

Abstract
Stroke is a devastating brain injury that is a leading cause of adult disability with limited treatment options. Using a rat model of middle cerebral artery occlusion (MCAO) to induce cerebral ischemia, we profiled microRNAs (miRNAs), small non-protein coding RNAs, in the ischemic cortex. Many miRNAs were confirmed by qPCR to be robustly upregulated 24 hours following MCAO surgery including miR-155, miR-297a, miR-466f, miR-466h, and miR-1224. In addition, we treated MCAO rats with valproic acid (VPA), a mood stabilizer and histone deacetylase inhibitor. This post-insult treatment was shown to improve neurological deficits and motor performance following MCAO. To provide mechanistic insight into the potential targets and pathways that may underlie these benefits, we profiled miRNAs regulated following this VPA treatment. Two promising post-insult VPA-regulated candidates were miR-331 and miR-885-3p. miR-331 was also regulated by VPA pre-treatment in rat cortical neuronal cultures subjected to oxygen-glucose deprivation, an in vitro ischemic model. The predicted targets of these miRNAs analyzed by Ingenuity Pathway Analysis (IPA) identified networks involved in hematological system development, cell death, and nervous system development. These predicted networks were further filtered using IPA and showed significant associations with neurological diseases including movement disorders, neurodegenerative disorders, damage to cerebral cortex, and seizure disorders among others. Collectively, these data support common disease mechanisms that may be under miRNA control and provide exciting directions for further investigations aimed at elucidating the miRNA mechanisms and targets that may yield new therapies for neurological disorders.
Keywords: Cerebral ischemia, valproic acid, microRNA, oxygen-glucose deprivation, neuroprotection
Introduction
Stroke is a severe and rapid loss of brain functions caused by either reduced blood supply (ischemia) or leakage of blood (hemorrhage). It is estimated that 795,000 strokes occur annually in the United States alone: this equates to one attack every 40 seconds. Ischemic stroke is the most common subtype and constitutes 87% of these events [1]. To date, intravenous administration of tissue plasminogen activator (t-PA), a serine protease that acts by breaking down blood clots, is the only Food and Drug Administration (FDA)-approved therapy for acute ischemic stroke. t-PA must be administered within 4.5 hours post-injury for its potential beneficial effects to warrant the accompanying risk of hemorrhage [2,3]. As a result of these limitations, extensive research efforts have been made to identify alternative treatment options for stroke.
One promising avenue is the use of histone deacetylase (HDAC) inhibitors: by increasing the acetylation of histone and non-histone proteins, HDAC inhibitors can reverse post-ischemia hypoacetylation, activate transcription, and enhance protective gene expression after experimental stroke [4,5]. For example, in a rat cerebral ischemia model, post-injury injections with HDAC inhibitors such as valproic acid (VPA), also known to be a mood stabilizer and anticonvulsant, significantly decreased brain infarct volume and improved neurological performance [6]. Proposed mechanisms for the beneficial effects of HDAC inhibition include neuroprotection, anti-inflammation, blood-brain barrier preservation, pro-angiogenesis, and pro-neurogenesis [7]. Clearly, HDAC inhibitors have promise as potential therapies to treat stroke. However, their mechanism(s) of action are highly complex and remain incompletely defined.
A novel direction for stroke research that is actively being pursued is the modulation of small non-coding RNAs called microRNAs (miRNAs). By binding to the 3’ UTR of complementary mRNAs, miRNAs can suppress gene expression and thereby regulate up to 75% of the human genome [8]. miRNAs are gaining ground as significant contributors to diseases ranging from cancer to neurological disorders [9,10]. miRNA changes have been observed in both brain and blood following experimental ischemic stroke in rats [11], and accumulating evidence implicates this class of molecules in regulating processes including hypoxic response, angiogenesis, and neurodegeneration [12,13]. Interestingly, HDAC inhibition has been shown to rapidly alter miRNA profiles in cancer [14], and chronic treatment with the HDAC inhibitor VPA modulated brain miRNA expression in rats [15]. In the present study, we therefore sought to profile miRNAs regulated in a rat cerebral ischemia model following protective VPA treatment to identify potential novel therapeutic miRNAs and their predicted targets.
Materials and methods
Middle cerebral artery occlusion (MCAO) and drug administration
All animal experiments were performed according to protocols approved by the National Institutes of Mental Health Animal Care and Use Committee. Male Sprague-Dawley rats (200-220 g, Charles River Laboratories, Wilmington, MA) were subjected to focal cerebral ischemia via right MCAO for 60 minutes as previously described [16]. Briefly, animals were placed under inhalational anesthesia (1.5% isoflurane in 70% N2O and 30% O2) and a 4-0 nylon suture with flame-rounded tip was advanced through the right common carotid artery and right internal carotid artery to occlude the origin of the right middle cerebral artery. After 60 minutes, the suture was withdrawn to initiate reperfusion. A rectal thermometer and heating blanket were used during surgery to maintain core body temperature at 37.0 ± 0.5 °C. Sham-operated rats underwent neck surgery without arterial occlusion. VPA (300 mg/kg, i.p., Sigma, St. Louis, MO) was administrated immediately after ischemic onset followed by another injection 12 hours later. One cohort of sham, MCAO, and MCAO+VPA rats was used for rotarod testing; the VPA-treated animals within this group were given additional injections once daily until sacrificed at day 3 after MCAO. A second cohort of animals was used for neurological scoring, microarray analysis, and qPCR confirmation and sacrificed at 24 hours. For MCAO+VPA animals, VPA plasma levels were quantified by MedTox Laboratories (St. Paul, MN). Animals that failed to show neurological deficits, developed hemorrhage, or had low plasma VPA levels (VPA-treated group) were excluded from further studies.
Accelerating rotarod test
An accelerating rotarod apparatus (San Diego Instruments, San Diego, CA) was used to measure motor skill learning and coordination in a subset of sham (N=6), MCAO (N=8), and MCAO+VPA (N=7) rats [17]. Three consecutive days before MCAO, rats received training sessions of three trials per day with a 30-minute interval. The longest time each rat remained on the rod as it was accelerated from 0 to 40 rpm within four minutes was recorded as baseline. One, two, and three days after MCAO, rats underwent three trials on the rotarod, and the best performance of each rat was recorded.
Neurological severity scoring
Immediately before sacrifice at 24 hours, rats were assessed for motor, sensory, and reflex performance using a modified 12-point neurological scoring system as described previously [17]. Seven tests of motor performance (flexion of forelimb or hind limb, head movement 10º to the vertical axis, inability to walk straight, circling towards the paralytic side, falling to the paralytic side, and immobility), two tests of sensation (visual and tactile placement and a proprioceptive test), and three reflex tests (pinna, corneal, and startle reflex) were evaluated. A score of 0 (normal) or 1 (abnormal) was given for each test by two investigators blinded to the treatment condition and then averaged for each animal (Sham N=4, MCAO N=4, MCAO+VPA N=7).
RNA isolation
Dissected brain tissue from the ipsilateral cortex of each animal was immediately placed in ice-cold lysis buffer (mirVana miRNA Isolation Kit, Ambion, Austin, TX), homogenized, and frozen at -80°C. The mirVana miRNA Isolation Kit was used for total RNA isolation per manufacturer’s instructions. RNA concentration and purity were determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Tech, Rockland, DE), with a 260/280 value >1.8 considered acceptable. RNA samples were further assessed for quality using a Bioanalyzer (Agilent Technologies, Foster City, CA) to ensure an RNA integrity number >7, and then stored at -80°C for further analysis.
miRNA microarray hybridization and analysis
Total RNA (1 μg) was labeled using the Flashtag Biotin RNA labeling kit (Genisphere, Hatfield, PA) as per manufacturer’s instructions. Biotin-labeled total RNA was then profiled using Affymetrix GeneChip miRNA arrays (Santa Clara, CA) according to the manufacturer’s protocol. Briefly, 5 sham samples, 6 MCAO samples, and 8 MCAO+VPA samples were hybridized on the same day under the same conditions. Hybridized arrays were then washed, stained, and scanned as instructed. RNA Spike Control Oligos were used in conjunction with miRNA quality control software to assess labeling efficiencies, and outliers from each group were identified and excluded using principle component analysis (PCA). Array data from 4 sham, 4 MCAO and 7 MCAO+VPA were analyzed using Partek Genomics Suite software (Chesterfield, MO).
Quantitative real-time PCR (qPCR)
Array data were confirmed by qPCR using the NCode miRNA First-Strand cDNA synthesis and qPCR kits by Invitrogen (Calsbad, CA) as per manufacturer’s instructions. Briefly, 1 μg total RNA was first polyadenylated and then reverse transcribed to generate first-strand cDNA. cDNA was diluted in DEPC-treated water and prepared for qPCR with Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen), miRNA-specific forward primers, a universal qPCR primer, and ROX reference dye. All reactions were run in triplicate using a 7500 Real Time PCR System (Applied Biosystems, Foster City, CA). Relative miRNA levels were normalized to U6 snRNA, a verified endogenous control. See Table 1 for primer sequences.
Table 1.
