

Abstract
Background
Oxidative stress and inflammation promote the development and progression of chronic kidney disease. Oxidative stress is associated with depletion of tissue glutathione (GSH), the most abundant endogenous intracellular antioxidant, but degradation of oral GSH by digestive enzymes limits its therapeutic use. We hypothesized that GSH repletion with F1, a novel oral GSH precursor containing cystine as a cysteine carrier, would restore tissue GSH and attenuate oxidative stress and inflammation, and thereby reduce the severity of interstitial nephropathy in chronic renal failure (CRF).
Methods
Male Sprague-Dawley rats (n=5-8) were assigned to 3 groups: Control (regular rat chow), CRF (rat chow containing 0.7% adenine), and F1-treated CRF (rat chow containing 0.7% adenine and F1, 0.5g/kg/day) for 2-weeks. Animals were switched to regular chow and euthanized after 2 additional weeks.
Results
Consumption of 0.7% adenine-containing diet caused azotemia; severe kidney swelling; heavy tubular and glomerular damage; massive tubulointerstitial nephropathy; impaired urinary concentrating capacity; severe anemia; increased markers of oxidative stress, plasma oxidized glutathione disulfide (GSSG); reduced GSH/GSSG ratio and manganese superoxide dismutase; increased expression of inflammatory mediators (cyclooxygenase-2, cytoplasmic NF-κB, p-IκBα, nuclear NF-κB p65), and 3-nitrotyrosine, p<0.05. Co-treatment with F1 significantly attenuated tubulointerstitial inflammation and edema, improved urinary concentrating capacity, azotemia and anemia, and normalized markers of tissue oxidative and nitrosative stress, p<0.05.
Conclusions
The novel oxidative stress modulator, F1, markedly attenuated oxidative stress indicators, inflammation, renal injury and dysfunction in the rat model of CRF. Studies to determine the effects of F1 in other models of acute and CRF are warranted.
Keywords: Glutathione precursor, oxidative stress modulator, inflammation, glutathione disulfide, p-IκBα, NF-κB, NF-κB p65, hematocrit
Introduction
Chronic kidney disease (CKD) is a global public health problem [1] associated with rising costs and high rates of premature morbidity and mortality [2]. In general, CKD is complicated by accelerated arteriosclerosis and cardiovascular risk factors (both traditional and non-traditional) [3], and therefore strategies directed at treating CKD may be beneficial in addressing these issues. CKD due to diabetes and hypertension, which account for approximately 75% of all cases of end-stage renal disease in the United States [4], are characterized by tubulointerstitial fibrosis and glomerulosclerosis [5-8].
Oxidative stress and inflammation are key driving forces in the development and progression of CKD and its sequelae including hypertension, interstitial fibrosis, uremia and anemia [9]. Thus, CKD in humans can be reproduced in the rodent model of chronic renal failure (CRF) by adenine; and this CRF model provides a unique opportunity to study the pathogenesis and effects of interventions that target disease progression due to the presence of chronic progressive tubulointerstitial nephritis, metabolic abnormalities and declining renal function [9].
Oxidative stress in CRF is associated with depletion of glutathione (GSH) which is the most abundant endogenous intracellular antioxidant that exists in tissues in millimolar concentrations and in bodily fluids in micromolar concentrations [10]. GSH or γ-glutamylcysteinylglycine is synthesized in cells from the amino acids glycine, L-glutamic acid and L-cysteine. L-cysteine provides a sulfhydryl group and acts as a proton donor to facilitate the biologic activity of glutathione. Two GSH moieties (2GSH) reduce free radicals and peroxides during conversion to its oxidized form glutathione disulfide (GSSG) [11,12]. Under normal physiologic conditions, the majority of the GSH pool exists as reduced GSH and a small portion is in the GSSG state, such that a reduced GSH/GSSG ratio signifies the presence of oxidative stress. Thus, GSH exerts potent antioxidant actions by scavenging reactive oxygen species (ROS) [13] and serving as the substrate for major antioxidant enzymes.
Although, the administration of antioxidants might be a seemingly suitable antidote to the detrimental effects of oxidative stress in CRF, high doses of conventional antioxidants as oral supplements have demonstrated variable benefit in attenuating uremic complications [14-18]. Indeed, GSH is an attractive candidate for the treatment of oxidative stress, however, its enzymatic degradation precludes its therapeutic use by oral supplementation [19]. Recently, the oral administration of a novel oxidative stress modulator, F1 containing cystine as a cysteine carrier has been shown to ameliorate oxidative stress and its consequences both in vitro [20] and in vivo [21], possibly in part by increasing intracellular GSH levels. The present study was designed to test the hypothesis that GSH repletion with F1 administration may attenuate oxidative stress and inflammation and thereby ameliorate the severity of tubulointerstitial nephropathy in the adenine-induced rat model of CRF.
