

Abstract
Lysine-specific demethylase 5A (KDM5A), an enzyme that removes activating H3K4 di- and trimethylation marks, plays critical roles in controlling transcription and chromatin architecture, yet its biological functions largely remain uncharacterized, particularly in the context of human cancer. In the present study, we found that the KDM5A gene was significantly amplified and over-expressed in various human tumors, including breast cancer. Reducing the expression of KDM5A by shRNA knockdown inhibited proliferation of KDM5A-amplified breast cancer cells. More importantly, we demonstrated that KDM5A over-expression was associated with breast cancer drug resistance. Furthermore, knockdown of KDM5A gene expression altered H3K4 methylation and induced upregulation of CDK inhibitors as well as genes mediating apoptotic cell death. Taken together, our study strongly links KDM5A histone demethylase activity to breast cancer proliferation and drug resistance, and suggests KDM5A is a potential target for breast cancer therapy.
Keywords: KDM5A, histone demethylases, gene amplification, drug-resistance
Introduction
Cancer has been traditionally viewed as a genetic disorder. However, it is increasingly apparent that epigenetic alterations, including histone modifications, DNA methylation, and microRNA dysregulation, play fundamental roles in cancer initiation and progression. Specifically, the use of systematic genome-wide discovery efforts has unexpectedly revealed a high frequency of cancer-specific alterations in genes involved in epigenetic histone modification in multiple tumor types [1-3]. The identification of these epigenetic modifier genes has raised important questions regarding the mechanisms by which they contribute to malignant transformation and progression. Furthermore, a better understanding of the intertwined relationship between genetic and epigenetic alterations in tumorigenesis is indisputably important for the development of new prognostic markers and therapeutic targets.
The epigenetic modifier gene KDM5A (Lysine-specific demethylase 5A, also known as RBP2 and JARID1A), encodes a lysine-specific histone 3 demethylase [4-6]. Histone lysine methylation is a principal chromatin-regulatory mechanism that influences fundamental nuclear processes [7]. Lysine (K) residues on the tails of histone H3 can accept up to three methyl groups to form mono-, di-, and trimethylated derivatives (me1, me2, and me3, respectively). Depending on the site and degree of methylation, lysine methylation can have different transcriptional and biological outcomes. Specifically, KDM5A can function as a transcriptional repressor through the demethylation of tri- and dimethylated histone H3 at lysine 4 (H3K4) active marks [4-6]. KDM5A has been shown to regulate the expression of multiple genes and has also been shown to be required for normal development [4-6]. Indeed, KDM5A was originally identified as the retinoblastoma-binding protein and was implicated in regulation of retinoblastoma target genes [8]. Mutations in the Drosophila KDM5A homolog lid result in severe defects in cell growth and differentiation and are homozygous lethal [9]. More recently, several studies have shown that dysregulation of KDM5A is associated with human cancer. KDM5A is over expressed in gastric cancer, and its inhibition triggers cellular senescence of gastric cancer cells [10]. In acute myeloid leukemia (AML), KDM5A has been shown to form a fusion protein with a nucleoporin 98 gene (NUP98), and overexpression of this fusion protein alone is sufficient to induce AML in murine models. Furthermore, genetic ablation of KDM5A decreases tumor formation and prolongs survival in pRB-defective mice [11]. Very recently, KDM5A was found to be a critical epigenetic factor for the development of drug resistance in lung cancer cells [12]. However, the role played by KDM5A in breast cancer remains poorly understood. In this study, we observed a significant amplification and over-expression of the KDM5A gene in various tumors, including breast cancer. We found that breast cancer cells with KDM5A gene amplification had intrinsic drug resistance properties and knocking down KDM5A with shRNAs improved the efficacy of epidermal growth factor receptor (EGFR) inhibitors against these breast cancer cells. Furthermore, increasing the expression of KDM5A in breast cancer led to global histone methylation level changes and altered the expression of a subset of key genes, including tumor suppressor p21 and apoptosis effector BAK1. Our findings suggest that genetic alteration of KDM5A may play a critical role in the pathogenesis of breast cancer.