Primers used for qPCR confirmation of array results of miRNAs listed below.
| Target | Primer Sequence |
|---|---|
| rno/mmu-miR-1224 | 5’-GTGAGGACTGGGGAGGTGGAG-3’ |
| mmu-miR-155 | 5’-TTAATGCTAATTGTGATAGGGGTA-3’ |
| mmu-miR-297a | 5’-ATGTATGTGTGCATGTGCATGT-3’ |
| rno-miR-331 | 5’-GCCCCTGGGCCTATCCTAGAA-3’ |
| mmu-miR-466f | 5’-TACGTGTGTGTGCATGTGCATG-3’ |
| mmu-miR-466h | 5’-TGTGTGCATGTGCTTGTGTGTA-3’ |
| hsa-miR-885-3p | 5’-AGGCAGCGGGGTGTAGTGGATA-3’ |
| U6 snRNA | 5’- CTCGCTTCGGCAGCACA-3’ (Forward) |
| 5’- AACGCTTCACGAATTTGCGT-3’ (Reverse) |
Primary cortical neuronal culture
Cortical neurons were prepared from 17-day-old Sprague-Dawley rat embryos as described previously with several modifications [18]. Briefly, cortices were dissected and cells were dissociated by trypsinization followed by treatment with horse serum and DNase. After repeated triturations, dissociated cells were resuspended in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 2.0 mM L-glutamine, and plated on poly-D-lysine coated cell culture plates at a density of 7.0 x 105 cells/ml. After 24 hours, media was changed to serum-free B27/neurobasal medium (Gibco, Carlsbad, CA) supplemented with 0.5 mM glutaMAX (Gibco), 1x antibiotic-antimycotic (Gibco), and 5 μM cytosine arabinofuranoside was added to arrest the growth of non-neuronal cells. A half medium change was completed every 4-5 days. Cultures were treated with 1 mM VPA at 9 or 10 days in vitro and subjected to oxygen-glucose deprivation (OGD) four days later.
OGD studies
Cortical neuronal cultures in 24- or 6-well plates were switched to glucose-free neurobasal medium supplemented with 0.5 mM GlutaMAX and deoxygenated by bubbling with 95% N2/5% CO2 for 30 minutes. Plates were then placed in a Modular Incubator Chamber (Billups-Rothenberg, Del Mar, CA) filled with a gas mixture of 95%N2/5% CO2 at 37°C for 3 hours. Control cells were maintained in neurobasal medium containing 25 mM glucose and 0.5 mM GlutaMAX for the same time interval under normoxic conditions. After 3 hours, all culture medium was replaced with a 1:1 mixture of fresh and conditioned glucose-containing B27/neurobasal medium. Cells were returned to normoxic conditions for 24 hours before cell viability analysis or harvesting for RNA isolation using the mirVana miRNA Isolation Kit. For VPA-treated cells, the drug was re-supplemented both at the beginning of OGD and immediately after reperfusion at a final concentration of 1 mM.
Measurement of cell viability
Cell viability was evaluated by the mitochondrial dehydrogenase activity to reduce 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), as previously described [19]. Cortical neurons in 24-well culture plates were incubated with 125 μg/ml MTT for 2 hours at 37°C. The medium was then aspirated, and the formazan product was dissolved in dimethylsulfoxide and quantified spectrophotometrically at 540 nm. Results are expressed as a percentage of viability of the control cultures.
Pathway analysis
Predicted mRNA targets of select miRNAs were identified using the miRWalk database (http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/index.html) [20]. Targets were generated from four independent sources: miRanda, miRDB, miRWalk, and Targetscan, and only mRNAs predicted concurrently by 3 or more of these databases (miRNA binding sites restricted to 3’UTR) were considered for further analysis. Final target lists were then uploaded to Ingenuity Pathway Analysis (IPA) software for functional interpretation using filters for relationships specific to central nervous system (CNS) tissues and cell lines with Fisher’s Exact Test and a p-value threshold set at 0.05.
Statistical analyses
Data are expressed as mean ± SEM. For rotarod data, two-way repeated measures ANOVA was performed to analyze the overall difference between treatment groups over time, and then Bonferroni corrected post-hoc comparisons were used to analyze the difference between treatment groups at each time point. Student's t-test and one-way ANOVA followed by Tukey’s post-hoc test were used to evaluate the difference between two groups and multiple groups, respectively. P<0.05 was considered statistically significant.
Results
VPA protects against cerebral ischemia
In order to determine the benefits of post-insult VPA treatment following cerebral ischemia in rats, we first evaluated the neurological severity score at 24 hours after MCAO. Compared to sham animals, MCAO surgery alone produced a significant impairment in neurological function (MCAO 6.13±0.31 vs. sham 0.25±0.25), and post-insult VPA treatment (MCAO+VPA 3.00±0.24) partially restored this deficit (Figure 1A). We also administered an accelerating rotarod test to assess motor skill learning and coordination up to three days after either sham or MCAO surgery. There was no significant difference between groups at baseline (sham: 131.2±13.4sec; MCAO: 111.0±9.1sec; MCAO+VPA 118.0±6.5sec) however following MCAO surgery there was a 70% reduction in time retained on the rotarod (39.5±5.1sec) when compared against sham (130.3±9.9sec) at 24 hours. This deficit persisted over days 2 (40.9±7.4sec) and 3 (47.5±8.7sec) as well. The post-insult VPA group demonstrated a significant improvement in coordination as measured by increased time spent on the rotarod compared against the MCAO group on days 2 (77.0±5.2sec) and 3 (93.7±13.6sec) (Figure 1B). We performed miRNA profiling on ipsilateral cortex from each group at 24 hours after MCAO to further assess these therapeutic benefits of post-insult VPA treatment. Principal component analysis (PCA) on these miRNA array results demonstrated a clear separation between sham and MCAO groups with a less extreme, but nonetheless distinct, separation between MCAO and MCAO+VPA groups (Figure 1C).
Figure 1.
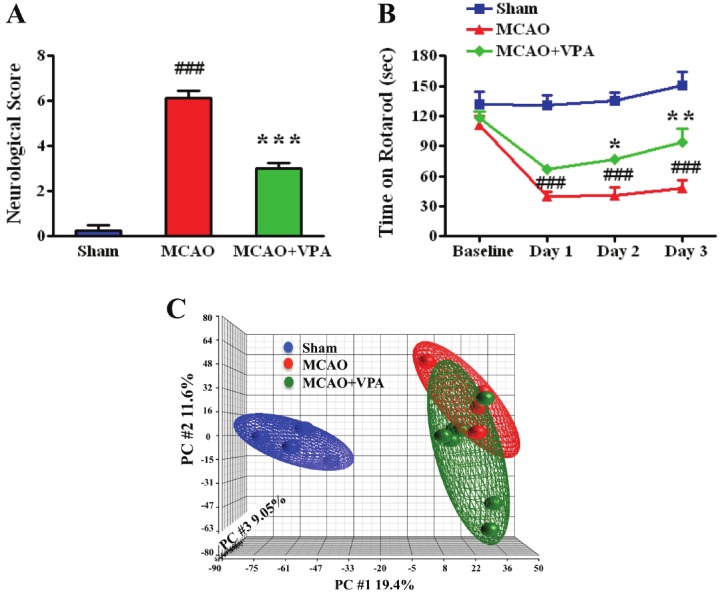
Post-insult VPA treatment protects against cerebral ischemia and alters miRNA expression. (A, B) Post-insult VPA (300 mg/kg) treatment significantly improved neurological function at 24 hours and rotarod performance at days 2 and 3 after ischemic insult. ### p<0.001 vs sham; *p<0.05, **p<0.01, ***p<0.001 vs MCAO. (C) Principle Component Analysis (PCA) after miRNA profiling comparing MCAO (n=4), sham (n=4), and MCAO+VPA (n=7).
MCAO-regulated miRNAs and their targets
Focusing initially on MCAO vs. sham-regulated miRNAs, there were 101 (FDR p<0.05, ±2 fold) differentially regulated miRNAs (Supplemental Table 1; human, mouse, and rat miRNAs considered). A heat map of these miRNAs along with a list of selected top MCAO-regulated miRNAs is provided in Figure 2. Bolded miRNAs are ones that were confirmed by qPCR (Figure 3) including miR-155, miR-297a, miR-466f, miR-466h, and miR-1224. Predicted targets of these confirmed miRNAs were obtained from miRWalk and are shown in Figure 4 as a Venn diagram where the numbers of overlapping targets are depic t ed accor d ing l y . N in e t y - f i ve (22+18+6+15+34) common targets of at least 3 miRNAs were then used for Ingenuity Pathway Analysis (IPA). IPA results revealed top canonical pathways, networks, and functions of these 95 common targets (Table 2).
Figure 2.

Differentially expressed miRNAs between MCAO vs. sham. (A) Heat map analysis showed upregulated miRNAs in red and downregulated in blue (FDR p<0.05, ±2 fold). (B) Accompanying list identifies selected top regulated miRNAs and their fold regulation (MCAO vs sham) where bolded miRNAs have been confirmed by qPCR.
Figure 3.

qPCR confirmation of five MCAO-upregulated miRNAs identified byarray (n=4; *p<0.05, **p<0.01). The relative quantity of each miRNA is shownfor the MCAO group compared with sham.
Figure 4.
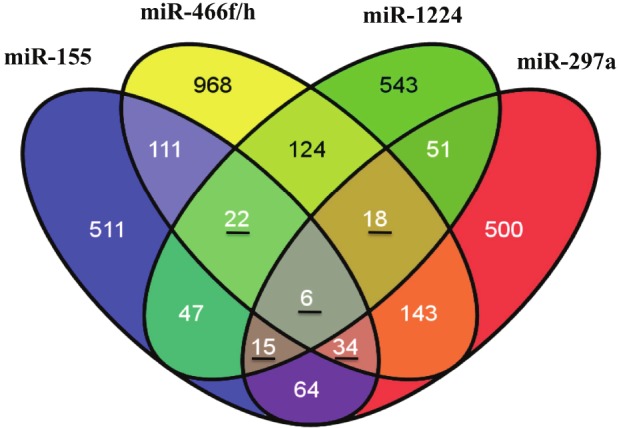
Top predicted targets of MCAO-regulatedmiRNAs. Predicted mRNA targets of qPCR confirmedmiRNAs were generated using four independent predictionalgorithms. Only mRNAs predicted concurrentlyby 3 or more databases were selected for comparison.The numbers of mRNAs targeted by eachmiRNA are listed in the appropriate section. Thosenumbers underlined are common mRNAs targeted by3 or more miRNAs that were further analyzed by IngenuityPathway Analysis.