Materials and methods
Antioxidant
The novel antioxidant, F1 (FT061452/RE39734™), is designed as a GSH precursor in which cysteine is replaced with cystine and selenomethonine [22]. F1, a tasteless and odorless powder was provided by Proimmune®, Rhinebeck, NY with the following formulation: L-cystine: 99.68 mg, glycine: 199.39 mg, selenomethionine: 1.54 mg and L-glutamine: 199.39 mg per 500 mg.
Animals
The experimental protocol was approved by the Institutional Animal Care and Use Committee of the University of California, Irvine, CA. Male Sprague-Dawley rats [23] with an average body weight of 250-300 g (Harlan Sprague-Dawley Inc., Indianapolis, IL) were housed in a climate-controlled vivarium with 12-h day and night cycles. Male SD rats were randomly assigned to: Control (fed regular rat chow), CRF (fed rat chow containing 0.7% adenine for 2 weeks), and F1-treated CRF (fed rat chow containing 0.7% adenine and F1, 0.5 g/Kg/day for 2 wks) groups and followed for an additional 2 weeks on regular diet (Prolab, RMH 2500), which provided 59.1% of the energy as carbohydrate, 12.1% as fat and 28.8% as protein. Animals were then euthanized by exsanguination using cardiac puncture under general anesthesia (sodium pentobarbital, 50 mg/kg IP).
Blood pressure was determined by tail-cuff plethysmography (CODA2, Kent Scientific Corporation, Torrington, CT) at 2 and 4 weeks. Conscious rats were placed in a restrainer on a warming pad and allowed to rest for 15min before blood pressure measurements. A tail-cuff was inflated around the rat tails and released several times to allow the animal to be conditioned to the procedure. Blood pressures were measured at 2 and 4 weeks.
Biochemical determination
Animals were placed in metabolic cages for timed urine collection for protein (Chondrex Inc., Redmond, WA) and creatinine determination and protein-to-creatinine ratio calculation. Plasma urea (Bioassay Systems, Hayward, CA), and Jaffe alkaline picrate assay-based creatinine (Bioassay Systems, Hayward, CA) for creatinine clearance were measured per manufacturer’s instructions. All laboratory determinations were made on samples taken between 9-11 am without overnight food restriction.
Histology
Kidneys were removed and weighed, and a section was fixed in 10% formalin, dehydrated in alcohol series, cleared in toluene and embedded in paraffin. The remainder was cleaned with PBS, snap-frozen in liquid nitrogen, and stored at -70°C until processed. Light microscopy studies were done on paraffin-embedded kidney sections stained with hematoxylin and eosin (H & E) or periodic acid-Schiff (PAS) and examined under a photomicroscope (Nikon Eclipse, Japan). Proliferation, mesangial expansion, and glomerulosclerosis (segmental collapse and obliteration of capillary lumen with accumulation of PAS-positive material in part or the whole glomerular tuft, with or without adhesion to Bowman’s capsule) were evaluated. Severity of glomerulosclerosis was evaluated using an index score that includes the percent of glomeruli showing sclerosis and the extension of the glomerulosclerosis within the glomeruli with a grade of 0 to +4, as previously described [24]. Briefly, glomeruli were graded from 0 to +4 as follows: grade 0, normal; grade +1, <25% involvement of the glomerular tuft; grade +2, 25% to 50% involvement of the glomerular tuft; grade +3, 50% to 75%; and grade +4, sclerosis occupying >75% of the glomerular tuft. The glomerulosclerosis score was obtained as follows: (1 × number of glomeruli with grade +1) + (2 × number of glomeruli with grade +2) + (3 × number of glomeruli with grade +3) + (4 × number of glomeruli with grade +4)/total number of glomeruli examined. All glomeruli suitable for analysis (range 50 to 61) were examined in each animal.
Tubulointerstitial damage (infiltration, fibrosis, tubular dilatation, or atrophy) was evaluated semi-quantitatively, in which the grading of 0 to 4 was done according to the extent of damaged tubulointerstitial area in the renal cortex as in previous investigations [25-27]: 0, normal; grade 1, <10%; grade 2, 10%-25%; grade 3, 25%-50%; grade 4, 50%-75%; and grade 5, 75% -100%. The extent of the damage was evaluated by visual selection of the injured areas in successive fields in the cortical and juxtamedullary areas of each animal using computer-assisted analysis of digitalized images (Nikon, NIS-Elements D 3.2 System Microscope and Microscope Digital Camera), with the GraphPad Prism 4 software. All histological determinations were done by a blinded observer.
Determination of reduced (GSH) and oxidized (GSSG)
Reduced (GSH) and oxidized (GSSG) levels were measured using the BIOXYTECH® GSH/GSSG-412™ assay kit (OXISResearch™, A division of Oxis Health Product, Inc., Portland, OR), as per manufacturer’s instructions. Briefly, reduced GSH and oxidized GSSG concentrations on separate tissue samples were measured by omission or addition of 1-methyl-2-vinlylpyridinium trifluromethanesulfonate, respectively and GSH/GSSG ratio was used as an indicator of redox balance.