Material and methods
Genomic array CGH
The isolation and culture of the SUM series of human breast cancer cell lines, Colo824, HCC1937, HCC1428, ZR-75-1 and non-tumorigenic mammary epithelial MCF10A cells have been described in detail previously [13,14]. Genomic array CGH experiments were done using the Agilent 244K human genome CGH microarray chip (Agilent Technologies, Palo Alto, CA, USA) as described previously [13]. Briefly, for each array, female DNA (Promega, Madison, WI) was used as a reference sample and labelled with Cy-3. The samples of interest were each labelled with Cy-5. Agilent’s CGH Analytics software was used to calculate various measurement parameters, including log2 ratio of total integrated Cy-5 and Cy-3 intensities for each probe. Array data have been posted at the NCBI GEO database (GEO accession: GSE28989, GSM718287, GSM718288, GSM718289, GSM718290).
Lentivirus-mediated KDM5A shRNA knockdown
KDM5A knockdown was achieved by using the Expression Arrest GIPZ lentiviral shRNAmir system (OpenBiosystems). Lentivirus was produced by transfecting 293FT cells with the combination of the lentiviral expression plasmid DNA and viral packaging mix (OpenBiosystems). Cells were infected with the virus by incubating with the mixture of growth medium and virus-containing supernatant (1:1 ratio), supplemented with polybrene at a final concentration of 5μg/ml. An equal volume of fresh growth medium was added after 24 hours and selection of stable cells was started after 48 hours. Cells expressing shRNA were selected with puromycin for 2-4 weeks for functional studies (cell proliferation and colony formation assays) and for 4 to 10 days after infection for protein and RNA extraction.
Examination of cell growth
Cell growth was assessed by using a Coulter counter or the MTT assay [15]. For the MTT assay, cells were seeded in 6-well plates at a density of 2×104 cells per well and allowed to attach overnight. At designated time points, thiazolyl blue tetrazolium bromide (MTT, Sigma Aldrich) was added to each well of cells (final 0.5 mg/ml) and incubated for 3-5 hours at 37° C. After removing the growth medium, DMSO was added to solubilize the blue MTT-formazan product and the samples were incubated for an additional 30 minutes at room temperature. Absorbance of the solution was read at a test wavelength of 570nm against a reference wavelength of 650nm.
Cell growth in soft agar
Soft agar assays were performed as previously described [13]. Briefly, dishes were coated with a 1:1 mix of the appropriate 2x medium for the cell line being studied and 1% Bactoagar. ZR-75-1, HCC1937 and SUM149 cells transduced with a control (Ctrl-sh) or with KDM5A shRNAs (sh#4 and sh#5) were plated at 1x105 cells/well in a 1:1 mixture of appropriate 2x medium and 0.3% Bactoagar. Cells were fed 3 times/week for 3-4 weeks, stained with 500μg/ml p-iodonitrotetrazolium violet (Sigma-Aldrich, St Louis, MO, USA) overnight, photographed (left panel), and counted with an automated mammalian cell colony counter (Oxford Optronix GELCOUNT, Oxford, United Kingdom).
Immunoblotting and antibodies
Whole cell lysates were prepared by scraping cells from the dishes into cold RIPA lysis buffer and sonicating for 10 seconds. After centrifugation at high speed in the cold, protein content was estimated with the Bradford method. A total of 20-100μg of total cell lysate was resolved by SDS-PAGE and transferred onto PVDF membrane. Antibodies used in the study included anti-KDM5A (Bethyl Laboratories A300-897A, Montgomery, TX, USA) and anti-β-Actin (Sigma-Aldrich A5441, St Louis, MO, USA), anti-phospho-EGFR (Tyr1068) antibody (Cell Signaling #2234, Danvers, MA, USA), anti-EGFR antibody (Cell Signaling #D38B1), anti-H3K4me3 (Abcam ab8580, Cambridge, MA, USA), p21 (Cell Signaling #3814) and BAK1 Cell Signaling #2947) antibodies.
Statistical analysis
Kendall’s tau was used to assess the statistical significance of the association between copy number and expression for each gene. Holm’s step-down procedure was used to adjust significance level for the large number of estimates to reduce the likelihood of false positive results. We used P = 0.01 as a cut-off for a statistically significant association between copy number and expression. For analyzing the results of cell growth, a two-tailed independent Student’s t-test was performed. A value of P < 0.05 was considered statistically significant.
