revealed that ferulic acid (20, Fig.6) was mainly glucuronidated by UGT1A1 on the two nucleophilic groups (i.e., carboxylic acid and hydroxyl group) [45]. UGT1A3 was able to glucuronidate the two positions at about the same rate, but with low efficiency. UGT1A6 and 1A8 were strictly involved in the formation of the ether glucuronide, whereas UGT1A7, 1A10 and 2B7 were principally responsible for glucuronidation at the carboxyl group.
Figure.6. Regioselective glucuronidation of phenolic compounds in literatures [43–48,60,62].

The chemical structures are randomly numbered (in parenthesis). * Percentage based on calculated intrinsic clearance (CLint) values. HLM, human liver microsomes; HIM, human intestine microsomes; (m)ugt1a1, mouse ugt1a1.
4.2. Preference of phenol glucuronidation over alcohol glucuronidation
We believed that phenolic hydroxyl groups are more prone to glucuronidation than alcoholic hydroxyl groups. When both phenolic and alcoholic hydroxyl groups are present in the same structure, glucuronidation occurs primarily at the phenolic (−OH) groups. This can be seen from the glucuronidation of a variety of UGT substrates such as SN38 (21, Fig.6) [46], ezetimibe (22, Fig.6) [47] and estradiol (23, Fig.6) [48].
5 Regioselective glucuronidation of phenolics
5.1. Hydroxycinnamic acid
Caffeic acid is a hydroxycinnamic acid that can be found in majority of plants (1, Fig.1). UGT1A1 and UGT1A9 were active in glucuronidation of caffeic acid (50 µM), albeit at a low rate o f 4 ~ 8 pmol/min/mg [13]. UGT1A1 specifically catalyzed the formation of caffeic acid-4-O-glucuronide, whereas UGT1A9 conjugated both 3-OH and 4-OH groups of caffeic acid [13]. Furthermore, kinetic profiling demonstrated that UGT1A1 and UGT1A9 had lower affinity and activity towards caffeic acid (Table 4), compared to its analogs such as ferulic and isoferulic acids [13].
Table 4.
Summary of kinetic constants for regioselective glucuronidation of phenolics
| Substrates | Position | Enzyme source |
Km(µM) | Vmax(pmol/min/mg) | Vmax/Km (µl/min/mg) |
|---|---|---|---|---|---|
| Caffeic acid [13] | 3-OH | UGT1A1 | N.D. | N.D. | N.D. |
| 4-OH | 4,240 | 178 | 0.04 | ||
| 3-OH | UGT1A9 | 1,900 | 206 | 0.11 | |
| 4-OH | 1,910 | 325 | 0.17 | ||
| Daphnetin [62] | 7-OH | UGT1A6 | 74.4 | 15,600 | 208 |
| 8-OH | 74.4 | 7,400 | 99 | ||
| 7-OH | UGT1A9 | 60.5 | 12,400 | 207 | |
| 8-OH | 44.3 | 8,100 | 183 | ||
| EGC [60] | 3’-OH | UGT1A1 | 470 | 500 | 1.1 |
| 3’-OH | UGT1A3 | 1,490 | 36 | 0.024 | |
| 3’-OH | UGT1A8 | 280 | 120 | 0.41 | |
| 3’-OH | UGT1A9 | 200 | 2,000 | 9.9 | |
| EGCG [60] | 3’-OH | UGT1A1 | 11 | 170 | 15 |
| 4”-OH | 8.0 | 980 | 123 | ||
| 3’-OH | UGT1A3 | N.D. | N.D. | N.D. | |
| 4”-OH | 54 | 580 | 11 | ||
| 3’-OH | UGT1A8 | N.D. | N.D. | N.D. | |
| 4”-OH | 28 | 5,590 | 200 | ||
| 3’-OH | UGT1A9 | 24 | 780 | 32 | |
| 4”-OH | 31 | 4,230 | 136 | ||
| 6,7-dihydroxyflavone* | 6-OH | UGT1A9 | 2.9 | 920 | 320 |
| 7-OH | 4.5 | 4,090 | 910 | ||
| 3,3’,4’-trihydroxyflavone* | 3’-OH | UGT1A1 | 1.84 | 7,810 | 4,250 |
| 4’-OH | 1.84 | 1,920 | 1,040 | ||
| 3’-OH | UGT1A10 | 16 | 740 | 50 | |
| 4’-OH | N.D. | N.D. | N.D. | ||
| Fisetin* | 3’-OH | UGT1A1 | 1.28 | 6,940 | 5,430 |
| 4’-OH | 1.04 | 1,410 | 1,350 | ||
| 3’-OH | UGT1A10 | 9.8 | 220 | 22 | |
| 4’-OH | 5.3 | 340 | 64 | ||
| Luteolin* | 7-OH | UGT1A1 | 0.69 | 690 | 1,000 |
| 3’-OH | 1.43 | 6,660 | 4,660 | ||
| 4’-OH | 0.78 | 1,520 | 1,950 | ||
| 7-OH | UGT1A10 | 5.1 | 1,050 | 210 | |
| 3’-OH | 8.6 | 170 | 20 | ||
| 4’-OH | 2.0 | 340 | 170 | ||
| Quercetin* | 3’-OH | UGT1A1 | 0.37 | 2,130 | 5,830 |
| 4’-OH | UGT1A10 | 10 | 280 | 28 | |
| Myricetin* | 3’-OH | UGT1A1 | 2.1 | 20,100 | 9,760 |
| 4’-OH | 1.7 | 1,330 | 800 | ||
| 4’-OH | UGT1A10 | 3.0 | 240 | 80 | |
| 6,3’,4’-tryhydroxyflavone* | 3’-OH | UGT1A1 | 0.9 | 4,060 | 4,510 |
| 4’-OH | 1.0 | 2,290 | 2,300 | ||
| 3’-OH | UGT1A10 | 12.8 | 320 | 20 | |
| 4’-OH | 8.7 | 830 | 100 |
Unpublished data; N.D., not detectable.