Tissue and nuclear protein preparation and Western blot analyses
Frozen kidney tissue was processed for determination of cyclooxygenase-2 (COX-2), nuclear factor-kappa B (NF-κB; Santa Cruz, CA), Mn-superoxide dismutase (Mn-SOD; Millipore, Billerica, MA), and phosphorylated (p)-IkBα (Cell Signaling, Denver, USA) abundance. GAPDH (Sigma-Aldrich, MO, USA) and Histone H1 (1:200; Santa Cruz, CA, USA) were used as housekeeping genes. Briefly, kidney tissue was homogenized in 1 ml of RIPA buffer (50 mM TrisHCl pH7.4, 150 mM NaCl, 2 mM EDTA, 1% NP-40, 0.1% SDS) and protease inhibitor cocktail (Sigma). The crude extract was centrifuged at 2,000 × g at 4°C for 15min to remove tissue debris. Protein concentration of the supernatant was measured prior to each Western blot analysis using a BCA Protein Assay Kit purchased from Pierce Biotechnology (Rockford, Ill., USA) following the manufacturer’s protocol. Nuclear proteins were prepared from kidney tissues using the CelLytic NuCLEAR Extraction kit (Sigma Aldrich) as per manufacturer’s instructions for nuclear NF-κB p65 rabbit mAb (Cell Signaling, Denver, USA). Aliquots of tissue and nuclear extract containing 50 μg of protein were fractionated on 8 and 4–20% Bis-Tris Gels (Invitrogen, Calif., USA and Novex, San Diego, CA) at 120V for 2h. After electrophoresis, proteins were transferred to Hybond-enhanced chemiluminescence (ECL) membrane (Amersham Life Science, Arlington Heights, Ill., USA). The membrane was incubated for 1h in blocking buffer (1 × Tris-buffered saline, 0.1% Tween-20, 5% TBST nonfat dry milk) and 0.05%-then overnight in the TBST buffer containing the primary antibody. Membranes were washed three times for 5 min in 1 × 0.05% TBS, Tween-20 prior to 2h incubation in blocking buffer plus diluted horseradish peroxidase-linked anti-mouse or rabbit IgG at 1:2000 (Sigma) or 1:1000 dilution (Amersham Life Science). The washing procedures were repeated before the membranes were developed with ECL agents and subjected to autoluminography for 10secs to 5mins.
Statistical analysis
Statistical comparisons were done with multigroup ANOVA analysis and Tukey post-tests with the help of a commercial statistical program (InstatR, Graph Pad, San Diego, CA). Two-tailed p values of p<0.05 were considered statistically significant. Data are given as mean ± standard deviation [23].
Results
Two weeks after the animals were fed adenine-containing diet, they developed CRF with significant loss of body weight and modest but significant increase in systolic blood pressure (p<0.05), which was sustained until the end of the study (Table 1). At completion of the 4-week study, urine output increased 4.5-fold and plasma urea increased 1.7-fold compared to controls, p<0.05. F1 administration did not significantly alter body weight or systolic blood pressure but significantly reduced plasma urea 1.4-fold and urine volume 1.3-fold compared to CRF rats, p<0.05 (Table 1). Plasma creatinine increased 3.6-fold and creatinine clearance was reduced ~5.0-fold in CRF animals compared to controls. These parameters were improved 1.8- and 3.1-fold, respectively with co-administration of F1, but the changes did not reach statistical significance.
Table 1.
Physiologic parameters of animals
| Control (n = 5-7) | CRF (n = 6-8) | CRF + F1 (n = 7) | |
|---|---|---|---|
| Body Weight (g) | 470±19 | 421±13* | 410±39* |
| Systolic blood pressure (mm Hg) at 2 weeks | 124±7 | 147±10* | 158±10* |
| Systolic blood pressure (mm Hg) at 4 weeks | 119±9 | 139±11* | 134±14* |
| Plasma urea (mg/dl) | 44.65±9.28 | 96.45±19.73* | 68.74±15.90** |
| Plasma creatinine (mg/dl) | 0.29±0.06 | 1.03±0.48 | 0.56±0.28 |
| Urine volume (ml) | 9.6±2.1 | 43.9±6.5* | 34.0±2.0* , ** |
| Creatinine clearance (ml/min) | 2.57±0.49 | 0.5±0.18 | 1.55±0.85 |
| Protein/creatinine ratio (g/mg) | 6.2±0.9 | 8.1±2.1 | 9.2±4.3 |
| Hematocrit (%) | 46±2 | 31±2* | 36±2* |
Data are mean ± SD:
p<0.05 compared to Control;
p<0.05 compared to CRF.