Table 2.
Pathway analysis of common MCAO-regulated miRNA targets. Common targets identified in Figure 4 were further analyzed by Ingenuity Pathway Analysis (IPA). Top networks, canonical pathways, and functions along with the predicted MCAO-regulated miRNA targets are listed below.
| Top Networks | Predicted Targets in Network |
|---|---|
| Cell Death, Cellular Development, Cellular Growth and Proliferation | AIFM1, APP, BAD, BCAR1, CCL2, CCL3, ceramide, CHAT, CXCR4, DCX, EIF5A2, EN1, FOXO1, FUS, GNAO1, IGF2, KRAS, LRP1, MAFB, NCAM1, NF1, NGFR, NME1, PLAT, PLD1, PRKACA, PURA, RARA, RPS6, RPS19, SMAD3, ST8SIA1, TPPP, TUBA3C/TUBA3D, YWHAZ |
| Molecular Transport, Behavior, Cell-To-Cell Signaling and Interaction | ACAT1, APP, ATN1, CA4, CALML4, Cd24a, CDK2AP1, CLOCK, CPLX2, DLG2, DLG4, ETV5, HMP19, HTT, IL6, IL6R, JUP, KCNA1, KCNAB1, KCNC3, MOBP, NEUROD2, PITPNM1, POU4F1, PRKACB, PTBP1, RUNX1T1, SLC12A2, SOX11, SP1, ST8SIA5, TAGLN3, TRH, TYRO3, ZIC3 |
|
| |
| Top Canonical Pathways | Predicted Targets in Pathway |
|
| |
| Prolactin Signaling | KRAS, SOCS7, SOS1, SP1 |
| Regulation of IL-2 Expression in Activated and Anergic T Lymphocytes | KRAS, NFAT5, SMAD3, SOS1 |
| TGF-β Signaling | KRAS, SMAD3, SMAD5, SOS1 |
| Mouse Embryonic Stem Cell Pluripotency | FZD5 ,KRAS, SMAD5, SOS1 |
| PPARα/RXRα Activation | CLOCK, KRAS, PRKAB2, SMAD3, SOS1 |
|
| |
| Nervous System Development and Functions | Predicted Targets implicated in Nervous System Development and Functions |
|
| |
| pathfinding of axons | NCAM1,SEMA5A |
| extension of neurites | DCX,NCAM1,PLD1,SP1 |
| arrest in migration of facial branchiomotor neurons | MAFB |
| branching of interneurons | DCX |
| delay in withdrawal of neurons | NCAM1 |
| depolarization of CA1 neuron | SLC12A2 |
| formation of basolateral amygdaloid nucleus | NEUROD2 |
| growth of retinal axons | SEMA5A |
| pathfinding of retinal axons | SEMA5A |
| quantity of postsynaptic density | NCAM1 |
| cytostasis of astrocytes | SMAD3 |
| entry into S phase of cortical neurons | ST8SIA1 |
| neuroprotection of striatal interneurons | IL6R |
| size of motor endplates | NCAM1 |
| proliferation of astrocytes | KRAS,NCAM1 |
| development of brain cells | NCAM1,NEUROD2 |
| dilation of cerebral ventricles | SOCS7 |
| excitation of CA3 neurons | SLC12A2 |
| miniature excitatory postsynaptic currents | CPLX2 |
| neuroprotection | IL6R,SP1 |
| abnormal morphology of hippocampus | CPLX2 |
| excitatory postsynaptic potential of cerebral cortex cells | CPLX2 |
| sprouting of mossy fibers | PLD1 |
| neuritogenesis of hippocampal neurons | NCAM1 |
| regeneration of motor neurons | IGF2 |
| thickness of cerebral cortex | SOCS7 |
| development of granule cells | NEUROD2 |
| survival of corticospinal neurons | IGF2 |
| cell viability of Schwann cells | IGF2 |
| excitatory postsynaptic potential of neurons | CPLX2 |
| long-term potentiation of mossy fibers | CPLX2 |
| maturation of synapse | SLC12A2 |
Profiling differentially expressed miRNAs between MCAO and MCAO+VPA
Identifying differentially expressed miRNAs between MCAO and MCAO+VPA groups highlights miRNAs that may contribute to beneficial effects of post-insult VPA treatment (Figures 1A and 1B). Analyzing differentially expressed miRNAs between MCAO and MCAO+VPA (unadjusted p<0.05, ±1.2 fold) identified 131 miRNAs (Supplemental Table 2; human, mouse, and rat miRNA considered). These miRNAs are represented in a heat map (Figure 5) where a list of the top regulated miRNAs is also provided. Hierarchical clustering of miRNAs differentially regulated comparing MCAO vs. MCAO+VPA segregated the three groups: MCAO, sham, and MCAO+VPA. We focused on two miRNA candidates, miR-331 and miR-885-3p, based on further analysis of their predicted targets (e.g. cytokines: CX3CL1, CXCL12; transporters: AQP3, SLC23A2, SLC25A10, TAP2; enzymes: DGAT1, GNAZ, GPD1, PADI3; and transcriptional regulators: GAS7, LZTS1, MTPN) and confirmed regulation by post-insult VPA treatment via qPCR (Figure 6A and 6B).
Figure 5.

Differentially expressed miRNAs between MCAO+VPA vs. MCAO. (A) Heat map analysis showed upregulated miRNAs in red and downregulated in blue. (B) Accompanying list identifies selected top MCAO+VPA regulated miRNAs compared with MCAO and their fold regulation (unadjusted p<0.05, ±1.2 fold).
Figure 6.
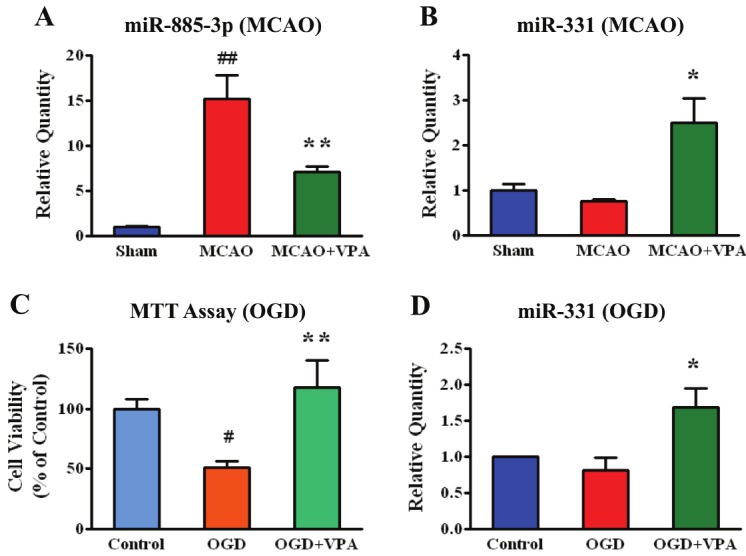
VPA regulated miRNAs after MCAO and oxygen-glucose deprivation(OGD). (A, B) qPCR confirmation of miRNAs regulated by post-MCAOtreatment with VPA (Sham, MCAO n=4, MCAO+VPA n=7; ##p<0.01 vsSham; *p<0.05, ** p<0.01 vs MCAO). (C) Cell viability measured by MTTassay following OGD with or without 1mM VPA pre-treatment for 4 days(n=14; #p<0.05 vs control, ** p<0.01 vs OGD). (D) Levels of miR-331measured by qPCR following OGD or OGD+VPA (n=5; *p<0.05).
VPA-regulated hsa-miR-885-3p and rno-miR-331
Candidates miR-885-3p and miR-331 were initially confirmed by qPCR to be regulated by post-insult VPA in the rat MCAO in vivo model (Figure 6A and 6B). Candidate miR-885-3p was upregulated following MCAO surgery alone where post-insult VPA treatment attenuated this upregulated response (Figure 6A). Candidate miR-331 was only upregulated by post-insult VPA treatment (Figure 6B). Using an OGD model of rat cortical neurons where cell viability was reduced following OGD and increased by VPA pretreatment (Figure 6C), miR-331 was also upregulated in this neuroprotective VPA treatment condition. We also tested miR-885-3p in this model, but did not see any significant changes in expression by either OGD or OGD+VPA (data not shown).
To suggest potential miRNA mediated mechanisms that may underlie the therapeutic benefits of post-insult VPA treatment, we considered targets of miR-331 and miR-885-3p using miRWalk. We analyzed targets of these miRNA candidates using IPA analysis and list the top networks, neurological diseases, and functions along with the associated miRNA targets (Tables 3 and 4). These networks, neurological diseases and functions will be considered in greater detail in the following discussion.
Table 3.