5.2. Flavonoids
5.2.1. Quick identification of the position of mono-glucuronide of flavones and flavonols
Evaluation of the regioselectivity requires the structural identification (or assignment of the conjugation site) of the glucuronide(s). Nuclear magnetic resonance (NMR) was necessary earlier on for this purpose, although it is not favored due to the exceeding difficulty in purifying the glucuronide isomers in a significant amount. It was recently reported that a “UV peak shift method” can be used to rapidly pinpoint glucuronidation site(s) of flavones and flavonols with hydroxyl groups at the 4'-, 3-, 5-, and/or 7-position(s) [49]. Briefly, glucuronidation of the 3- and 4'-OH groups results in band I (please define the λmax) hypsochromic shifts (or blue shifts) of 13–30 and 5–10 nm, respectively. Glucuronidation of the 5-OH group causes a band II (please define the λmax) hypsochromic shift of 5–10 nm. In contrast, glucuronidation of the 7-OH group does not cause any change in band I or II. It was acknowledged in the paper that UV spectrum change associated with glucuronidation of 6-OH was unpredictable [49]. Also differentiation between 7-OH and 3’-OH positions was impossible using the method, since no changes in band I or II were observed in glucuronidation of either group. However, 7-O-glucuronide is often the earlier-eluting isomer than 3’-O- glucuronide in a reversed-phase HPLC column, and this elution order was used to distinguish these two glucuronide [49,50]. Inspired by this fact, this research team compared the elution order of several 7-O-glucuronides and 6-O-glucuronides (i.e., 7-hydroxyflavone-7-O-glucuronide < 6-hydroxyflavone-6-O-glucuronide; 3,7-dihydroxyflavone-7-O-glucuronide < 3,6-dihydroxy flavone-6-O-glucuronide; 5,7-dihydroxyflavone-7-O-glucuronide < 5,6-dihydroxyflavone- 6-O-glucuronide; 7,4’-dihydroxyflavone-7-O-glucuronide < 6,4’-dihydroxyflavone- 6-O-glucuronide; 3,7,4’-trihydroxyflavone-7-O-glucuronide < 3,6,4’-trihydroxyflavone- 6-O-glucuronide; 7-hydroxy-4’-methoxyflavone-7-O-glucuronide < 6-hydroxy-4’-methoxyflavone- 6-O-glucuronide; baicalein-7-O-glucuronide < baicalein-6-O-glucuronide), and found that 7-O-glucuronide was often eluted earlier than 6-O-glucurondie. Therefore, we believe that this elution order could complement the “UV peak shift method” to identify the glucuronidation position (between 6-OH and 7-OH) of flavones and flavonols.
5.2.2. Flavones/flavonols/flavanones
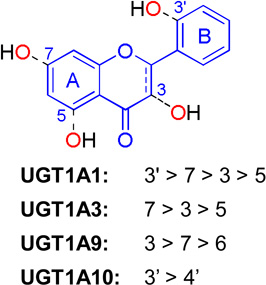
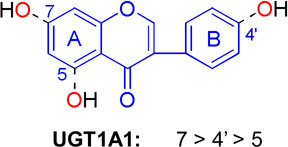
Flavones and flavonols share the same structural backbone of 2-phenylchromen-4-one (Table 5). The backbone of flavanones slightly differs from that of flavones in that the bond between C2–C3 is saturated (Table 5). Extensive glucuronidation studies [30,33,50–57] including a number of compounds from these three subclasses of flavonoids tended to render a generalized regioselectivity for various UGT isoforms (Table 5). Based on the intrinsic clearance values (CLint = Vmax/Km) or glucuronidation rates at a low concentration, the regioselectivity order is 3'-OH > 7-OH > 3-OH > 5-OH for UGT1A1, and 7-OH > 3- OH > 5-OH for UGT1A3. UGT1A9 preferentially glucuronidates 3-OH group, followed by 7-OH and 6-OH. UGT1A10 tends to metabolize 4’-OH group more efficiently than 3’-OH (Table 5).
Table 5.
Generalized regioselectivity of UGT1A1, 1A3, 1A9 or 1A10 towards four flavonoid subclasses: flavonols, flavones, flavanones, and isoflavones.
| Backbone structure | Supporting compounds | |||||
|---|---|---|---|---|---|---|
 |
Flavones | Chrysin [30,51] | Flavones | 3,7-dihydroxyflavone [52,53] | ||
| 6,7-dihydroxyflavone* | Galangin [52] | |||||
| Apigenin [51] | Resokaempferol [52] | |||||
| Baicalein [57] | Kaempferol [52] | |||||
| Wogonin [54] | 3,3',4'-trihydroxyflavone* | |||||
| Oroxylin A [54] | Fisetin* | |||||
| 6,3’,4’-trihydroxyflavone* | Quercetin*[56] | |||||
| Luteolin* [55] | Myricetin* [50] | |||||
| Flavanone | Hesperetin [33] | |||||
 |
Isoflavone | Daidzein [1,59] | ||||
| Genistein [1,59] | ||||||
| Formononetin [1,59] | ||||||
| Biochanin A [1,59] | ||||||
| Prunetin [1,58,59] | ||||||
| Glycitein [1,59] | ||||||
Please refer to Table 4 for kinetic parameters.