Control: rats fed regular rat chow for 4 weeks, CRF: fed 0.7% adenine for 2 weeks followed by regular rat chow for 2 weeks, CRF + F1: fed 0.7% adenine + F1 (0.5g/kg/day) for 2 weeks followed by regular rat chow for 2 weeks.
As expected, the CRF animals had marked renal swelling, discoloration and deformity. F1 dramatically attenuated the observed gross morphologic injury to the kidneys of CRF rats, Figure 1. On H & E stains of kidney sections, both tubulointerstitium and glomeruli of control rats appeared normal, Figure 2 A and B, while CRF rats had considerable damage consisting of severe inflammatory cell infiltration, vascular congestion, interstitial fibrosis and adhesions of glomerular tuft to Bowman’s capsule, Figure 2C and D. These changes were improved in the animals treated with F1, Figure 2E and F.
Figure 1.
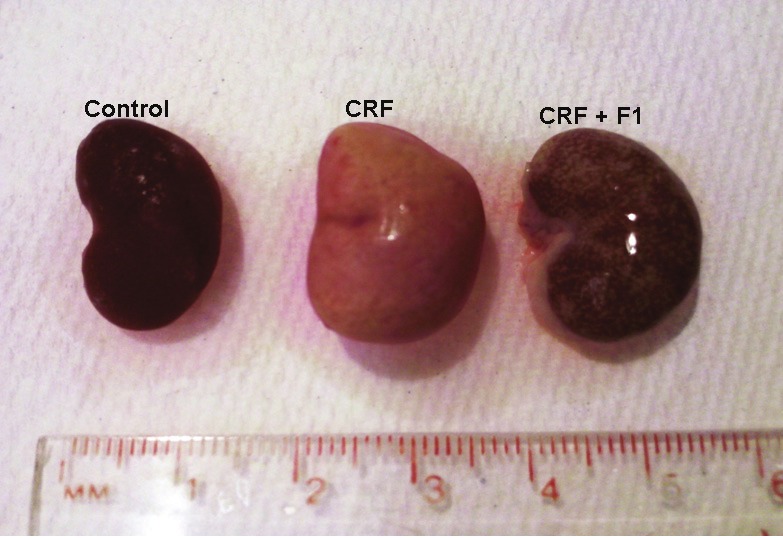
The figure shows gross massive swelling, discoloration and deformity of the kidneys following adenine-induced CRF at 4 weeks compared to Control rats fed regular rat chow. Simultaneous administration of F1 has prevented this renal damage. CRF: Chronic Renal Failure rats were fed 0.7% adenine for 2 weeks followed by regular rat chow for 2 weeks; CRF + F1: animals were fed 0.7% adenine and F1, 0.5g/kg/day simultaneously for 2 weeks followed by regular rat chow for 2 additional weeks.
Figure 2.
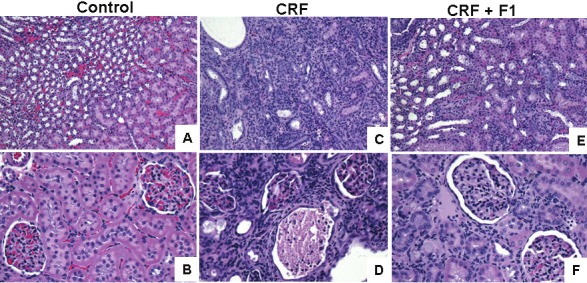
Hematoxylin & Eosin stains of kidney medulla and cortex. Tubulointerstitium (magnification × 100) and glomeruli (magnification × 200) appeared normal in Control rats, but were significantly abnormal in CRF rats. The changes were improved in CRF + F1-treated rats.
Renal injury was semi-quantified on PAS-stains of kidney sections. The tubulointerstitial damage of the adenine-treated CRF animals: grade 4, 75-100%, score 0.83 ± 0.08 was essentially undetectable in the controls and appreciably less in the CRF + F1 group: grade 3, 25-50%, score 0.47 ± 0.10, p<0.0001, Figure 3A. The tubulointerstitial damage was accompanied by prominent glomerulosclerosis: grade +3, 50-75%, score 0.65 ± 0.04 in the CRF animals compared to controls: grade 1, 0-25%, score 0.05±0.02, p<0.001 that was significantly attenuated in the CRF + F1 group: grade 2, 25-50%, score 0.43 ± 0.02, p<0.001, Figure 3B.
Figure 3.