Pathway analysis of post-insult VPA-regulated miR-331 targets. Predicted mRNA targets of miR-331 were generated using four independent prediction algorithms. Only mRNAs predicted concurrently by 3 or more databases were selected and further analyzed by Ingenuity Pathway Analysis to identify top canonical networks. These top networks were then filtered for disease and functions where both direct (BOLDED) and indirect miRNA targets associated with each network, disease or function are included.
| Top Networks: rno-miR-331 | Predicted Targets in Network |
|---|---|
| Cellular Movement, Hematological System Development and Function, Immune Cell Trafficking | APP, ATP6V1A, Ca2+, CCL20, CDKN2B, COL1A1, CX3CL1, DGAT1, ERBB2, FGF2, FLOT2, FUS, FYN, GLI1, GRIA1, HMOX1, HNRNPU, IMPA2, ITGA6, LZTS1, NOG, NPTX1, NUCB2, POLR2C, PRDX2, pregnenolone, PTPN5, RARA, S100A6, SERPINE1, SMAD1, ST3GAL6, TGFA, TGFB1, TUBA3C/TUBA3D |
| Cell Death, Hereditary Disorder, Neurological Disease | ADRBK1, ARF3, ATP5B, BBC3, CIT, DLG4, DRD2, FDFT1, FURIN, FXYD6, Gapdh, GAS7, GCLC, GLUL, GRIK2, GRIN2D, GRK5, HAP1, HTT, KCNAB1, KCNJ4, KCNJ12, LIN7A, NCS1, NFE2L2, NGF, NSF, PKM2, SDHA, SERPINF1, SGK1, SLC7A8, STX1B, TAF4, VSNL1 |
| Cellular Assembly and Organization, Nervous System Development and Function, Cellular Movement | BDNF, CAMK4, CDH13, CSPG4, CXCL12, CXCR4, CYGB, DUSP1, EFNB1, EIF2AK2, EIF2S1, EIF4EBP1, ELK1, EPHB2, ethanol, GABA, GNAO1, GRIP1, GTP, KSR1, MAPK1, MAPK14, MCF2L, MTOR, NTRK3, PDE4A, PRKCZ, RAB3A, SKIL, SYN1, SYN2, SYT12, THRA, VAT1, VGF |
|
| |
| Top Neurological Diseases: rno-miR-331 | Predicted Targets in Disease |
|
| |
| movement disorder | APP, ATP6V1A, BDNF, Ca2+, CAMK4, CIT, CX3CL1, DRD2, EPHB2, ethanol, FDFT1, FGF2, GLI1, GNAO1, GRIK2, GRIN2D, GRK5, HAP1, HMOX1, HNRNPU, HTT, KCNAB1, KCNJ4, MTOR, NGF, NTRK3, PDE4A, PKM2, PRDX2, PTPN5, RAB3A, SDHA, SGK1, SYN1, SYN2, VSNL1 |
| disorder of basal ganglia | APP, ATP6V1A, BDNF, Ca2+, CAMK4, CX3CL1, DRD2, EPHB2, FDFT1, FGF2, GNAO1, GRIK2, GRIN2D, HAP1, HMOX1, HNRNPU, HTT, KCNAB1, KCNJ4, NGF, PDE4A, PKM2, PRDX2, PTPN5, RAB3A, SDHA, SGK1, SYN1, SYN2, VSNL1 |
| neuromuscular disease | APP, ATP6V1A, BDNF, Ca2+, CAMK4, CX3CL1, DRD2, EPHB2, FDFT1, FGF2, FURIN, GNAO1, GRIK2, GRIN2D, HAP1, HMOX1, HNRNPU, HTT, KCNAB1, KCNJ4, MTOR, NGF, PDE4A, PKM2, PRDX2, PTPN5, RAB3A, SDHA, SGK1, SYN1, SYN2, VSNL1 |
| Huntington's disease | ATP6V1A, BDNF, Ca2+, CAMK4, CX3CL1, DRD2, EPHB2, FDFT1, FGF2, GNAO1, GRIK2, GRIN2D, HAP1, HMOX1, HNRNPU, HTT, KCNAB1, KCNJ4, NGF, PKM2, PRDX2, PTPN5, RAB3A, SDHA, SGK1, VSNL1 |
| dementia | APP, BDNF, Ca2+, CXCL12, CXCR4, DRD2, EIF2AK2, EIF2S1, EIF4EBP1, FDFT1, FGF2, GRIN2D, HMOX1, HNRNPU, HTT, MTOR, NFE2L2, NGF, NPTX1, NTRK3, PRKCZ, SERPINE1, TGFB1, TUBA3C/TUBA3D |
| neurodegenerative disorder | APP, BDNF, Ca2+, CXCL12, CXCR4, DRD2, EIF2AK2, EIF2S1, EIF4EBP1, FDFT1, FGF2, FLOT2, GRIN2D, HMOX1, HNRNPU, HTT, MTOR, NFE2L2, NGF, NPTX1, NTRK3, PRKCZ, TGFB1, TUBA3C/TUBA3D |
| schizophrenia | APP, BDNF, CIT, CXCL12, DLG4, DRD2, ELK1, FXYD6, GCLC, GRIA1, GRIK2, GRIN2D, IMPA2, NCS1, NPTX1, NSF, pregnenolone, S100A6, SYN2, VGF, VSNL1 |
| damage of hippocampus | ADRBK1, APP, BBC3, FGF2, NGF |
| damage of cerebral cortex | APP, BBC3, CX3CL1, FGF2, NGF |
| bipolar disorder | BDNF, CIT, DLG4, DRD2, GRIA1, GRIK2, GRIN2D, IMPA2, NCS1, pregnenolone, SYN1, SYN2, THRA, VGF |
| damage of brain | ADRBK1, APP, BBC3, BDNF, CX3CL1, FGF2, HTT, NFE2L2, NGF |
|
| |
| Top Functions: rno-miR-331 | Predicted Targets for Purported Functions |
|
| |
| cell death of brain | ADRBK1, APP, BBC3, BDNF, Ca2+, CXCL12, DLG4, DRD2, ethanol, FGF2, GCLC, GNAO1, HTT, KSR1, MAPK14, NCS1, NFE2L2, NGF, NPTX1, NTRK3, PTPN5, SDHA, SERPINE1, SERPINF1, TAF4, TGFA, TGFB1 |
| long-term potentiation | APP, BDNF, Ca2+, CAMK4, DLG4, DRD2, EFNB1, EPHB2, ethanol, FYN, GRIA1, GRIK2, GRIN2D, HTT, KCNAB1, KSR1, MAPK1, NGF, NTRK3, PTPN5, RAB3A |
| cell death of central nervous system cells | ADRBK1, APP, BBC3, BDNF, Ca2+, CXCL12, DRD2, ethanol, GCLC, GNAO1, HTT, KSR1, MAPK1, MAPK14, NCS1, NFE2L2, NGF, NPTX1, NTRK3, PTPN5, SERPINE1, SERPINF1, TGFA, TGFB1 |
| growth of plasma membrane projections | APP, BDNF, Ca2+, CAMK4, CXCL12, EFNB1, EIF4EBP1, ELK1, EPHB2, ethanol, FGF2, FYN, GAS7, GNAO1, GRK5, HAP1, HTT, MAPK1, MTOR, NGF, NOG, NPTX1, NTRK3, PDE4A, SGK1, SKIL, SYN1 |
| synaptic depression | APP, BDNF, Ca2+, CAMK4, DLG4, DRD2, EPHB2, ethanol, GRIA1, HTT, MTOR, RAB3A, SYN1, SYN2, VGF |
| neurotransmission | APP, BDNF, Ca2+, CAMK4, DLG4, DRD2, EPHB2, ERBB2, ethanol, FGF2, GABA, GRIA1, GRIK2, GRIN2D, HAP1, HTT, KCNAB1, NCS1, NPTX1, NSF, PRKCZ, RAB3A, SYN1, SYN2 |
| synaptic transmission | APP, BDNF, Ca2+, CAMK4, DLG4, DRD2, EPHB2, ethanol, FGF2, GABA, GRIA1, GRIK2, GRIN2D, HAP1, HTT, NCS1, NPTX1, NSF, PRKCZ, RAB3A, SYN1, SYN2 |
| cell death of cortical neurons | APP, BBC3, BDNF, Ca2+, DRD2, GCLC, GNAO1, HTT, KSR1, MAPK14, NCS1, NFE2L2, NGF, NPTX1, NTRK3, PTPN5, SERPINE1, TGFA |
| growth of neurites | APP, BDNF, Ca2+, CAMK4, CXCL12, EFNB1, EIF4EBP1, ELK1, EPHB2, ethanol, FGF2, GAS7, GNAO1, GRK5, HAP1, HTT, MAPK1, MTOR, NGF, NOG, NPTX1, NTRK3, PDE4A, SGK1, SKIL, SYN1 |
| cell death of cerebral cortex cells | ADRBK1, APP, BBC3, BDNF, Ca2+, DRD2, GCLC, GNAO1, HTT, KSR1, MAPK14, NCS1, NFE2L2, NGF, NPTX1, NTRK3, PTPN5, SERPINE1, TGFA, TGFB1 |
| branching of neurites | APP, BDNF, Ca2+, CAMK4, CXCL12, DLG4, DRD2, EFNB1, EIF4EBP1, EPHB2, FGF2, HTT, MTOR, NGF, NTRK3, SGK1, SKIL, TGFB1 |
| necrosis | ADRBK1, APP, BBC3, BDNF, Ca2+, CAMK4, CIT, CSPG4, CX3CL1, CXCL12, CXCR4, DLG4, DRD2, DUSP1, EFNB1, EIF2AK2, EIF4EBP1, ELK1, EPHB2, ERBB2, ethanol, FDFT1, FGF2, FURIN, FUS, FYN, GABA, GCLC, GLI1, GNAO1, GRIK2, HAP1, HMOX1, HTT, ITGA6, KSR1, MAPK1, MAPK14, MCF2L, MTOR, NCS1, NFE2L2, NGF, NOG, NPTX1, NTRK3, PDE4A, PKM2, PRDX2, PRKCZ, PTPN5, RARA, S100A6, SDHA, SERPINE1, SERPINF1, SGK1, SKIL, TAF4, TGFA, TGFB1, THRA, VSNL1 |
| apoptosis of neurons | APP, BBC3, BDNF, Ca2+, CXCL12, DRD2, DUSP1, ethanol, FGF2, GCLC, GNAO1, GRIK2, HAP1, HMOX1, HTT, KSR1, MAPK14, NFE2L2, NGF, NPTX1, NTRK3, PRDX2, SERPINF1, TGFA, TGFB1 |
| morphogenesis of neurites | APP, BDNF, Ca2+, CAMK4, CIT, CXCL12, DLG4, DRD2, EFNB1, EIF4EBP1, EPHB2, FGF2, GAS7, HTT, LZTS1, MTOR, NGF, NTRK3, SGK1, SKIL, TGFB1 |
Table 4.