5.2.3. Isoflavones
The regioselective glucuronidation of isoflavone prunetin is isoform-, organ-, and species-dependent, as reviewed by Wu et al [1]. Characterization on glucuronidation of another 5 isoflavones (i.e., daidzein, genistein, formononetin, biochanin A and glycitein) suggests that UGT1A1 glucuronidates this class of flavonoids following a strict regioselective pattern, namely, 7-OH > 4’-OH > 5-OH (Table 5) [58, 59].
5.2.4. Catecins
(−)-Epigallocatechin (EGC) (24, Fig.6) and (−)-epigallocatechin gallate (EGCG) (25, Fig.6) are major green tea polyphenols with antioxidant and anticancer activities. Liu et al.[60] studied glucuronidation of EGC and EGCG in human, mouse, and rat liver microsomes and in 9 human UGT isoforms expressed in insect cells. EGCG-4”-O-glucuronide was the major EGCG glucuronide formed in all incubations, whereas EGC-3’-O-glucuronide was the major EGC glucuronide formed in all incubations (Table 4). UGT1A1, 1A8 and 1A9 displayed high glucuronidation activities towards EGCG. Interesting, UGT1A8 had the highest Vmax/Km value with EGCG but low activity with EGC (Table 4).
5.3. Anthraquinone
Emodin (26, Fig.6) is a major active anthraquinone present in the rhubarb. Emodin 3-O-glucuronide was the only glucuronide formed, when incubating emodin with a panel of microsomal fractions prepared from mice, rats, guinea pigs, dogs, and humans [61]. The 1-OH and 8-OH groups of emodin were not metabolized at all, probably due to the formation of an intramolecular hydrogen bond between the neighboring carbonyl and 1-OH or 8-OH groups (26, Fig.6).
5.4. Coumarin
Daphnetin (27, Fig.6) is a dihydroxycoumarin being used in China for the treatment of coagulation disorders. UGT1A6 and UGT1A9 can rapidly conjugate both hydroxyl groups and generate 7-O-glucuronide and 8-O-glucuronide [62]. Although less efficient, UGT1A3, 1A4, 1A7, 1A8, and 1A10 selectively glucuronidated the 7-OH position of daphnetin [62].
5.5. Other phenolic compounds
In addition to those phenolics discussed above, regioselectivities of various human UGT isoforms against other phenolic compounds such as resveratrol (both trans- and cis-forms), raloxifene and morphine were summarized in a recent review [1], to which one can refer for details.
6 Structural basis for regioselectivity of UGT1A1 towards flavonoids
To explore the molecular basis of the substrate regioselectivity, a homology model of UGT1A1 was generated using a plant UGT structure as the template. Structural models were constructed using the program Modeller 9v5 [63] with the standard protocol (numbers of models were set to 50). The sequence alignment of the target protein to the template protein (i.e., UGT71G1 (PDB code: 2ACW)) is shown in Fig.7A. The co-crystalized sugar donor (UDP-glucose) was copied to the homology model as a block residue. Molecular docking of the substrates to the UGT1A1 model with the program GOLD (CCDC, Cambridge, UK) was performed using distant constraints, similar to our earlier publications [19,21]. The distance constraints (2.5~4 Å) were set between the potential glucuronidation site (OH group) and His39 (the catalytic residue or general base). The final UGT1A1 model was composed of two N- and C-terminal domains with α/α/α folds (Fig.7B). The N- and C-terminal domains contain central stranded parallel β sheets flanked by α helices on both sides. The two domains pack very tightly and form a deep cleft where the aglycone and cofactor are bound for reaction. The catalytic dyad His39-Asp151 [18] is located at the helix N1α (at its N-terminus) and the strand Nβ4, respectively (Fig.7B). Nα5-2 packs to Nα3-2, and facilitates specific interactions (e.g. salt bridge) between the two regions (Fig.7B), which might play roles in the entry of the aglycone and in the departure of the product [23,26]. It was also proposed that residues in Nα3-2 area might undergo certain local conformational changes when substrate binds, and when product leaves [23]. Residues between Nα5-1and Nα5-2, as well as residues within loop N2 were positioned too far from the acceptor substrates to enable direct interactions (Fig.7B). The binding of the cofactor UDPGA to human UGTs has been extensively reviewed [1,18,64], and hence is not discussed here.
Figure.7. Homology model of UGT1A1.

Panel A: Sequence alignment between human UGT1A1 and plant UGT71G1 (PDB code: 2ACW) used for homology modeling. The aligned sequence identity is 13.7%. The full length UGT1A1 sequence (Accession: Q5DT03) was downloaded from the SwissProt database (http://www.uniprot.org/). Approximately 450 amino acids long sequence of human 1A1 (excluding N-terminal signal peptide, C-terminal transmembrane domain and cytosolic tail) was aligned to UGT71G1 with the aid of predicted secondary structures [67]. The sequence aligning approaches used in modeling human UGT was discussed [1]. Similar to an early work [25], helices Nα3-1 and Nα3-3 were not modeled in this work and were shown as random coils in the final generated structures. The best model with the lowest objective function values (DOPE) were selected for further investigation. The quality of the model was validated using PROCHECK (http://www.jcsg.org/prod/scripts/validation/sv_final.cgi). The 44-aa signature motif of UGTs is enclosed by a box. Residues which were predicted to be in contact with aglycone are highlighted in red whereas the catalytic dyad residues (His39-Asp151) are highlighted in grey. Panel B: Ribbon diagram of the structure model of human UGT1A1. Regions forming the substrate binding pocket in N-/C- terminus are shown in red. The copied UDP-glucose (in black) and co-crystallized kaempferol (in blue) from vVGT1 (PDB code: 2C1Z) are shown as stick models. Loop N2 (shown in purple) is positioned far away from the catalytic histidine, and does not participate in binding small substrates such as flavonols.