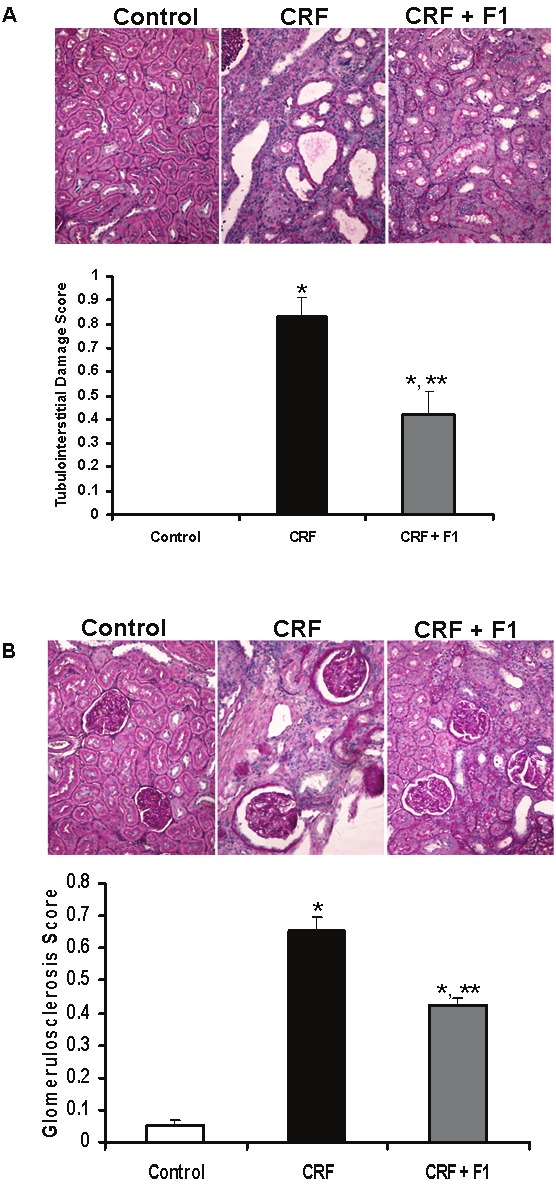
Representative micrographs show PAS-stained kidney sections from Control, CRF and CRF + F1-treated animals and quantitative data. A. Tubulointerstitial damage was undetectable in Control rats, prominent in CRF animals and significantly reduced in CRF + F1 rats. B. PAS staining of renal cortex showed significantly increased glomerulosclerosis in CRF rats compared to Control rats that was prevented with co-administration of F1. PAS: Periodic Acid Schiff. Magnification × 200, N=5-8, *p<0.001 vs. Control, **p<0.001 vs. CRF.
We examined the level of oxidative stress induced by adenine as determined by plasma GSSG level and GSH/GSSG ratio. Plasma GSSG was significantly increased in CRF compared to control (4.88 ± 1.79 vs. 2.43 ± 1.29), and plasma GSH/GSSG was also significantly reduced (2.48 ± 0.67 vs. 1.76 ± 0.30), p<0.05. These alterations were essentially blocked with administration of F1 (2.19 ± 0.56 and 3.32 ± 0.90, respectively), p<0.05, Figures 4A and 4B. As a result of the increased oxidative stress induced by adenine, we examined the natural mitochondrial antioxidant response reflected in the level of MnSOD. We observed a significant reduction in Mn-SOD/GAPDH expression compared to control (0.19 ± 0.08 vs. 0.51 ± 0.27) that was prevented with F1 treatment (0.57 ± 0.25) supporting the beneficial effect of F1, p<0.05, Figure 5.
Figure 4.

Assay of A. Plasma GSSG and B. GSH/GSSG ratio was performed on plasma from Control, CRF and CRF + F1-treated rats. F1 administration blocked the increase in GSSG and reduced GSH/GSSG ratio for individual animals. GSSG: oxidized glutathione disulfide, GSH: reduced glutathione, N=5-8, bars = means, *p<0.05.
Figure 5.
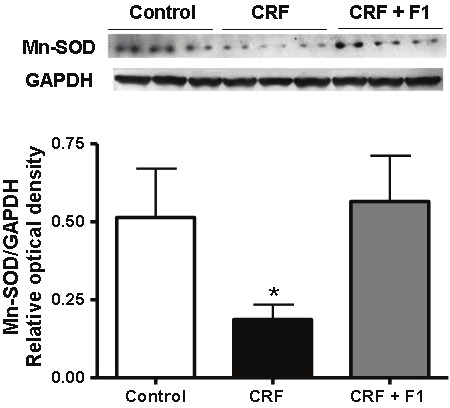
Representative Western blot and quantitative data of Mn-SOD and GAPDH protein expression from Control, CRF and CRF + F1 treated animal groups. Expression of Mn-SOD was significantly depressed in CRF and this reduction was prevented by F1 co-administration. Mn-SOD: manganese superoxide dismutase, N=5-8, *p<0.05
We investigated the effect of CRF and F1 administration on the oxidative stress-induced release of inflammatory mediators. Western blot analysis of whole kidney tissue demonstrated dramatically enhanced expression of COX-2 in CRF rats compared to control rats (1.40 ± 0.05 vs. 0.16 ± 0.06) that was normalized with F1 co-administration (0.02 ± 0.01), p<0.05, Figure 6.
Figure 6.