Results
KDM5A is significantly amplified and over expressed in human tumors
To identify genomic aberrations in human breast cancer, we first performed genomic PCR and Agilent oligonucleotide array-based comparative genomic hybridization (CGH) on a panel of breast cancer cell lines and 50 primary human breast cancers. We observed that the KDM5A gene is located within a focal peak region (12p13.3) of gain/amplification in approximately 15% of breast cancers (Figure 1A). Of the fifty-one breast cancer lines examined, nine also showed KDM5A gain/amplification: Colo824, ZR-75-1, HCC1937, HCC1428, SUM-149, HCC3153, HCC2185, HCC1187 and HBL100. To obtain further support for the involvement of KDM5A amplification in human tumors, we searched the published array-CGH database that contains a collection of 3131 copy-number profiles across different solid and liquid cancers. Using the CGH analysis program, Genomic Identification of Significant Targets in Cancer (GISTIC), we saw a significant gain/amplification (~23%) of KDM5A across the entire data set of 3131 tumors [16]. Thus, KDM5A is significantly amplified in various tumors, including breast cancer. To measure expression levels of KDM5A, we performed quantitative RT-PCR (qRT-PCR) and Western blot assays in our panel of breast cancer cell lines. As expected, cell lines with KDM5A gene gain/amplification, Colo824, ZR-75-1, HCC1937, HCC1428 and SUM149 cells, showed higher mRNA and protein levels of KDM5A than the ones without the gene amplification (P<0.001) (Figure 1B and C). Thus, KDM5A gene amplification correlates with increased expression at both mRNA and protein levels in a subset of breast cancer cells.
Figure 1.
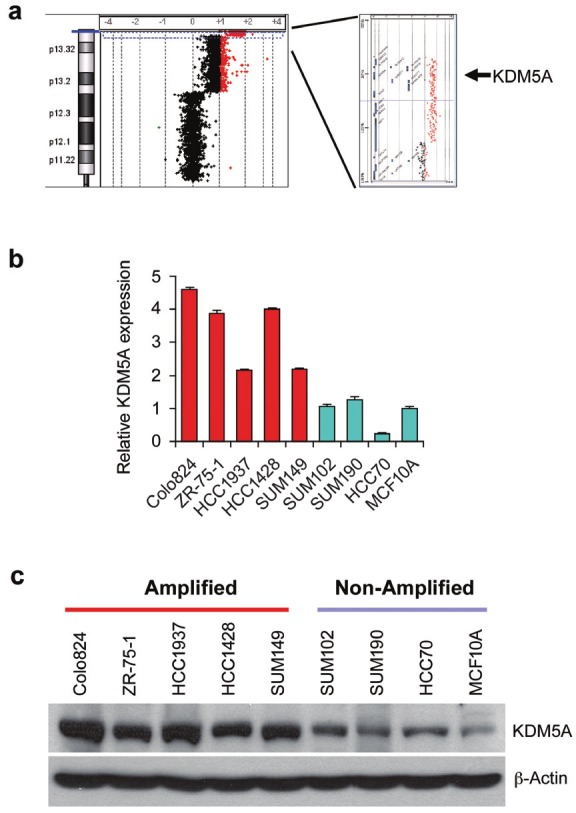
KDM5A is amplified and over-expressed in human breast cancer. (A) The representative array-CGH image showing chromosome 12p and KDM5A amplification in one breast cancer sample. (B) The mRNA expression level of KDM5A was examined by qRT-PCR assays in breast cancer cells with KDM5A gain/amplification (Colo824, ZR-75-1, HCC1937, HCC1428 and SUM149) or without the gain/amplification (SUM102, SUM190 and HCC70). mRNA expression level in the MCF10A cells was arbitrarily set as 1. Significance was set as P<0.05 by the student’s t-test (P<0.05). (C) KDM5A protein levels were analyzed by Western blot in eight breast cancer cell lines with or without gene amplification, as well as in MCF10A control cells.
Knockdown of KDM5A inhibits proliferation of KDM5A amplified breast cancer cells
To assess the contribution of endogenous KDM5A to breast cancer transformation, we knocked down KDM5A using a shRNA approach in breast cancer cells with or without KDM5A amplification. We obtained five pGIPZ-KDM5A shRNA expression constructs, and identified the two that most effectively knocked down KDM5A expression in ZR-75-1, HCC1937, SUM149 and SUM102 cells (Figure 2A). KDM5A knockdown caused significant growth inhibition of ZR-75-1, HCC1937 and SUM149 cells, all of which harbor KDM5A amplification (p<0.05). In contrast, there was no significant inhibition on the growth of SUM102 cells or the non-tumorigenic human mammary epithelial cells MCF10A, neither of which harbors the KDM5A gene amplification (Figure 2B). Furthermore, knockdown of KDM5A suppressed anchorage-independent growth of ZR-75-1, HCC1937 and SUM-149 cells (Figure 2C). Taken together, these data suggest that KDM5A may play an important role in the in vitro proliferation and maintenance of transformed phenotypes of breast cancer cells with KDM5A gene amplificaiton.