In this modeling scheme, residues predicted to form part of the substrate pockets were highlighted (in red) in the sequence alignment diagram (Fig.7A). The substrate binding pocket was almost entirely formed by the N-terminal residues, although some C-terminal residues also contributed to its formation. The pocket was primarily formed by loop N1, Nα1, Nα3-2, loop N4, Nα5-1, Nα5-2, loop C1 and loop C5 (Fig.7B). This is consistent with the topological arrangement of β strands (3-2-1-4-5-6-7) of the enzymes. β-Strands Nβ2, Nβ3, Nβ6 and Nβ7 twisted far away from the core Nβ1 where the catalytic histidine was situated. Within the active pocket, loopN4 was located at the bottom of the pocket (far from the bound cofactor); Nα5-1 residues formed the wall in the end of the pocket; loop N1 and Nα3-2 lined the entrance of the pocket; loops C1 and C5 covered the top of the pocket; and the residues in the N-terminus of Nα5-2 occupied the right side of the pocket. The cofactor was present at the left side of the pocket. It was suggested that UDPGA binds first to the enzyme, followed by binding of aglycone substrates [65]. The residues predicted to be in contact with flavonoids are mainly hydrophobic: Phe105, Ile110, Phe153, Phe170, Phe217, Tyr230, Phe237, Met310 and Phe394. There were also several polar residues in the binding pocket (i.e., His39, Ser178, Gln182, and Asp396).
As summarized in Table 5, UGT1A1 can actively metabolize the hydroxyl groups at different positions of flavonols, following a regioselective order of 3’-OH > 7-OH > 3-OH. In particular, UGT1A1 predominantly produces quercetin 3’-O-glucuronide. However, molecular docking studies of UGT1A1 with quercetin show that quercetin can be positioned in different orientations in the active site, allowing each hydroxyl group (3-OH, 7-OH, and 3’-OH) for glucuronidation (Fig.8B–D). This is probably contributed by a large binding pocket as showed in Fig.8A. Positioning of 3-OH, 7-OH or 3’-OH groups inside of the active pocket gave similar fitting scores (Table 3), suggesting that binding affinity of quercetin to UGT1A1 using these three different catalytic modes were not divergent. The latter is consistent with the fact that regiospecific glucuronidation of several flavonols displayed close (or almost identical) Km values (Table 4) [52]. It is noteworthy that 5-OH of quercetin can also be properly positioned for glucuronidation; but formation of quercetin 5-O-glucuronide was not detected. On the other hand, UGT1A1 does possess the ability to glucuronidate the 5-OH group in several flavones (in the absence of 3’-OH or 7-OH groups) such as 5-hydroxylfavone [52] and 3,5-dihydroxyfavone (unpublished data), which indicates that it is inadequate to use of the formation of intermolecular hydrogen bond as the sole criterion to explain the slow or nonexistent formation of 5-O-glucuronide [66].
Figure.8. The substrate binding pocket of human UGT1A1 (homology model) and its interaction with quercetin.

Panel A: Molecular surface diagram of the substrate binding pocket. The copied UDP-glucose is shown as stick models. Panel B–D, stereo diagrams showing interactions between quercetin in distinct (and regioselective) catalytic modes (panel B, 3’-OH; panel C, 7-OH, panel D, 3-OH) and side chains in the substrate-binding pocket. Dashed lines indicate potential hydrogen bonds.
7 Concluding remarks
The phase II metabolizing enzymes SULTs and UGTs possess the ability to conjugate polyphenols at different positions with a distinct regioselectivity, which is recognized as a unique property of the enzymes. The mechanisms determining the regioselectivity are complex, one of which is found to be the size and shape of the substrate-binding pocket. Large binding site (e.g., those of SULT1E1 and UGT1A1) allows flexible orientation of the substrate, rendering different hydroxyl groups for conjugation. The unique shape (with a hump structure) of SULT1A3 active site restricts the positioning of flavones and flavonols to the 7-OH catalytic mode, which uncovers the structural basis for this enzyme’s highly selective activity toward 7-OH group. In addition, the location of the bound substrate relative to the enzyme cofactor (either UDPGA or PAPS) and the catalytic residue (histidine) is important; a phenolic (OH) group has to be positioned near the cofactor and catalytic residue for conjugation activity. The molecular structures of the enzymes can be used to explain regioselective metabolism regarding the binding property, but predicting the turnover at different positions remains a particularly difficult task. This limitation might contributed by the static nature of the crystal structure, which is not an appropriate reflection of the native dynamic status of enzymes in action. Further elucidation of structural elements responsible for the metabolic turnover of the substrates would be necessary. We hope that this review will attract structural biologists to take what we believe is an important problem worthy of attention, because conjugation is a major detoxification pathway for humans.
Acknowledgement
This work was supported by grants from the National Institutes of Health [GM070737] to MH.
Abbreviations used
- UGTs
UDP-glucuronosyltransferases
- SULTs
sulfotransferases
- UDPGA
uridine diphosphoglucuronic acid
- PAPS
3'-phosphoadenosine 5'-phosphosulfate
- NMR
Nuclear magnetic resonance.