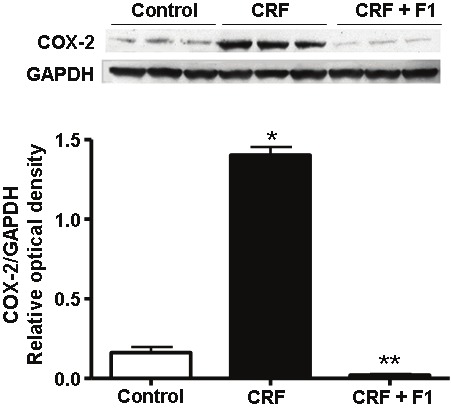
Representative Western blot and quantitative data of COX-2 and GAPDH protein expression from control, CRF and CRF + F1-treated animal groups. Expression of COX-2 was significantly augmented in CRF and this increase was prevented with F1 treatment. COX-2: cyclooxygenase-2, N=5-8, *p<0.05 compared to Control, **p<0.05 compared to CRF rats.
Oxidative stress is a primary activator of the heterodimeric NF-κB. Upon activation, release of cytoplasmic p-IκBα allows for translocation of NF-κB to the nucleus to enhance transcription of NF-kB-dependent genes and consequent release of proinflammatory cytokines in the pathogenesis of inflammation and fibrosis [28]. There were considerable increases in the expression of NF-κB (0.66 ± 0.25) and p-IκBα (0.57 ± 0.08) in adenine-induced CRF compared to control (0.24 ± 0.06 and 0.29 ± 0.04, respectively) that were significantly inhibited with F1 (0.18 ± 0.05 and 0.32 ± 0.04, respectively), p<0.05, Figure 7A and 7B. In addition, there was a large increase in nuclear NF-kB p65 subunit compared to control (0.68 ± 0.12 vs. 0.17 ± 0.05) that was significantly attenuated by F1 treatment (0.49 ± 0.02), p<0.05, Figure 7C.
Figure 7.
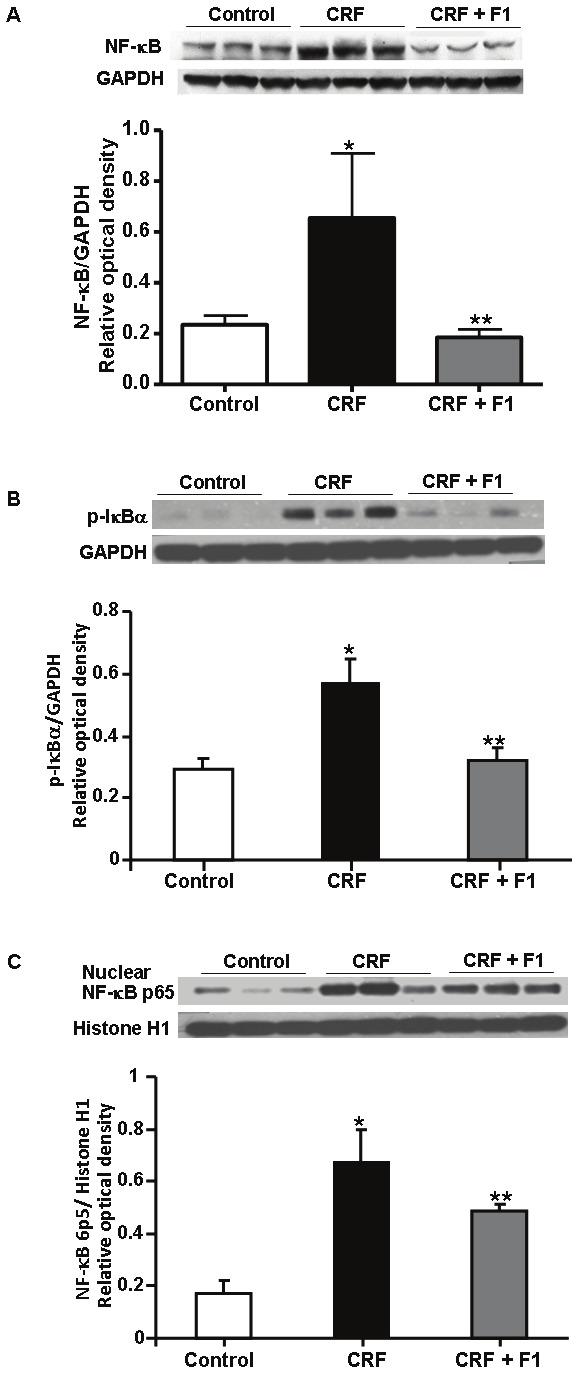
Representative Western blot and quantitative data for A. NF-κB/GAPDH, B. p- IκBα/GAPDH from whole kidney and C. nuclear NF-κB p65/Histone H1 abundance from nuclear protein of Control, CRF and CRF + F1 treated animal groups. Expression of NF-κB, p-IκBα and NF-κB was significantly enhanced in CRF and this increase was prevented with F1 administration. NF-κB: Nuclear factor-kappa B, p-IκBα: phophorylated-I kappa Bα. N=5-8, *p<0.05 compared to Control, **p<0.05 compared to CRF rats.