Figure 2.

Reducing KDM5A expression by shRNA knockdown resulted in decreased cell proliferation and colony formation in soft agar. (A) Knockdown of KDM5A in four breast cancer cell lines with two different shRNAs was confirmed by Western blot assays. (B) shRNA-mediated knockdown of KDM5A inhibits cell growth in breast cancer cells with gene amplification. Cells (ZR-75-1, HCC1937 and SUM149 with KDM5A amplification, SUM102 without the amplification, as well as non-tumorigenic MCF10A) infected with control (Ctrl-sh) or KDM5A shRNAs (sh#4 and sh#5) were plated at equal density and selected with puromycin for 4 weeks. Surviving cells were stained with Crystal Violet (left panel) or counted (right panel). Relative growth was shown as the mean ± SD of triplicate determinations (*P<0.05 and ** P<0.01, Student’s t test). (C) Knockdown of KDM5A impeded the anchorage-independent growth of breast cancer cells. Relative colony number (right panel) was shown as the mean ± SD of triplicate determinations (*P<0.05 and ** P<0.01, Student’s t test).
KDM5A is strongly associated with breast cancer drug resistance
KDM5A was recently identified as an important factor that is positively associated with EGFR inhibitor (erlotinib)-resistant phenotypes in lung cancer [12]. EGFR is over expressed in approximately 60% of basal breast cancers and correlates with poor prognosis, but has yet to emerge as a good therapeutic target in basal breast cancer [17]. We therefore sought to examine the EGFR family drug sensitivities of basal breast cancer cell lines with or without KDM5A gene amplification and overexpression. We found that HCC1937 and SUM149 cells (with KDM5A amplification) exhibited significantly higher EGFR inhibitor IC50 values as compared to SUM102 and MCF10A cells (without KDM5A amplification), although all cell lines expressed high-levels of EGFR protein (Figure 3A and data not shown). Next, we treated HCC1937, SUM149 and SUM102 breast cancer cell lines as well as the MCF10A line with 2μM or 4μM erlotinib for six, nine, twelve, and thirty days. Western blot with anti-phospho-EGFR (P-1068) antibody showed that erlotinib suppressed EGFR kinase activity in all of the treated cell lines (Figure 3A). As seen in Figure 3B, a subpopulation in the three cancer cell lines survived the drug treatments, even beyond thirty days. As expected based on the IC50 values of the EGFR inhibitors, HCC1937 and SUM149 cells had more drug-tolerant cells than SUM102 cells, whereas no drug-tolerant MCF10A cells were detected after treatment for thirty days (Figure 3B). These data suggest that breast cancer cells with KDM5A gene amplification are intrinsically more resistant to EGFR inhibitors than cells without KDM5A amplification.
Figure 3.

KDM5A is associated with breast cancer drug resistance. (A) EGFR inhibitor erlotinib (ERL) suppressed EGFR kinase activity in HCC1937, SUM149 and SUM102 breast cancer cell lines and MCF10A control line. Cells were treated with 4 μM erlotinib or vehicle for 1 hour. Protein extracts were immunoblotted with anti-phospho-EGFR (Tyr1068) and anti-EGFR antibodies. (B) Breast cancer cell lines HCC1937, SUM149 and SUM102 as well as the control MCF10A line were plated and left either untreated (Ctrl) or treated with 2 and 4 μM erlotinib for 30 days. Cells were fixed and stained with Crystal Violet or counted. Each experiment was performed in triplicate, and a representative image is presented. (C) Drug-tolerant subpopulation of SUM149 and SUM102 cells had increased KDM5A expression. Cells were plated and treated with 4 μM erlotinib for 6, 9 and 30 days with media/drug changes every two days and then isolated total RNA and protein. Protein extracts were immunoblotted with a KDM5A antibody. (D) KDM5A knockdown reduced the number of drug-tolerant cells in SUM149 and HCC1937. Stable KDM5A-knockdown and control HCC1937 and SUM149 cells were treated with the indicated concentration of erlotinib for 30 days. Cell counting was shown as the mean ± SD of triplicate determinations (*P<0.05 and ** P<0.01, Student’s t test)
To determine whether the drug-tolerant subpopulation has increased KDM5A expression, we treated SUM149 and SUM102 cells with erlotinib for six, nine and thirty days and then isolated total RNA and protein. qRT-PCR and immunoblotting experiments revealed that both mRNA and protein expression of KDM5A were increased in drug-tolerant cells as compared to parental control cells (Figure 3C). Thus, similar to the study done in lung cancer cells, KDM5A expression underwent up-regulation in the drug-tolerant subpopulations of breast cancer cells [12]. Next, to determine whether suppressing KDM5A in breast cancer cells circumvents erlotinib resistance, we challenged stable KDM5A-knockdown HCC1937 and SUM149 cell lines with erlotinib for thirty days. KDM5A knockdown significantly reduced the number of drug-tolerant cells in both cancer cell lines (Figure 3D). Taken together, our data reveal a strong association between KDM5A expression and breast cancer drug resistance.