References
- 1.Wu B, Kulkarni K, Basu S, Zhang S, Hu M. First-pass metabolism via UDP-glucuronosyltransferase: a barrier to oral bioavailablity of phenolics. J Pharm Sci. 2011;100(9):3655–3681. doi: 10.1002/jps.22568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crozier A, Jaganath IB, Clifford MN. Dietary phenolics: chemistry, bioavailability and effects on health. Nat Prod Rep. 2009;26(8):1001–1043. doi: 10.1039/b802662a. [DOI] [PubMed] [Google Scholar]
- 3.Gao S, Hu M. Bioavailability challenges associated with development of anti-cancer phenolics. Mini Rev Med Chem. 2010;10(6):550–567. doi: 10.2174/138955710791384081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ibrahim KE, Midgley JM, Crowley JR, Williams CM. The Mammalian Metabolism of R-(-)-m-Synephrine. J Pharm Pharmacol. 1983;35(3):144–147. doi: 10.1111/j.2042-7158.1983.tb04293.x. [DOI] [PubMed] [Google Scholar]
- 5.Gumbhir K. An Investigation of Pharmacokinetics of Phenylephrine and its Metabolites in Humans. University of Missouri-Kansas City: Pharmaceutical Sciences; 1993. p. 216. [Google Scholar]
- 6.Hengstmann JH, Goronzy J. Pharmacokinetics of 3H-Phenylephrine in Man. Eur J Clin Pharmacol. 1982;21:335–341. doi: 10.1007/BF00637623. [DOI] [PubMed] [Google Scholar]
- 7.Allali-Hassani A, Pan PW, Dombrovski L, Najmanovich R, Tempel W, Dong A, Loppnau P, Martin F, Thornton J, Edwards AM, Bochkarev A, Plotnikov AN, Vedadi M, Arrowsmith CH. Structural and chemical profiling of the human cytosolic sulfotransferases. PLoS Biol. 2007;5(5):e97. doi: 10.1371/journal.pbio.0050097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teubner W, Meinl W, Florian S, Kretzschmar M, Glatt H. Identification and localization of soluble sulfotransferases in the human gastrointestinal tract. Biochem J. 2007;404(2):207–215. doi: 10.1042/BJ20061431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riches Z, Stanley EL, Bloomer JC, Coughtrie MW. Quantitative evaluation of the expression and activity of five major sulfotransferases (SULTs) in human tissues: the SULT "pie". Drug Metab Dispos. 2009;37(11):2255–2261. doi: 10.1124/dmd.109.028399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackenzie PI, Bock KW, Burchell B, Guillemette C, Ikushiro S, Iyanagi T, Miners JO, Owens IS, Nebert DW. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenetics Genomics. 2005;15:677–685. doi: 10.1097/01.fpc.0000173483.13689.56. [DOI] [PubMed] [Google Scholar]
- 11.Ohno S, Nakajin S. Determination of mRNA expression of human UDP-glucuronosyltransferases and application for localization in various human tissues by real-time reverse transcriptase-polymerase chain reaction. Drug Metab Dispos. 2009;37(1):32–40. doi: 10.1124/dmd.108.023598. [DOI] [PubMed] [Google Scholar]
- 12.Huang C, Chen Y, Zhou T, Chen G. Sulfation of dietary flavonoids by human sulfotransferases. Xenobiotica. 2009;39(4):312–322. doi: 10.1080/00498250802714915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong CC, Meinl W, Glatt HR, Barron D, Stalmach A, Steiling H, Crozier A, Williamson G. In vitro and in vivo conjugation of dietary hydroxycinnamic acids by UDP-glucuronosyltransferases and sulfotransferases in humans. J Nutr Biochem. 2010;21(11):1060–1068. doi: 10.1016/j.jnutbio.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 14.Nakano H, Ogura K, Takahashi E, Harada T, Nishiyama T, Muro K, Hiratsuka A, Kadota S, Watabe T. Regioselective monosulfation and disulfation of the phytoestrogens daidzein and genistein by human liver sulfotransferases. Drug Metab Pharmacokinet. 2004;19(3):216–226. doi: 10.2133/dmpk.19.216. [DOI] [PubMed] [Google Scholar]
- 15.Miksits M, Sulyok M, Schuhmacher R, Szekeres T, Jäger W. In-vitro sulfation of piceatannol by human liver cytosol and recombinant sulfotransferases. J Pharm Pharmacol. 2009;61(2):185–191. doi: 10.1211/jpp/61.02.0007. [DOI] [PubMed] [Google Scholar]
- 16.Wu B, Morrow JK, Singh R, Zhang S, Hu M. Three-dimensional quantitative structure-activity relationship studies on UGT1A9-mediated 3-O-glucuronidation of natural flavonols using a pharmacophore-based comparative molecular field analysis model. J Pharmacol Exp Ther. 2011;336(2):403–413. doi: 10.1124/jpet.110.175356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu JH, Li HT, Liu MC, Zhang JP, Li M, An XM, Chang WR. Crystal structure of human sulfotransferase SULT1A3 in complex with dopamine and 3'-phosphoadenosine 5'-phosphate. Biochem Biophys Res Commun. 2005;335(2):417–423. doi: 10.1016/j.bbrc.2005.07.091. [DOI] [PubMed] [Google Scholar]