In uremia, reactive nitrogen species-induced nitrosative stress may participate in the pathogenesis of tissue injury and inflammation through the interaction between superoxide anion and nitric oxide, and 3-nitrotyrosine is an in vivo biomarker of this process [29,30]. There was an increase in 3-nitrotyrosine expression in CRF rats compared to controls (0.48 ± 0.09 vs. 0.07 ± 0.02) that was normalized with F1 administration (0.09 ± 0.04), p<0.05, Figure 8.
Figure 8.
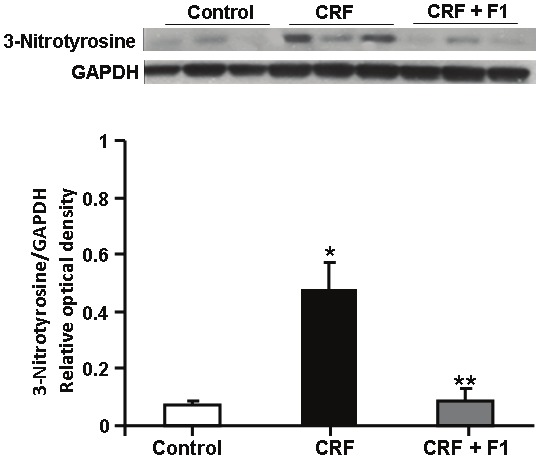
Representative Western blot and quantitative data of 3-Nitrotyrosine/GAPDH protein from whole kidney of Control, CRF and CRF + F1-treated rats. Expression of 3-nitrotyrosine was upregulated in CRF and ameliorated by F1 administration. N= 5-8, *p<0.0.05 compared to Control, **p<0.05 compared to CRF.
The development of anemia is a nearly universal complication of progressive renal failure caused by a combination of shortened erythrocyte lifespan and diminished erythropoiesis due to reduced erythropoietin production by the diseased kidney and erythropoietin resistance caused by the prevailing inflammation. The untreated CRF group exhibited significant anemia (hematocrit of 31% vs. 46% in controls, p<0.05) confirming the result of an earlier study in this model[9]. Administration of F1 resulted in partial amelioration of anemia (hematocrit to 36%) in the CRF animals, Table 1.
Figure 9 depicts a summary of our results. It shows the overall effects of oxidative and nitrosative stress in the development of tubulointerstitial fibrosis induced by adenine and factors that were significantly ameliorated by F1 administration.
Figure 9.
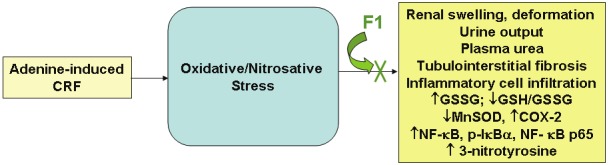
The figure shows a schematic of results of the study showing the effects of adenine to induce components of oxidative stress, and the effects of F1 to significantly attenuate these alterations. CRF: chronic renal failure, GSSG: oxidized glutathione, GSH/GSSG: reduced glutathione/oxidized glutathione ratio, MnSOD: manganese superoxide dismutase, COX-2: cyclooxygenase-2, NF-κB: nuclear factor-kappa B, p-IκBα: phosphorylated-I kappa Bα, NF-κB p65: nuclear factor-kappa B p65 subunit. Please see text for details.
Discussion
This is the first study to show that modulation of a GSH precursor by replacement of cysteine with cystine in F1 can cause significant attenuation of kidney tissue damage, marked by gross edema and interstitial fibrosis, azotemia, markers of oxidative stress, and inflammation in the adenine-induced animal model of CRF with severe progressive chronic interstitial nephropathy. In addition, we observed a modest improvement of plasma creatinine, creatinine clearance and anemia. These results are in agreement with previous works indicating that effective amelioration of excess oxidative stress and inflammation can reduce CRF progression [26,31,32]. In particular, melatonin, which has been shown to possess potent antioxidant and anti-inflammatory properties, significantly reduced glomerulosclerosis and tubulointerstitial injury in rats following renal ablation [31]. Also, Gum Arabic, which has traditionally been used in Middle Eastern countries to alleviate the severity of CRF in humans, was recently shown to improve glomerular, tubular and interstitial lesions, and renal dysfunction, and increase renal SOD activity in the adenine-induced model of CRF [33]. Although, the exact mechanism(s) for these observations have not been identified, it has been suggested that the beneficial effects of these therapies may possibly involve direct antioxidant and/or anti-inflammatory activity. Recently, the use of bardoxolone methyl, which stimulates expression of endogenous antioxidant and cytoprotecitve molecules via activation of the Keap 1-Nrf2 pathway, has been shown to improve renal function in patients with CRF [34].