Pathway analysis of post-insult VPA-regulated miR-885-3p targets. Predicted mRNA targets of miR-885-3p were generated using four independent prediction algorithms. Only mRNAs predicted concurrently by 3 or more databases were selected and further analyzed by Ingenuity Pathway Analysis to identify top canonical networks. These top networks were then filtered for disease and functions where both direct (BOLDED) and indirect miRNA targets associated with each network, disease or function are included.
| Top Networks: hsa-miR-885-3p | Predicted Targets in Network |
|---|---|
| cell death, cellular movement, cellular assembly and organization | AKT1, CALML3, CAMK2A, CASP3, CTSB, CXCL12, DNAJC5, ENSA, FAIM2, GNAO1, GRIN1, HDAC5, HYOU1, INPPL1, LRPAP1, MAPT, MMP2, NCAM1, NUMB, PEBP1, PRKCA, PTEN, RAD51D, RPS6KA3, SLC1A1, SLC6A1, SMAD4, SP1, ST8SIA1, STIP1, TFAP2A, THBS1, TNFRSF1B, TPPP, VEGFA |
| behavior, cellular development, nervous system development and function | ADRA2B, ADRBK1, AGRN, ARC, CHRNB2, CNTN2, CSNK1E, EFNB2, ELK1, EN1, GAS7, GFAP, GIT1, GPC1, GRM1, HAP1, HIP1, HTT, IGFBP5, KCNA1, KCNIP3, KCNJ2, LIF, NGFR, NOTCH1, OLIG2, OPRL1, PDE1B, PMP22, SLC25A22, SP6, STX12, TGFBR2, THBS2, ULK1 |
| organismal development, reproductive system development and function, tissue development | AIM1, ALDOC, ANGPT2, ANTXR1, APP, ascorbic acid, ATP6V1A, BTC, CD82, CDON, CEBPB, CLDN3, COL5A1, COL5A3, CYB561, DAP, DCN, DERL1, DGAT1, EIF2S1, ERBB2, ERBB4, GHR, KCND2, KLRK1, LCN2, PQLC1, SETDB1, SQRDL, TMEM39A, TUBB3, UCK1, UPP1, VDR, VWF |
|
| |
| Top Neurological Diseases: hsa-miR-885-3p | Predicted Targets in Disease |
|
| |
| movement disorder | ADRA2B, AKT1, APP, ascorbic acid, ATP6V1A, CAMK2A, CASP3, CHRNB2, CTSB, DNAJC5, GFAP, GIT1, GNAO1, GRIN1, GRM1, HAP1, HDAC5, HIP1, HTT, KCNA1, LIF, MAPT, NGFR, OPRL1, PDE1B, PEBP1, PMP22, SETDB1, SLC1A1, SLC6A1, SP1, ST8SIA1, VEGFA |
| tauopathy | ADRA2B, AGRN, APP, ascorbic acid, CAMK2A, CASP3, CTSB, CXCL12, DCN, EIF2S1, GFAP, GRIN1, GRM1, HTT, LRPAP1, MAPT, NGFR, NOTCH1, PRKCA, PTEN, SLC1A1, TGFBR2, VDR |
| neurodegenerative disorder | ADRA2B, AGRN, APP, ascorbic acid, CAMK2A, CASP3, CTSB, CXCL12, DCN, EIF2S1, GFAP, GRIN1, GRM1, HTT, LRPAP1, MAPT, NGFR, NOTCH1, PRKCA, PTEN, SLC1A1, TGFBR2, VDR, VEGFA |
| Alzheimer's disease | ADRA2B, AGRN, APP, ascorbic acid, CAMK2A, CASP3, CTSB, CXCL12, DCN, EIF2S1, GFAP, GRIN1, GRM1, HTT, LRPAP1, MAPT, NGFR, NOTCH1, PRKCA, PTEN, TGFBR2, VDR |
| hyperactive behavior | APP, CAMK2A, CHRNB2, DGAT1, ERBB4, GNAO1, HTT, MAPT, PDE1B, SLC6A1, TGFBR2 |
| seizures | APP, CNTN2, GNAO1, GRIN1, GRM1, HTT, HYOU1, KCNA1, NGFR, PMP22, PTEN, SLC1A1, SLC6A1, ST8SIA1, TNFRSF1B |
| neurodegeneration | APP, CASP3, DNAJC5, ERBB2, GRIN1, HAP1, HTT, MAPT, NGFR, SLC1A1, ST8SIA1, TGFBR2, VEGFA |
| neurological signs | ADRA2B, AKT1, APP, ATP6V1A, CAMK2A, CASP3, CHRNB2, GFAP, GIT1, GNAO1, GRIN1, HAP1, HDAC5, HTT, KCNA1, MAPT, PDE1B, PMP22, SETDB1, SLC1A1, SP1, ST8SIA1 |
| epilepsy | CHRNB2, CTSB, GRIN1, HTT, KCNA1, NGFR, SLC1A1, SLC25A22, SLC6A1 |
| tremor | CTSB, GNAO1, GRM1, HIP1, HTT, KCNA1, LIF, PMP22, SLC6A1 |
| ataxia | APP, CHRNB2, DNAJC5, GRIN1, GRM1, HTT, KCNA1, NGFR, SLC1A1, SLC6A1 |
| damage of brain | ADRBK1, APP, ascorbic acid, GRIN1, HTT, HYOU1, THBS1, VWF |
|
| |
| Top Functions: hsa-miR-885-3p | Predicted Targets for Purported Functions |
|
| |
| neuronal cell death | ADRBK1, AGRN, AKT1, APP, ascorbic acid, CAMK2A, CASP3, CTSB, CXCL12, DNAJC5, EFNB2, ELK1, EN1, ERBB2, FAIM2, GFAP, GHR, GNAO1, GRIN1, GRM1, HAP1, HDAC5, HIP1, HTT, HYOU1, INPPL1, KCNIP3, LCN2, LIF, LRPAP1, MAPT, NGFR, PRKCA, PTEN, SLC1A1, SP1, ST8SIA1, STIP1, TNFRSF1B, VEGFA |
| behavior | ADRBK1, APP, ARC, ascorbic acid, CAMK2A, CASP3, CHRNB2, CNTN2, DGAT1, DNAJC5, EFNB2, EN1, ERBB4, GHR, GIT1, GNAO1, GRIN1, GRM1, HAP1, HTT, KCNA1, KCNIP3, LRPAP1, NCAM1, NGFR, NOTCH1, OLIG2, OPRL1, PDE1B, PRKCA, PTEN, SETDB1, SLC6A1, ST8SIA1, THBS2, TNFRSF1B, VDR, VEGFA |
| necrosis | ADRBK1, AGRN, AKT1, ANGPT2, ANTXR1, APP, ascorbic acid, BTC, CAMK2A, CASP3, CEBPB, COL5A3, CTSB, CXCL12, DCN, DNAJC5, EFNB2, ELK1, EN1, ERBB2, ERBB4, FAIM2, GFAP, GHR, GNAO1, GPC1, GRIN1, GRM1, HAP1, HDAC5, HIP1, HTT, HYOU1, IGFBP5, INPPL1, KCNIP3, KLRK1, LCN2, LIF, LRPAP1, MAPT, MMP2, NCAM1, NGFR, NOTCH1, NUMB, PDE1B, PEBP1, PMP22, PRKCA, PTEN, RAD51D, SLC1A1, SMAD4, SP1, ST8SIA1, STIP1, TFAP2A, TGFBR2, THBS1, THBS2, TNFRSF1B, TUBB3, VDR, VEGFA |
| organization of cytoskeleton | ADRBK1, AGRN, AKT1, ANGPT2, ANTXR1, APP, CALML3, CD82, CHRNB2, CNTN2, CXCL12, EFNB2, ELK1, ERBB2, ERBB4, GAS7, GFAP, GHR, GIT1, GRIN1, HAP1, HTT, INPPL1, KCNJ2, LCN2, LRPAP1, MAPT, NCAM1, NGFR, NOTCH1, NUMB, PMP22, PRKCA, PTEN, SP1, ST8SIA1, STIP1, THBS1, TPPP, TUBB3, ULK1, VEGFA, VWF |
| quantity of neurons | ANGPT2, APP, ascorbic acid, CASP3, CHRNB2, CNTN2, EN1, ERBB2, ERBB4, GNAO1, HTT, KCNIP3, LIF, MAPT, NGFR, NOTCH1, NUMB, OLIG2, SLC1A1, SMAD4, TFAP2A |
| differentiation of cells | ADRA2B, AGRN, AKT1, ANGPT2, APP, ascorbic acid, BTC, CASP3, CD82, CDON, CEBPB, CNTN2, CTSB, CXCL12, DCN, EFNB2, ELK1, EN1, ERBB2, ERBB4, GAS7, GHR, GNAO1, GPC1, HDAC5, HTT, IGFBP5, KLRK1, LCN2, LIF, MAPT, MMP2, NCAM1, NGFR, NOTCH1, NUMB, OLIG2, PMP22, PRKCA, PTEN, RPS6KA3, SETDB1, SMAD4, SP1, SP6, ST8SIA1, TFAP2A, TGFBR2, THBS1, THBS2, TNFRSF1B, TUBB3, ULK1, VDR, VEGFA |
| development of neurons | APP, CAMK2A, CXCL12, EN1, ERBB4, HTT, KCNJ2, LIF, NCAM1, NGFR, NOTCH1, OLIG2, PMP22, STIP1, THBS1, THBS2, VEGFA |
| microtubule dynamics | AGRN, AKT1, ANGPT2, APP, CALML3, CD82, CHRNB2, CNTN2, CXCL12, EFNB2, ELK1, ERBB2, ERBB4, GAS7, GHR, GIT1, GRIN1, HAP1, HTT, INPPL1, KCNJ2, LCN2, LRPAP1, MAPT, NCAM1, NGFR, NOTCH1, NUMB, PMP22, PRKCA, PTEN, SP1, ST8SIA1, STIP1, THBS1, TPPP, TUBB3, ULK1, VEGFA |
| morphology of nervous system | AGRN, AKT1, APP, CASP3, CDON, CNTN2, CTSB, CXCL12, DNAJC5, EN1, ERBB2, ERBB4, FAIM2, GFAP, GIT1, GRIN1, HAP1, HTT, HYOU1, KCNA1, KCND2, LIF, LRPAP1, NCAM1, NGFR, NOTCH1, NUMB, OLIG2, PMP22, PTEN, TFAP2A, TNFRSF1B, UPP1, VEGFA |