Knockdown of KDM5A alters H3K4 methylation and induces up-regulation of CDK inhibitors and genes mediating apoptotic cell death
Because KDM5A is the key histone demethylase that specifically targets H3K4me3 and me2 active marks, the possibility exists that knocking down KDM5A in breast cancer cells would result in increased H3K4me3/me2 levels, and consequently the up-regulation of a specific set of genes. Thus, we first sought to examine the global H3K4me3 methylation status in KDM5A-knockdown SUM149 cells. As expected, shRNA-mediated inhibition of KDM5A expression in SUM149 cells resulted in increased H3K4me3 levels (Figure 4). Next, to identify genes with altered expression upon KDM5A knockdown, we performed a genomewide expression profiling analysis. Knockdown of KDM5A in SUM149 cells yielded 208 upregulated genes and 188 down-regulated genes with at least a two-fold change relative to control (data not shown). Previous studies demonstrated that KDM5A can inhibit the expression of cyclin-dependent kinase inhibitor p21 via its H3K4 demethylase activity in gastric cancer cells [10]. Our expression profiling analysis and Western blot experiments (Figure 4A), which showed p21 up-regulation with KDM5A knockdown, corroborate this finding and suggest that p21 is a KDM5A target gene in breast cancer cells.
Figure 4.
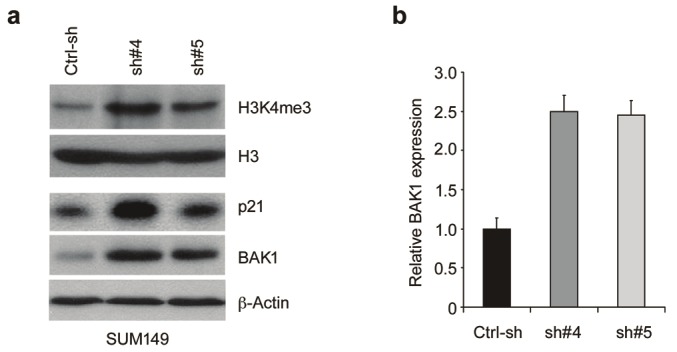
Knockdown of KDM5A altered H3K4 methylation and induced up-regulation of CDK inhibitors and genes mediating apoptotic cell death. (A) KDM5A was knocked down in SUM149 cells and the whole lysate was harvested for Western blot analysis. (B) mRNA levels of BAK1 were examined by real-time RT-PCR after knocking down KDM5A in SUM149 cells. The baseline for the cells infected with control shRNA was arbitrarily set as 1.
Bioinformatic analyses of the results obtained from the genome-wide expression profiling study were performed with the Pathway-Express (PE) and Onto-Express (OE) programs [18]. In SUM149 cells, the pathways most affected by KDM5A knockdown included those involved in the regulation of transcription, organismal development, oxidation reduction and apoptosis (data not shown). Of particular interest is the apparent inverse relationship in expression between KDM5A and BAK1 (BCL2-antagonist/killer 1). BAK1 plays a key role in trigging apoptosis and its altered expression may help explain the drug resistance phenotypes associated with KDM5A amplification and overexpression [19]. To validate these array-based observations, we examined the expression of BAK1 by quantitative RT-PCR and Western blot in SUM149 cells following KDM5A knockdown (Figure 4). Depletion of KDM5A in SUM149 cells resulted in up-regulation of BAK1, indicating that KDM5A regulates the expression of this target gene. Thus, KDM5A may regulate a subset of genes involved in various functional pathways in breast cancer.