- 18.Miley MJ, Zielinska AK, Keenan JE, Bratton SM, Radominska-Pandya A, Redinbo MR. Crystal structure of the cofactor-binding domain of the human phase II drug-metabolism enzyme UDP-glucuronosyltransferase 2B7. J Mol Biol. 2007;369(2):498–511. doi: 10.1016/j.jmb.2007.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shao H, He X, Achnine L, Blount JW, Dixon RA, Wang X. Crystal structures of a multifunctional triterpene/flavonoid glycosyltransferase from Medicago truncatula. Plant Cell. 2005;17:3141–3154. doi: 10.1105/tpc.105.035055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Offen W, Martinez-Fleites C, Yang M, Kiat-Lim E, Davis BG, Tarling CA, Ford CM, Bowles DJ, Davies GJ. Structure of a flavonoid glucosyltransferase reveals the basis for plant natural product modification. EMBO J. 2006;25:1396–1405. doi: 10.1038/sj.emboj.7600970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Modolo LV, Escamilla-Trevino LL, Achnine L, Dixon RA, Wang X. Crystal Structure of Medicago truncatula UGT85H2 - Insights into the Structural Basis of a Multifunctional (Iso)flavonoid Glycosyltransferase. J Mol Biol. 2007;370:951–963. doi: 10.1016/j.jmb.2007.05.036. [DOI] [PubMed] [Google Scholar]
- 22.Brazier-Hicks M, Offen WA, Gershater MC, Revett TJ, Lim EK, Bowles DJ, Davies GJ, Edwards R. Characterization and engineering of the bifunctional N- and O-glucosyltransferase involved in xenobiotic metabolism in plants. Proc Natl Acad Sci. 2007;104:20238–20243. doi: 10.1073/pnas.0706421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Modolo LV, Li L, Pan H, Blount JW, Dixon RA, Wang X. Crystal structures of glycosyltransferase UGT78G1 reveal the molecular basis for glycosylation and deglycosylation of (iso)flavonoids. J Mol Biol. 2009;392(5):1292–1302. doi: 10.1016/j.jmb.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 24.Li C, Wu Q. Adaptive evolution of multiple-variable exons and structural diversity of drug metabolizing enzymes. BMC Evol Biol. 2007;7:69. doi: 10.1186/1471-2148-7-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Locuson CW, Tracy TS. Comparative modelling of the human UDPglucuronosyltransferases: insights into structure and mechanism. Xenobiotica. 2007;37(2):155–168. doi: 10.1080/00498250601129109. [DOI] [PubMed] [Google Scholar]
- 26.Laakkonen L, Finel M. A molecular model of the human UGT1A1, its membrane orientation and the interactions between different parts of the enzyme. Mol Pharmacol. 2010;77(6):931–939. doi: 10.1124/mol.109.063289. [DOI] [PubMed] [Google Scholar]
- 27.Fujiwara R, Nakajima M, Yamamoto T, Nagao H, Yokoi T. In silico and in vitro approaches to elucidate the thermal stability of human UDP-glucuronosyltransferase (UGT) 1A9. Drug Metab Pharmacokinet. 2009;24(3):235–244. doi: 10.2133/dmpk.24.235. [DOI] [PubMed] [Google Scholar]
- 28.van Dorp EL, Morariu A, Dahan A. Morphine-6-glucuronide: potency and safety compared with morphine. Expert Opin Pharmacother. 2008;9(11):1955–1961. doi: 10.1517/14656566.9.11.1955. [DOI] [PubMed] [Google Scholar]
- 29.Sorich MJ, Smith PA, Miners JO, Mackenzie PI, McKinnon RA. Recent advances in the in silico modeling of UDP glucuronosyltransferase substrates. Curr Drug Metab. 2008;9(1):60–69. doi: 10.2174/138920008783331167. [DOI] [PubMed] [Google Scholar]
- 30.Galijatovic A, Otake Y, Walle UK, Walle T. Extensive metabolism of the flavonoid chrysin by human Caco-2 and Hep G2 cells. Xenobiotica. 1999;29(12):1241–1256. doi: 10.1080/004982599237912. [DOI] [PubMed] [Google Scholar]
- 31.Otake Y, Hsieh F, Walle T. Glucuronidation versus oxidation of the flavonoid galangin by human liver microsomes and hepatocytes. Drug Metab Dispos. 2002;30(5):576–581. doi: 10.1124/dmd.30.5.576. [DOI] [PubMed] [Google Scholar]
- 32.Ung D, Nagar S. Variable sulfation of dietary polyphenols by recombinant human sulfotransferase (SULT) 1A1 genetic variants and SULT1E1. Drug Metab Dispos. 2007;35(5):740–746. doi: 10.1124/dmd.106.013987. [DOI] [PubMed] [Google Scholar]
- 33.Brand W, Boersma MG, Bik H, Hoek-van den Hil EF, Vervoort J, Barron D, Meinl W, Glatt H, Williamson G, van Bladeren PJ, Rietjens IM. Phase II metabolism of hesperetin by individual UDP-glucuronosyltransferases and sulfotransferases and rat and human tissue samples. Drug Metab Dispos. 2010;38(4):617–625. doi: 10.1124/dmd.109.031047. [DOI] [PubMed] [Google Scholar]
- 34.Ruefer CE, Gerhäuser C, Frank N, Becker H, Kulling SE. In vitro phase II metabolism of xanthohumol by human UDP-glucuronosyltransferases and sulfotransferases. Mol Nutr Food Res. 2005;49(9):851–856. doi: 10.1002/mnfr.200500057. [DOI] [PubMed] [Google Scholar]
- 35.Vaidyanathan JB, Walle T. Glucuronidation and sulfation of the tea flavonoid (−)-epicatechin by the human and rat enzymes. Drug Metab Dispos. 2002;30(8):897–903. doi: 10.1124/dmd.30.8.897. [DOI] [PubMed] [Google Scholar]