| apoptosis | ADRBK1, AGRN, AKT1, ALDOC, ANGPT2, ANTXR1, APP, ARC, ascorbic acid, BTC, CAMK2A, CASP3, CEBPB, COL5A3, CSNK1E, CTSB, CXCL12, DAP, DCN, DNAJC5, EFNB2, EIF2S1, ELK1, EN1, ERBB2, ERBB4, FAIM2, GHR, GNAO1, GRIN1, GRM1, HAP1, HDAC5, HIP1, HTT, HYOU1, IGFBP5, INPPL1, KCNIP3, LCN2, LIF, LRPAP1, MAPT, MMP2, NCAM1, NGFR, NOTCH1, NUMB, PDE1B, PEBP1, PRKCA, PTEN, RPS6KA3, SMAD4, SP1, ST8SIA1, STIP1, TFAP2A, TGFBR2, THBS1, THBS2, TNFRSF1B, VDR, VEGFA |
| cell death | ADRBK1, AGRN, AKT1, ALDOC, ANGPT2, ANTXR1, APP, ARC, ascorbic acid, BTC, CAMK2A, CASP3, CD82, CEBPB, CLDN3, COL5A3, CSNK1E, CTSB, CXCL12, DAP, DCN, DNAJC5, EFNB2, EIF2S1, ELK1, EN1, ERBB2, ERBB4, FAIM2, GFAP, GHR, GNAO1, GPC1, GRIN1, GRM1, HAP1, HDAC5, HIP1, HTT, HYOU1, IGFBP5, INPPL1, KCNIP3, KLRK1, LCN2, LIF, LRPAP1, MAPT, MMP2, NCAM1, NGFR, NOTCH1, NUMB, PDE1B, PEBP1, PMP22, PRKCA, PTEN, RAD51D, RPS6KA3, SLC1A1, SMAD4, SP1, ST8SIA1, STIP1, TFAP2A, TGFBR2, THBS1, THBS2, TNFRSF1B, TUBB3, VDR, VEGFA |
| differentiation of neurons | ADRA2B, AGRN, AKT1, APP, CDON, CEBPB, CNTN2, CXCL12, EN1, GAS7, HTT, LIF, MAPT, NCAM1, NGFR, NOTCH1, NUMB, OLIG2, PRKCA, TFAP2A, TUBB3, ULK1, VEGFA |
| formation of plasma membrane projections | AGRN, APP, CHRNB2, CNTN2, CXCL12, EFNB2, ERBB2, ERBB4, GAS7, GIT1, GRIN1, HTT, KCNJ2, LCN2, MAPT, NCAM1, NGFR, NOTCH1, NUMB, PMP22, PRKCA, PTEN, ST8SIA1, STIP1, ULK1 |
| development of central nervous system | APP, CASP3, CD82, CDON, CHRNB2, CXCL12, EN1, ERBB2, ERBB4, FAIM2, GFAP, GRIN1, HTT, LIF, NCAM1, NGFR, NOTCH1, NUMB, OLIG2, PMP22, PTEN, RPS6KA3, STIP1, TGFBR2, THBS1, THBS2, VEGFA |
| sprouting | AGRN, ANGPT2, APP, CHRNB2, CXCL12, DCN, ERBB4, GIT1, HTT, LCN2, NCAM1, NGFR, NOTCH1, NUMB, PTEN, SMAD4, THBS1, ULK1, VEGFA |
Discussion
The physiological response to ischemic stroke is complex and involves a multitude of processes (for review see [7]) that are not completely understood, nor are effective clinical treatments currently available. Therapeutic application of HDAC inhibitors for CNS disorders such as stroke is being considered in experimental models [21]; however, the precise mechanisms of action remain unclear. We used an HDAC inhibitor, VPA, and showed that it has beneficial effects following MCAO injury by improving neurological score and motor coordination. Our study first identified miRNA response profiles following ischemic stroke and presented pathway analysis based on these ischemia-regulated miRNA targets. We then profiled the miRNA response following post-insult VPA treatment to identify miRNAs that may contribute to these beneficial effects. We will first discuss the MCAO-regulated miRNAs followed by the post-insult VPA-regulated miRNAs. Together we will provide analysis of the miRNA regulated predicted networks, pathways, functions, and neurological diseases. We will also address cell-type specificity for selected miRNAs and how this may impact their function. We suggest that future in silico algorithms need to account for cell-type specific expression of both miRNAs and their targets to improve their biological significance. Finally, we will consider limitations and future directions for this research.
MCAO-regulated miRNAs
In this study, we identified and validated several miRNAs: miR-155, miR-297a, miR-466f, miR-466h, miR-1224 in the ischemic cortex 24 hours after MCAO. A previous study showed that miR-155 is downregulated following permanent MCAO in both hippocampus and peripheral blood, suggesting that it may be a key indicator of the injury response following cerebral ischemia [22]. miR-155 is enriched in hematopoetic cells [23], suggesting that it may have broad relevance to the inflammatory response after cerebral ischemia. In addition, miR-155 is upregulated following micoglia activation by lipopolysaccharide (LPS) where knock down of miR-155 upregulates one its targets: suppressor of cytokine signaling 1 (SOCS-1) and decreases the production of nitric oxide and the expression of inflammatory cytokines and inducible nitric oxide synthase [24]. Furthermore, treating neuronal primary cultures with conditioned medium obtained from microglia cells where miR-155 is blocked before microglia activation decreases neuronal cell death attributed to microglial activation. Therefore, miR-155’s effects are supported to be pro-inflammatory and perhaps suppression of miR-155 may lead to beneficial effects following cerebral ischemia that would act directly on microglia and provide both a cell- and injury- specific response therapy. Further support for miR-155’s inflammatory effects include the finding that treatment with a natural antioxidant, Resveratrol, reduces the upregulation of miR-155 following LPS stimulation [25]. Genetic risk for cardiovascular disease is also associated with the polymorphism (+1166/A/C) in the human angiotensin II type 1 receptor (AT1R) gene, which occurs in the 3’UTR and interferes with the miR-155 binding site [26]. Importantly, this study supports that a polymorphism in a miRNA-binding site can disrupt miRNA regulation of susceptibility genes leading to increased risk for cardiovascular disease.
Sparse data is available for the other MCAO-regulated miRNA candidates and their potential functions in cerebral ischemia. These may represent previously uncharacterized miRNAs that mediate the cerebral ischemic response. Interestingly, robust upregulation of the miR-297-669 cluster, which includes candidates miR-297a, miR-466h, and miR-466f, is observed following nutrient depletion in Chinese hamster ovary (CHO) cells. This may be linked to proapoptotic pathways, as suppression of miR-466h upregulates anti-apoptotic genes (bcl2l2, dad1, birc6 and stat5a), increases cell viability, and decreases caspase 3/7 activation in CHO cells [27]. The role of this miRNA cluster in facilitating OGD-induced cell death in cerebral ischemia warrants further investigation. Candidate miR-1224 is upregulated following LPS treatment in the spleens of mice where it is shown to negatively regulate tumor necrosis factor-α (TNF- α) via the transcription factor Specificity Protein 1 (Sp1) [28]. Further investigation is required to determine the exact function of miR-1224 in brain, particularly whether it may be involved in neuroinflammation in response to stroke.