Discussion
In the present study, we demonstrated that the H3K4 demethylase KDM5A is amplified and over expressed in various tumors, including breast cancer. Knockdown of KDM5A with shRNAs inhibited the growth of breast cancer cells harboring the KDM5A amplification. Furthermore, breast cancer cells with KDM5A gene amplification have intrinsic drug resistance properties and knocking down KDM5A improves the efficacy of EGFR inhibitors against these breast cancer cells. Our finding that KDM5A up-regulation alters H3K4 methylation status, and thus may repress the expression of a set of key genes including CDK inhibitors as well as genes mediating apoptotic cell death, provides a potential mechanism for KDM5A mediated drug resistance. Our study points to an important role for the histone demethylase KDM5A in human breast cancer, and this protein represents a potential target for the development of novel anticancer drugs.
A growing body of evidence indicates that amplification, translocation or mutation of histone methyltransferases and demethylases is linked to the development of many human cancers. For example, we originally identified and cloned the histone demethylase GASC1 gene from an amplified region at 9p24 in esophageal cancer [20]. Later studies showed GASC1 amplification in other tumor types, including lymphoma, medulloblastoma, lung and breast cancers [21-23]. We subsequently demonstrated that stable over-expression of GASC1 in the non-tumorigenic MCF10A cell line induces transformed phenotypes whereas knockdown in tumor cells inhibits proliferation, supporting a role for GASC1 as a transforming oncogene [13]. Houvras et al. revealed that the histone methyltransferase SETDB1 is recurrently amplified in melanoma and cooperates with oncogenic BRAF in accelerating oncogenesis [24]. Amplification and translocation of NSD1, 2 and 3 methyltransferase genes has been found in breast and lung cancers, and leukemia [25-29]. Very recently, Kuo et al. demonstrated that NSD2, via H3K36me2 catalysis, promotes transcription and cell transformation [30]. Here, we identified and investigated a frequently amplified region of DNA located on chromosome 12p13.3. Integration of copy number and gene expression data revealed the KDM5A gene as a candidate oncogene responsible for driving recurrent 12p13.3 amplification (data not shown). Furthermore, we validated the biologic effect of KDM5A upregulation by showing that KDM5A suppression impedes cell proliferation and anchorage-independent growth in breast cancer cell lines with KDM5A amplification. Our studies, together with others, indicate that genetic alteration in components of the histone modification machinery plays a central role in cancer initiation and progression.
Histone lysine methylation is a key regulator of gene transcription and chromatin architecture. In the case of H3K4 methylation, this mark is generally associated with active transcription [31]. KDM5A is capable of removing the H3K4me3/me2 mark from histones, which makes it a potential player in the downregulation of tumor suppressors. Indeed, previous studies revealed that KDM5A can inhibit the expression of p16, p21, and p27 via its H3K4 demethylase activity in gastric cancer cells [10]. In this study, we demonstated that upregulation of KDM5A alters H3K4 methylation status and may regulate a subset of genes, including p21 and BAK1, a protein that effects apoptosis-triggering cues [19]. Apoptosis is a predominant mechanism by which targeted or chemotherapeutic agents kill cancer cells. Genetic or epigenetic perturbations resulting in a defective execution of an apoptotic response could potentially result in drug-tolerant tumor cells [32]. Thus, although we cannot rule out the possibility that other target genes regulated by KDM5A are involved in drug resistance, our findings suggest that BAK1 might be an important downstream mediator of this phenotype. Furthermore, targeting histone demethylases is currently an active frontier in novel epigenetic drug development [33,34]. Given that KDM5A is amplified and over expressed in various tumors, and plays a critical role in mediating transforming and drug resistance phenotypes, KDM5A may represent a potentially excellent target for the development of novel anticancer drugs.
Acknowledgements
This work was supported by grants from the Department of Defense Breast Cancer Program (BC086177 and BC083945) to Zeng-Quan Yang, and a grant from the Susan G. Komen for the Cure Career Catalyst Grant (KG081416) to Julie L Boerner. We thank Dr. Stephen P Ethier for providing the SUM breast cancer cell lines and his continuous encouragement. We thank Michele L Dziubinski and Xiaogang Wang for technical assistance on the cell cultures.