- 36.Miksits M, Maier-Salamon A, Aust S, Thalhammer T, Reznicek G, Kunert O, Haslinger E, Szekeres T, Jaeger W. Sulfation of resveratrol in human liver: evidence of a major role for the sulfotransferases SULT1A1 and SULT1E1. Xenobiotica. 2005;35(12):1101–1119. doi: 10.1080/00498250500354253. [DOI] [PubMed] [Google Scholar]
- 37.Itäaho K, Alakurtti S, Yli-Kauhaluoma J, Taskinen J, Coughtrie MW, Kostiainen R. Regioselective sulfonation of dopamine by SULT1A3 in vitro provides a molecular explanation for the preponderance of dopamine-3-O-sulfate in human blood circulation. Biochem Pharmacol. 2007;74(3):504–510. doi: 10.1016/j.bcp.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Pai TG, Oxendine I, Sugahara T, Suiko M, Sakakibara Y, Liu MC. Structure-function relationships in the stereospecific and manganese-dependent 3,4-dihydroxyphenylalanine/tyrosine-sulfating activity of human monoamine-form phenol sulfotransferase, SULT1A3. J Biol Chem. 2003;278(3):1525–1532. doi: 10.1074/jbc.M203108200. [DOI] [PubMed] [Google Scholar]
- 39.Gamage NU, Duggleby RG, Barnett AC, Tresillian M, Latham CF, Liyou NE, McManus ME, Martin JL. Structure of a human carcinogen-converting enzyme, SULT1A1. Structural and kinetic implications of substrate inhibition. J Biol Chem. 2003;278(9):7655–7662. doi: 10.1074/jbc.M207246200. [DOI] [PubMed] [Google Scholar]
- 40.Pedersen LC, Petrotchenko E, Shevtsov S, Negishi M. Crystal structure of the human estrogen sulfotransferase-PAPS complex: evidence for catalytic role of Ser137 in the sulfuryl transfer reaction. J Biol Chem. 2002;277(20):17928–17932. doi: 10.1074/jbc.M111651200. [DOI] [PubMed] [Google Scholar]
- 41.Rehse PH, Zhou M, Lin SX. Crystal structure of human dehydroepiandrosterone sulphotransferase in complex with substrate. Biochem J. 2002;364(Pt 1):165–171. doi: 10.1042/bj3640165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu J, Li H, Zhang J, Li M, Liu MY, An X, Liu MC, Chang W. Crystal structures of SULT1A2 and SULT1A1 *3: insights into the substrate inhibition and the role of Tyr149 in SULT1A2. Biochem Biophys Res Commun. 2010;396(2):429–434. doi: 10.1016/j.bbrc.2010.04.109. [DOI] [PubMed] [Google Scholar]
- 43.Basu NK, Kole L, Basu M, McDonagh AF, Owens IS. Targeted inhibition of glucuronidation markedly improves drug efficacy in mice - a model. Biochem Biophys Res Commun. 2007;360(1):7–13. doi: 10.1016/j.bbrc.2007.05.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mazur A, Lichti CF, Prather PL, Zielinska AK, Bratton SM, Gallus-Zawada A, Finel M, Miller GP, Radomińska-Pandya A, Moran JH. Characterization of human hepatic and extrahepatic UDP-glucuronosyltransferase enzymes involved in the metabolism of classic cannabinoids. Drug Metab Dispos. 2009;37(7):1496–1504. doi: 10.1124/dmd.109.026898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X, Shang L, Wu Y, Abbas S, Li D, Netter P, Mohamed O, Wang H, Magdalou J. Identification of the human UDP-glucuronosyltransferase isoforms involved in the glucuronidation of the phytochemical ferulic acid. Drug MetabPharmacokinet. 2011 doi: 10.2133/dmpk.dmpk-10-rg-125. (in press) [DOI] [PubMed] [Google Scholar]
- 46.Hanioka N, Ozawa S, Jinno H, Ando M, Saito Y, Sawada J. Human liver UDP-glucuronosyltransferase isoforms involved in the glucuronidation of 7-ethyl-10-hydroxycamptothecin. Xenobiotica. 2001;31(10):687–699. doi: 10.1080/00498250110057341. [DOI] [PubMed] [Google Scholar]
- 47.Ghosal A, Hapangama N, Yuan Y, Achanfuo-Yeboah J, Iannucci R, Chowdhury S, Alton K, Patrick JE, Zbaida S. Identification of human UDP-glucuronosyltransferase enzyme(s) responsible for the glucuronidation of ezetimibe (Zetia) Drug Metab Dispos. 2004;32(3):314–320. doi: 10.1124/dmd.32.3.314. [DOI] [PubMed] [Google Scholar]
- 48.Soars MG, Ring BJ, Wrighton SA. The effect of incubation conditions on the enzyme kinetics of udp-glucuronosyltransferases. Drug Metab Dispos. 2003;31(6):762–767. doi: 10.1124/dmd.31.6.762. [DOI] [PubMed] [Google Scholar]
- 49.Singh R, Wu B, Tang L, Liu Z, Hu M. Identification of the position of mono-O-glucuronide of flavones and flavonols by analyzing shift in online UV spectrum (lambdamax) generated from an online diode array detector. J Agric Food Chem. 2010;58(17):9384–9395. doi: 10.1021/jf904561e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davis BD, Brodbelt JS. Regioselectivity of human UDP-glucuronosyl-transferase 1A1 in the synthesis of flavonoid glucuronides determined by metal complexation and tandem mass spectrometry. J Am Soc Mass Spectrom. 2008;19(2):246–256. doi: 10.1016/j.jasms.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh R, Wu B, Tang L, Hu M. Uridine Diposphate Glucuronosyltransferases Isoform-Dependent Regiospecificity Of Glucuronidation Of Flavonoids. J Agric Food Chem. 2011 doi: 10.1021/jf1041454. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu B, Xu B, Hu M. Regioselective Glucuronidation of Flavonols by Six Human UGT1A Isoforms. Pharm Res. 2011;28(8):1905–1918. doi: 10.1007/s11095-011-0418-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tang L, Ye L, Singh R, Wu B, Lv C, Zhao J, Liu Z, Hu M. Use of glucuronidation fingerprinting to describe and predict mono- and dihydroxyflavone metabolism by recombinant UGT isoforms and human intestinal and liver microsomes. Mol Pharm. 2010;7(3):664–679. doi: 10.1021/mp900223c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou Q, Zheng Z, Xia B, Tang L, Lv C, Liu W, Liu Z, Hu M. Use of isoform-specific UGT metabolism to determine and describe rates and profiles of glucuronidation of wogonin and oroxylin A by human liver and intestinal microsomes. Pharm Res. 2010;27(8):1568–1583. doi: 10.1007/s11095-010-0148-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boersma MG, van der Woude H, Bogaards J, Boeren S, Vervoort J, Cnubben NH, van Iersel ML, van Bladeren PJ, Rietjens IM. Regioselectivity of phase II metabolism of luteolin and quercetin by UDP-glucuronosyl transferases. Chem Res Toxicol. 2002;15(5):662–670. doi: 10.1021/tx0101705. [DOI] [PubMed] [Google Scholar]
- 56.Chen YK, Chen SQ, Li X, Zeng S. Quantitative regioselectivity of glucuronidation of quercetin by recombinant UDP-glucuronosyltransferases 1A9 and 1A3 using enzymatic kinetic parameters. Xenobiotica. 2005;35(10–11):943–954. doi: 10.1080/00498250500372172. [DOI] [PubMed] [Google Scholar]
- 57.Zhang L, Lin G, Zuo Z. Involvement of UDP-glucuronosyltransferases in the extensive liver and intestinal first-pass metabolism of flavonoid baicalein. Pharm Res. 2007;24(1):81–89. doi: 10.1007/s11095-006-9126-y. [DOI] [PubMed] [Google Scholar]
- 58.Joseph TB, Wang SW, Liu X, Kulkarni KH, Wang J, Xu H, Hu M. Disposition of flavonoids via enteric recycling: enzyme stability affects characterization of prunetin glucuronidation across species, organs, and UGT isoforms. Mol Pharm. 2007;4(6):883–894. doi: 10.1021/mp700135a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang L, Singh R, Liu Z, Hu M. Structure and concentration changes affect characterization of UGT isoform-specific metabolism of isoflavones. Mol Pharm. 2009;6(5):1466–1482. doi: 10.1021/mp8002557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu H, Meng X, Li C, Sang S, Patten C, Sheng S, Hong J, Bai N, Winnik B, Ho CT, Yang CS. Glucuronides of tea catechins: enzymology of biosynthesis and biological activities. Drug Metab Dispos. 2003;31(4):452–461. doi: 10.1124/dmd.31.4.452. [DOI] [PubMed] [Google Scholar]
- 61.Liu W, Tang L, Ye L, Cai Z, Xia B, Zhang J, Hu M, Liu Z. Species and gender differences affect the metabolism of emodin via glucuronidation. AAPS J. 2010;12(3):424–436. doi: 10.1208/s12248-010-9200-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liang SC, Ge GB, Liu HX, Zhang YY, Wang LM, Zhang JW, Yin L, Li W, Fang ZZ, Wu JJ, Li GH, Yang L. Identification and characterization of human UDP-glucuronosyltransferases responsible for the in vitro glucuronidation of daphnetin. Drug Metab Dispos. 2010;38(6):973–980. doi: 10.1124/dmd.109.030734. [DOI] [PubMed] [Google Scholar]
- 63.Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 64.Radominska-Pandya A, Bratton SM, Redinbo MR, Miley MJ. The crystal structure of human UDP-glucuronosyltransferase 2B7 C-terminal end is the first mammalian UGT target to be revealed: the significance for human UGTs from both the 1A and 2B families. Drug Metab Rev. 2010;42(1):133–144. doi: 10.3109/03602530903209049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luukkanen L, Taskinen J, Kurkela M, Kostiainen R, Hirvonen J, Finel M. Kinetic characterization of the 1A subfamily of recombinant human UDP-glucuronosyltransferases. Drug Metab Dispos. 2005;33(7):1017–1026. doi: 10.1124/dmd.105.004093. [DOI] [PubMed] [Google Scholar]
- 66.Zhang L, Lin G, Zuo Z. Position preference on glucuronidation of mono-hydroxylflavones in human intestine. Life Sci. 2006;78(24):2772–2780. doi: 10.1016/j.lfs.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 67.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 68.Dombrovski L, Dong A, Bochkarev A, Plotnikov AN. Crystal structures of human sulfotransferases SULT1B1 and SULT1C1 complexed with the cofactor product adenosine-3'-5'-diphosphate (PAP) Proteins. 2006;64(4):1091–1094. doi: 10.1002/prot.21048. [DOI] [PubMed] [Google Scholar]
- 69.Lee KA, Fuda H, Lee YC, Negishi M, Strott CA, Pedersen LC. Crystal structure of human cholesterol sulfotransferase (SULT2B1b) in the presence of pregnenolone and 3'-phosphoadenosine 5'-phosphate. Rationale for specificity differences between prototypical SULT2A1 and the SULT2BG1 isoforms. J Biol Chem. 2003;278(45):44593–44599. doi: 10.1074/jbc.M308312200. [DOI] [PubMed] [Google Scholar]