MCAO-regulated miRNA predicted targets were evaluated using IPA
Top molecular networks generated from common MCAO-regulated miRNA predicted targets generated by IPA included: (1) cell death, cellular development, cellular growth and proliferation and (2) molecular transport, behavior, cell-to-cell signaling and interaction (Table 2). The first network includes many predicted mRNA targets that are well established to play a role in cell death mechanisms also implicated in ischemia. Some examples include apoptosis-inducing factor 1, mitochondrial (AIFM1) and BCL2-associated agonist of cell death (BAD). BAD has recently been implicated in the rapamycin-induced autophagy mechanism found to protect against hypoxia-ischemia [29]. The second network may represent some new candidates that underlie the miRNA-regulated response to cerebral ischemia. For instance, carbonic anhydrase 4 (CA4) is a member of a family of zinc metalloenzymes that catalyze the reversible hydration of carbon dioxide. CAs also provide an important role in buffering the brain against acute changes in pH and are shown to be implicated in the asphyxia-compensatory-response that protects neonates from pathological changes associated with loss of oxygen [30]. Therefore, investigating these predicted targets in this second network may provide new compensatory mechanisms for protecting against cerebral ischemia and suggests another advantage for miRNA research. We have also listed miRNA predicted targets for top canonical pathways and nervous system development and functions that may also be characterized in future studies to contribute to the miRNA-regulated response mechanisms following cerebral ischemia.
Post-insult VPA-regulated miRNAs
Our study identified and validated miR-885-3p and miR-331 following protective VPA treatment in ischemic cortex 24 hours after MCAO. Candidate miR-885-3p was upregulated by MCAO in our study while post-insult VPA treatment decreased its expression. Interestingly, miR-885-3p is involved in the regulation of cell viability, apoptosis and/or autophagy in squamous cell carcinoma cells upon cisplatin exposure via regulation of anti-apoptotic protein BCL2 [31]. If miR-885-3p is able to exert similar BCL2 regulatory effects in brain, the previously observed upregulation of neuronal BCL2 by VPA treatment [19] may occur, at least in part, through downregulation of miR-885-3p. Candidate miR-885-3p was not regulated in our neuronal OGD in vitro model of ischemia. This may be due to its enriched expression in astrocytes [23]. This demonstrates a clear example of why cell-type specific expression of miRNAs is an important consideration that may impact their function and role in cerebral ischemia.
Candidate miR-331 was upregulated following post-insult VPA treatment in vivo and also following treatment with VPA in our neuronal OGD in vitro model of ischemia. This response profile may represent a compensatory response of the post-insult VPA treatment that may be exploited further for therapeutic benefits. In support of this, using our OGD model where cell viability was reduced following OGD and increased by VPA pretreatment, miR-331 was also upregulated in this neuroprotective VPA treatment condition. Interestingly, miR-331 is expressed in both neurons and astrocytes [23]. This may be one of the contributing factors as to why miR-331 is regulated by VPA in our neuronal OGD model. Previous reports have shown that miR-331 is upregulated in whole brain following MCAO in rats [11]. This may be due to processing the entire brain versus only including the ipsilateral cortex, as was performed in our study where we found miR-331 to not be regulated following MCAO alone. Candidate miR-331 is best characterized for its role in cancer where it has been shown to target ERBB2, a tyrosine kinase receptor frequently overexpressed in prostate cancer [32,33]. The potential functions of miR-885-3p and miR-331 in cerebral ischemia have not been extensively characterized. Therefore, we identified their predicted targets and performed pathway analysis using IPA.
Post-insult VPA-regulated miRNA targets evaluated using IPA
This analysis provided top networks, neurological diseases, and functions potentially regulated by our candidate miRNAs. Top networks for candidate miR-331 include: (1) cellular movement, hematological system development and function, immune cell trafficking; (2) cell death, hereditary disorder, neurological disease and (3) cellular assembly and organization, nervous system development and function, cellular movement. Briefly considering some interesting predicted targets, chemokine C-X3-C motif ligand 1 (CX3CL1) has been shown to be neuroprotective in permanent focal cerebral ischemia [34] while protein tyrosine phosphatase non-receptor type 5 (PTPN5) has been shown to be regulated by cerebral ischemia [35], and PTPN5 dysregulation has been implicated in numerous neuropsychiatric disorders including stroke, Alzheimer’s disease, and Huntington’s disease [36]. PTPN5 is one of many targets implicating miR-331 in numerous neurological diseases listed in Table 3. This type of analysis highlights another benefit of miRNA predictive target analysis where common targets, such as PTPN5, APP, ATP6V1A, BDNF, and MTOR, among others, may be dysregulated across multiple neurological diseases. Collectively this may suggest common functions to these neurological diseases such as cell death, necrosis, long-term potentiation, and synaptic transmission as listed in Table 3 that are under miRNA control. Therefore, characterizing key candidate miRNAs that can most effectively modulate these common mechanisms may provide novel therapeutics for neurological diseases.
Top networks for candidate miR-885-3p include (1) cell death, cellular movement, cellular assembly and organization; (2) behavior, cellular development, nervous system development and function; and (3) organismal development, reproductive system development and function, tissue development. Briefly considering some interesting predicted targets of miR-885-3p, vascular endothelial growth factor A (VEGFA) promotes angiogenesis, vasculogenesis, endothelial cell growth, and inhibits apoptosis. VEGF is regulated at the mRNA and protein level following cerebral ischemia [37], can be further regulated after chronic VPA treatment in a rat model of cerebral ischemia to promote functional recovery [38], and may be further modulated by miR-885-3p. RAC-alpha serine/threonine-protein kinase (AKT1), another predicted target of miR-885-3p, is involved in cell survival where it is shown that activation of both VEGF and AKT1 is necessary to promote neuronal survival mediated by hypoxic preconditioning [39]. Histone deacetylase 5 (HDAC5) is also a target of miR-885-3p, and VPA [4], where its function in the CNS has been implicated in modulating angiogenesis [40] and cocaine addiction [41]. In terms of top neurological diseases predicted for miR-885-3p from our analysis, movement disorder, tauopathy, neurodegenerative disorder, Alzheimer’s disease, and seizures were some of those represented (Table 4). Some of the common predicted targets for these neurological diseases include: α2B adrenoceptor (ADRA2B), amyloid precursor protein (APP), chemokine (C-X-C motif) ligand 12 (CXCL12), calcium/calmodulin-dependent protein kinase type II alpha chain (CAMK2A), and phosphatase and tensin homolog (PTEN). Some of the functions targeted by miR-885-3p predicted targets that may be implicated in these neurological diseases include neuronal cell death, behavior, necrosis, and organization of cytoskeleton. Future studies will be required to evaluate the therapeutic potential of targeting these functions through miR-885-3p in neurological diseases.
Limitations, future directions, and conclusions
The major limitations of this study include validation of miRNA targets and characterization of miRNA biological function. Validation of miRNA targets has been reviewed previously [42] and includes using multiple algorithms to predict miRNA binding sites, determining miRNA/mRNA co-expression, identifying altered protein expression by miRNA, and characterizing miRNA impact on biological function. Future in silico algorithms for predicting miRNA targets should consider cell- and tissue- specific co-expression of miRNAs and their targets. This comprehensive expression analysis paired with miRNA binding site prediction algorithms is currently not available. Adding this expression analysis will improve the biological validity of these prediction algorithms. In the mean time, to infer biological function, we used literature support, predicted targets, pathway analysis, and tissue-specific expression. Future investigations will need to validate each candidate miRNA in experimental models of cerebral ischemia. Additional studies should elucidate the miRNA regulatory networks, pathways, and functions presented in our study and exploit them for therapeutic use. Exploring miRNA mechanisms underlying cerebral ischemia and following HDAC inhibition may provide common networks to facilitate recovery that have currently been unrecognized. In addition, elucidation of pan-HDAC inhibitors versus targeting specific isoforms in producing post-insult benefits and regulating miRNA mechanisms is also warranted. These studies will help clarify which isoform-specific inhibition hold the greatest therapeutic promise and likewise which miRNA mechanisms may underlie these benefits.
In summary, our study provides novel evidence identifying miRNAs involved with the injury response to cerebral ischemia, as well as involved with the beneficial effects following post-insult VPA treatment. We have analyzed these miRNA candidates based on their predictive targets to infer functions, networks, pathways, and diseases. Collectively, these array studies identify miRNA mechanisms that may contribute to the endogenous compensatory mechanisms following MCAO injury, as well as those therapeutic mechanisms facilitated via HDAC inhibition. These results have broad implications for the field of neurology by suggesting another layer of controls of gene expression to modulate pathways and mechanisms that will provide new insights for stroke research.
Acknowledgments
We would like to acknowledge the support of the Intramural Research Program of the NIMH, NIH, and by the Hsu family foundation. We would also like to acknowledge the technical support kindly provided by Weiwei Wu for microRNA array processing and the editorial suggestions by Peter Leeds.