References
- 1.Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fullgrabe J, Kavanagh E, Joseph B. Histone onco-modifications. Oncogene. 2011;30:3391–3403. doi: 10.1038/onc.2011.121. [DOI] [PubMed] [Google Scholar]
- 3.Varier RA, Timmers HT. Histone lysine methylation and demethylation pathways in cancer. Biochim Biophys Acta. 2011;1815:75–89. doi: 10.1016/j.bbcan.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Christensen J, Agger K, Cloos PA, Pasini D, Rose S, Sennels L, Rappsilber J, Hansen KH, Salcini AE, Helin K. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell. 2007;128:1063–1076. doi: 10.1016/j.cell.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 5.Lopez-Bigas N, Kisiel TA, Dewaal DC, Holmes KB, Volkert TL, Gupta S, Love J, Murray HL, Young RA, Benevolenskaya EV. Genome-wide analysis of the H3K4 histone demethylase RBP2 reveals a transcriptional program controlling differentiation. Mol Cell. 2008;31:520–530. doi: 10.1016/j.molcel.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasini D, Hansen KH, Christensen J, Agger K, Cloos PA, Helin K. Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and Polycomb-Repressive Complex 2. Genes Dev. 2008;22:1345–1355. doi: 10.1101/gad.470008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Defeo-Jones D, Huang PS, Jones RE, Haskell KM, Vuocolo GA, Hanobik MG, Huber HE, Oliff A. Cloning of cDNAs for cellular proteins that bind to the retinoblastoma gene product. Nature. 1991;352:251–254. doi: 10.1038/352251a0. [DOI] [PubMed] [Google Scholar]
- 9.Gildea JJ, Lopez R, Shearn A. A screen for new trithorax group genes identified little imaginal discs, the Drosophila melanogaster homologue of human retinoblastoma binding protein 2. Genetics. 2000;156:645–663. doi: 10.1093/genetics/156.2.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeng J, Ge Z, Wang L, Li Q, Wang N, Bjorkholm M, Jia J, Xu D. The histone demethylase RBP2 Is overexpressed in gastric cancer and its inhibition triggers senescence of cancer cells. Gastroenterology. 2010;138:981–992. doi: 10.1053/j.gastro.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Lin W, Cao J, Liu J, Beshiri ML, Fujiwara Y, Francis J, Cherniack AD, Geisen C, Blair LP, Zou MR, Shen X, Kawamori D, Liu Z, Grisanzio C, Watanabe H, Minamishima YA, Zhang Q, Kulkarni RN, Signoretti S, Rodig SJ, Bronson RT, Orkin SH, Tuck DP, Benevolenskaya EV, Meyerson M, Kaelin WG Jr, Yan Q. Loss of the retinoblastoma binding protein 2 (RBP2) histone demethylase suppresses tumorigenesis in mice lacking Rb1 or Men1. Proc Natl Acad Sci USA. 2011;108:13379–13386. doi: 10.1073/pnas.1110104108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, Wong KK, Brandstetter K, Wittner B, Ramaswamy S, Classon M, Settleman J. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu G, Bollig-Fischer A, Kreike B, van de Vijver MJ, Abrams J, Ethier SP, Yang ZQ. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene. 2009;28:4491–4500. doi: 10.1038/onc.2009.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu J, Liu S, Liu G, Dombkowski A, Abrams J, Martin-Trevino R, Wicha MS, Ethier SP, Yang ZQ. Identification and functional analysis of 9p24 amplified genes in human breast cancer. Oncogene. 2012;31:333–341. doi: 10.1038/onc.2011.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 16.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burness ML, Grushko TA, Olopade OI. Epidermal growth factor receptor in triple-negative and basal-like breast cancer: promising clinical target or only a marker? Cancer J. 2011;16:23–32. doi: 10.1097/PPO.0b013e3181d24fc1. [DOI] [PubMed] [Google Scholar]
- 18.Khatri P, Sellamuthu S, Malhotra P, Amin K, Done A, Draghici S. Recent additions and improvements to the Onto-Tools. Nucleic Acids Res. 2005;33:W762–765. doi: 10.1093/nar/gki472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cartron PF, Juin P, Oliver L, Meflah K, Vallette FM. Impact of proapoptotic proteins Bax and Bak in tumor progression and response to treatment. Expert Rev Anticancer Ther. 2003;3:563–570. doi: 10.1586/14737140.3.4.563. [DOI] [PubMed] [Google Scholar]