Supporting Information
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Ho PM, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Rosamond WD, Sorlie PD, Stafford RS, Turan TN, Turner MB, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Del Zoppo GJ, Saver JL, Jauch EC, Adams HP Jr. Expansion of the time window for treatment of acute ischemic stroke with intravenous tissue plasminogen activator: a science advisory from the American Heart Association/American Stroke Association. Stroke. 2009;40:2945–2948. doi: 10.1161/STROKEAHA.109.192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, Machnig T, Schneider D, von Kummer R, Wahlgren N, Toni D. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- 4.Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32:591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langley B, Brochier C, Rivieccio MA. Targeting histone deacetylases as a multifaceted approach to treat the diverse outcomes of stroke. Stroke. 2009;40:2899–2905. doi: 10.1161/STROKEAHA.108.540229. [DOI] [PubMed] [Google Scholar]
- 6.Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- 7.Wang ZF, Fessler EB, Chuang DM. Beneficial effects of mood stabilizers lithium, valproate and lamotrigine in experimental stroke models. Acta Pharmacol Sin. 2011;32:1433–1445. doi: 10.1038/aps.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 9.Visone R, Croce CM. MiRNAs and cancer. Am J Pathol. 2009;174:1131–1138. doi: 10.2353/ajpath.2009.080794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kocerha J, Kauppinen S, Wahlestedt C. microRNAs in CNS disorders. Neuromolecular Med. 2009;11:162–172. doi: 10.1007/s12017-009-8066-1. [DOI] [PubMed] [Google Scholar]
- 11.Jeyaseelan K, Lim KY, Armugam A. MicroRNA expression in the blood and brain of rats subjected to transient focal ischemia by middle cerebral artery occlusion. Stroke. 2008;39:959–966. doi: 10.1161/STROKEAHA.107.500736. [DOI] [PubMed] [Google Scholar]
- 12.Fasanaro P, Greco S, Ivan M, Capogrossi MC, Martelli F. microRNA: emerging therapeutic targets in acute ischemic diseases. Pharmacol Ther. 2010;125:92–104. doi: 10.1016/j.pharmthera.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 13.Saugstad JA. MicroRNAs as effectors of brain function with roles in ischemia and injury, neuroprotection, and neurodegeneration. J Cereb Blood Flow Metab. 2010;30:1564–1576. doi: 10.1038/jcbfm.2010.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott GK, Mattie MD, Berger CE, Benz SC, Benz CC. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res. 2006;66:1277–1281. doi: 10.1158/0008-5472.CAN-05-3632. [DOI] [PubMed] [Google Scholar]
- 15.Zhou R, Yuan P, Wang Y, Hunsberger JG, Elkahloun A, Wei Y, Damschroder-Williams P, Du J, Chen G, Manji HK. Evidence for selective microRNAs and their effectors as common long-term targets for the actions of mood stabilizers. Neuropsychopharmacology. 2009;34:1395–1405. doi: 10.1038/npp.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z, Leng Y, Tsai LK, Leeds P, Chuang DM. Valproic acid attenuates blood-brain barrier disruption in a rat model of transient focal cerebral ischemia: the roles of HDAC and MMP-9 inhibition. J Cereb Blood Flow Metab. 2011;31:52–57. doi: 10.1038/jcbfm.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai LK, Wang Z, Munasinghe J, Leng Y, Leeds P, Chuang DM. Mesenchymal stem cells primed with valproate and lithium robustly migrate to infarcted regions and facilitate recovery in a stroke model. Stroke. 2011;42:2932–2939. doi: 10.1161/STROKEAHA.110.612788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marinova Z, Ren M, Wendland JR, Leng Y, Liang MH, Yasuda S, Leeds P, Chuang DM. Valproic acid induces functional heat-shock protein 70 via Class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J Neurochem. 2009;111:976–987. doi: 10.1111/j.1471-4159.2009.06385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leng Y, Chuang DM. Endogenous alphasynuclein is induced by valproic acid through histone deacetylase inhibition and participates in neuroprotection against glutamate-induced excitotoxicity. J Neurosci. 2006;26:7502–7512. doi: 10.1523/JNEUROSCI.0096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dweep H, Sticht C, Pandey P, Gretz N. miRWalk--database: prediction of possible miRNA binding sites by "walking" the genes of three genomes. J Biomed Inform. 2011;44:839–847. doi: 10.1016/j.jbi.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 21.Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 22.Liu DZ, Tian Y, Ander BP, Xu H, Stamova BS, Zhan X, Turner RJ, Jickling G, Sharp FR. Brain and blood microRNA expression profiling of ischemic stroke, intracerebral hemorrhage, and kainate seizures. J Cereb Blood Flow Metab. 2010;30:92–101. doi: 10.1038/jcbfm.2009.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, Lin C, Socci ND, Hermida L, Fulci V, Chiaretti S, Foa R, Schliwka J, Fuchs U, Novosel A, Muller RU, Schermer B, Bissels U, Inman J, Phan Q, Chien M, Weir DB, Choksi R, De Vita G, Frezzetti D, Trompeter HI, Hornung V, Teng G, Hartmann G, Palkovits M, Di Lauro R, Wernet P, Macino G, Rogler CE, Nagle JW, Ju J, Papavasiliou FN, Benzing T, Lichter P, Tam W, Brownstein MJ, Bosio A, Borkhardt A, Russo JJ, Sander C, Zavolan M, Tuschl T. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cardoso AL, Guedes JR, Pereira de Almeida L, Pedroso de Lima MC. miR-155 modulates microglia-mediated immune response by downregulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology. 2012;135:73–88. doi: 10.1111/j.1365-2567.2011.03514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tili E, Michaille JJ, Adair B, Alder H, Limagne E, Taccioli C, Ferracin M, Delmas D, Latruffe N, Croce CM. Resveratrol decreases the levels of miR-155 by upregulating miR-663, a microRNA targeting JunB and JunD. Carcinogenesis. 2010;31:1561–1566. doi: 10.1093/carcin/bgq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin MM, Buckenberger JA, Jiang J, Malana GE, Nuovo GJ, Chotani M, Feldman DS, Schmittgen TD, Elton TS. The human angiotensin II type 1 receptor +1166 A/C polymorphism attenuates microrna-155 binding. J Biol Chem. 2007;282:24262–24269. doi: 10.1074/jbc.M701050200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Druz A, Chu C, Majors B, Santuary R, Betenbaugh M, Shiloach J. A novel microRNA mmu-miR-466h affects apoptosis regulation in mammalian cells. Biotechnol Bioeng. 2011;108:1651–1661. doi: 10.1002/bit.23092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niu Y, Mo D, Qin L, Wang C, Li A, Zhao X, Wang X, Xiao S, Wang Q, Xie Y, He Z, Cong P, Chen Y. Lipopolysaccharide-induced miR-1224 negatively regulates tumour necrosis factor-alpha gene expression by modulating Sp1. Immunology. 2011;133:8–20. doi: 10.1111/j.1365-2567.2010.03374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carloni S, Buonocore G, Longini M, Proietti F, Balduini W. Inhibition of rapamycin-induced autophagy causes necrotic cell death associated with Bax/Bad mitochondrial translocation. Neuroscience. 2012;203:160–169. doi: 10.1016/j.neuroscience.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 30.Nogradi A, Domoki F, Degi R, Borda S, Pakaski M, Szabo A, Bari F. Up-regulation of cerebral carbonic anhydrase by anoxic stress in piglets. J Neurochem. 2003;85:843–850. doi: 10.1046/j.1471-4159.2003.01721.x. [DOI] [PubMed] [Google Scholar]
- 31.Huang Y, Chuang AY, Ratovitski EA. Phospho-DeltaNp63alpha/miR-885-3p axis in tumor cell life and cell death upon cisplatin exposure. Cell Cycle. 2011;10:3938–3947. doi: 10.4161/cc.10.22.18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Epis MR, Barker A, Giles KM, Beveridge DJ, Leedman PJ. The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNA miR-331-3p in prostate cancer cells. J Biol Chem. 2011;286:41442–41454. doi: 10.1074/jbc.M111.301481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Epis MR, Giles KM, Barker A, Kendrick TS, Leedman PJ. miR-331-3p regulates ERBB-2 expression and androgen receptor signaling in prostate cancer. J Biol Chem. 2009;284:24696–24704. doi: 10.1074/jbc.M109.030098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cipriani R, Villa P, Chece G, Lauro C, Paladini A, Micotti E, Perego C, De Simoni MG, Fredholm BB, Eusebi F, Limatola C. CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J Neurosci. 2011;31:16327–16335. doi: 10.1523/JNEUROSCI.3611-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braithwaite SP, Xu J, Leung J, Urfer R, Nikolich K, Oksenberg D, Lombroso PJ, Shamloo M. Expression and function of striatal enriched protein tyrosine phosphatase is profoundly altered in cerebral ischemia. Eur J Neurosci. 2008;27:2444–2452. doi: 10.1111/j.1460-9568.2008.06209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goebel-Goody SM, Baum M, Paspalas CD, Fernandez SM, Carty NC, Kurup P, Lombroso PJ. Therapeutic implications for striatal-enriched protein tyrosine phosphatase (STEP) in neuropsychiatric disorders. Pharmacol Rev. 2012;64:65–87. doi: 10.1124/pr.110.003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hayashi T, Abe K, Suzuki H, Itoyama Y. Rapid induction of vascular endothelial growth factor gene expression after transient middle cerebral artery occlusion in rats. Stroke. 1997;28:2039–2044. doi: 10.1161/01.str.28.10.2039. [DOI] [PubMed] [Google Scholar]
- 38.Wang Z, Tsai LK, Munasinghe J, Leng Y, Fessler EB, Chibane F, Leeds P, Chuang DM. Chronic valproate treatment enhances postischemic angiogenesis and promotes functional recovery in a rat model of ischemic stroke. Stroke. 2012 doi: 10.1161/STROKEAHA.112.652545. epub, DOI: 10.1161/STROKEAHA.112.652545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci. 2002;22:6401–6407. doi: 10.1523/JNEUROSCI.22-15-06401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Urbich C, Rossig L, Kaluza D, Potente M, Boeckel JN, Knau A, Diehl F, Geng JG, Hofmann WK, Zeiher AM, Dimmeler S. HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood. 2009;113:5669–5679. doi: 10.1182/blood-2009-01-196485. [DOI] [PubMed] [Google Scholar]
- 41.Taniguchi M, Carreira MB, Smith LN, Zirlin BC, Neve RL, Cowan CW. Histone deacetylase 5 limits cocaine reward through cAMP-induced nuclear import. Neuron. 2012;73:108–120. doi: 10.1016/j.neuron.2011.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuhn DE, Martin MM, Feldman DS, Terry AV Jr, Nuovo GJ, Elton TS. Experimental validation of miRNA targets. Methods. 2008;44:47–54. doi: 10.1016/j.ymeth.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.