- 20.Yang ZQ, Imoto I, Fukuda Y, Pimkhaokham A, Shimada Y, Imamura M, Sugano S, Nakamura Y, Inazawa J. Identification of a novel gene, GASC1, within an amplicon at 9p23-24 frequently detected in esophageal cancer cell lines. Cancer Res. 2000;60:4735–4739. [PubMed] [Google Scholar]
- 21.Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, Mack S, Kongkham PN, Peacock J, Dubuc A, Ra YS, Zilberberg K, McLeod J, Scherer SW, Sunil Rao J, Eberhart CG, Grajkowska W, Gillespie Y, Lach B, Grundy R, Pollack IF, Hamilton RL, Van Meter T, Carlotti CG, Boop F, Bigner D, Gilbertson RJ, Rutka JT, Taylor MD. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. 2009;41:465–472. doi: 10.1038/ng.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vinatzer U, Gollinger M, Mullauer L, Raderer M, Chott A, Streubel B. Mucosa-associated lymphoid tissue lymphoma: novel translocations including rearrangements of ODZ2, JMJD2C, and CNN3. Clin Cancer Res. 2008;14:6426–6431. doi: 10.1158/1078-0432.CCR-08-0702. [DOI] [PubMed] [Google Scholar]
- 23.Italiano A, Attias R, Aurias A, Perot G, Burel-Vandenbos F, Otto J, Venissac N, Pedeutour F. Molecular cytogenetic characterization of a metastatic lung sarcomatoid carcinoma: 9p23 neocentromere and 9p23-p24 amplification including JAK2 and JMJD2C. Cancer Genet Cytogenet. 2006;167:122–130. doi: 10.1016/j.cancergencyto.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 24.Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ, Ferre F, Bourque C, Burke CJ, Turner L, Uong A, Johnson LA, Beroukhim R, Mermel CH, Loda M, Ait-Si-Ali S, Garraway LA, Young RA, Zon LI. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471:513–517. doi: 10.1038/nature09806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia MJ, Pole JC, Chin SF, Teschendorff A, Naderi A, Ozdag H, Vias M, Kranjac T, Subkhankulova T, Paish C, Ellis I, Brenton JD, Edwards PA, Caldas C. A 1 Mb minimal amplicon at 8p11-12 in breast cancer identifies new candidate oncogenes. Oncogene. 2005;24:5235–5245. doi: 10.1038/sj.onc.1208741. [DOI] [PubMed] [Google Scholar]
- 26.Gelsi-Boyer V, Orsetti B, Cervera N, Finetti P, Sircoulomb F, Rouge C, Lasorsa L, Letessier A, Ginestier C, Monville F, Esteyries S, Adelaide J, Esterni B, Henry C, Ethier SP, Bibeau F, Mozziconacci MJ, Charafe-Jauffret E, Jacquemier J, Bertucci F, Birnbaum D, Theillet C, Chaffanet M. Comprehensive profiling of 8p11-12 amplification in breast cancer. Mol Cancer Res. 2005;3:655–667. doi: 10.1158/1541-7786.MCR-05-0128. [DOI] [PubMed] [Google Scholar]
- 27.Yang ZQ, Albertson D, Ethier SP. Genomic organization of the 8p11-p12 amplicon in three breast cancer cell lines. Cancer Genet Cytogenet. 2004;155:57–62. doi: 10.1016/j.cancergencyto.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 28.Yang ZQ, Streicher KL, Ray ME, Abrams J, Ethier SP. Multiple interacting oncogenes on the 8p11-p12 amplicon in human breast cancer. Cancer Research. 2006;66:11632–11643. doi: 10.1158/0008-5472.CAN-06-2946. [DOI] [PubMed] [Google Scholar]
- 29.Pole JC, Courtay-Cahen C, Garcia MJ, Blood KA, Cooke SL, Alsop AE, Tse DM, Caldas C, Edwards PA. High-resolution analysis of chromosome rearrangements on 8p in breast, colon and pancreatic cancer reveals a complex pattern of loss, gain and translocation. Oncogene. 2006;25:5693–5706. doi: 10.1038/sj.onc.1209570. [DOI] [PubMed] [Google Scholar]
- 30.Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, Xi Y, Park BH, Shi X, Garcia BA, Li W, Gozani O. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell. 2011;44:609–620. doi: 10.1016/j.molcel.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eissenberg JC, Shilatifard A. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol. 2010;339:240–249. doi: 10.1016/j.ydbio.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilting RH, Dannenberg JH. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist Updat. 2012 doi: 10.1016/j.drup.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 33.Natoli G, Testa G, De Santa F. The future therapeutic potential of histone demethylases: A critical analysis. Curr Opin Drug Discov Devel. 2009;12:607–615. [PubMed] [Google Scholar]
- 34.Grant S. Targeting histone demethylases in cancer therapy. Clin Cancer Res. 2009;15:7111–7113. doi: 10.1158/1078-0432.CCR-09-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]