Consumption of 0.7% adenine-containing diet resulted in severe CRF due to crystal-induced tubulointerstitial nephritis, marked swelling, discoloration and deformity of the kidney, loss of urinary concentrating ability (polyuria), azotemia (elevated plasma urea), anemia, a significant rise in blood pressure, and minimal proteinuria, consistent with the interstitial, as opposed to glomerular nature of kidney disease in this rodent model.
NF-κB has a pivotal role in the induction as well as resolution of inflammation through regulation of hundreds of genes [35,36]. Although, there are several common drugs that may influence its activation [37-39], there are currently no therapeutics that specifically block NF-κB and thus interference with NF-κB may be a target of oxidative stress modulators. From our studies, F1 administration significantly attenuated expression of tissue NF-κB and activation of NF-κB as evidenced by reduction in p-IκBα as well as increased nuclear translocation of NF-κB p65 suggesting that the effects of F1 may be far reaching at the level of gene transcription. As we have demonstrated, administration of F1 for 2 weeks during concurrent exposure to adenine attenuated infiltration of inflammatory cells.
GSH, the most abundant intracellular freeradical scavenger performs its major functions to maintain redox balance in the cytosol where up to 85% to 90% of total body GSH resides, with a smaller portion functioning in organelles such as mitochondria, nuclear matrix, and peroxisomes [40]. GSH, as a whole molecule, cannot be directly taken up by mammals into cells and is largely metabolized in the gastrointestinal tract into its constituent amino acids, glycine, glutamic acid and cysteine. Ultimately, GSH is re-assimilated intracellularly under the regulation of two cytosolic, ATP-dependent enzymes. A priori, it might seem paradoxical that cystine, the oxidized form of cysteine, in combination with glutamic acid and glycine would improve the redox state. This paradox appears to be due to the pleiotropic nature of cystine/cysteine as cystine functions as a stable “cysteine carrier”, necessary for translocation to the intracellular milieu via the cystine-glutamate transporter [40]. Once in the cytoplasm, it is decoupled to two cysteines, via substrate-specific enzymes, oxidoreductases and thioltransferases, which supply the intracellular cysteine necessary for GSH and protein biosynthesis [41]. Thus, the processes that achieve GSH synthesis require special conditions that are only present within the cell.
Administration of oral GSH preparations is limited by their ability to provide adequate substrate to accommodate intracellular GSH synthesis. Intravenous GSH administration is expensive, short-lived, and impractical, as the transiently higher plasma concentrations are largely independent of intracellular GSH activity, where much of the oxidative reactions take place. N-acetyl-L-cysteine (NAC), a GSH precursor which despite showing initial promise [11], has not demonstrated consistent clinical efficacy [42,43]. Thus, a GSH precursor with cystine as the physiologic cysteine carrier, could create a more optimal Cys/CySS redox state, and improved cellular delivery of antioxidant substrate to attenuate oxidative stress to a greater degree than traditional GSH precursors, such as NAC. Although, NAC administration was not a part of this study, we have previously shown that F1 is more effective than NAC-based GSH antioxidants in attenuating spermineinduced oxidative stress in vascular smooth muscle cells [44].
In this study, we have demonstrated that F1 has a universal effect to target several pathways in the process of oxidative stress and inflammation resulting in measureable improvements in pathologic parameters associated with CRF. These findings support the need for additional studies to clarify the mechanistic pathways and examine the clinical efficacy of cysteine/cystine based GSH antioxidants for the prevention/treatment of oxidative stress-induced medical conditions including CRF-related atherosclerosis. It is also possible that F1 might act by modifying the antioxidant response element and/or other pathways yet to be clearly elucidated such as modulation of Nrf2 and Keap 1, which have been shown to be important in protecting against oxidative tissue injury [45]. Indeed, further studies are needed to assess the effect and mechanisms of continued F1 administration after cessation of adenine exposure and in various other models of CRF such as diabetic nephropathy, hypertensive nephrosclerosis, obstructive nephropathy, and glomerulonephritis.
Acknowledgements
This research was supported in part by the NIH-NIMHD Accelerating Excellence in Translational Science Grant (U54 M007598 formerly U54 RR026138) and P20MD00182.
References
- 1.Levey AS, Atkins R, Coresh J, Cohen EP, Collins AJ, Eckardt KU, Nahas ME, Jaber BL, Jadoul M, Levin A, Powe NR, Rossert J, Wheeler DC, Lameire N, Eknoyan G. Chronic kidney disease as a global public health problem: approaches and initiatives - a position statement from Kidney Disease Improving Global Outcomes. Kidney Int. 2007;72:247–259. doi: 10.1038/sj.ki.5002343. [DOI] [PubMed] [Google Scholar]
- 2.Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, Van Lente F, Levey AS. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038–2047. doi: 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- 3.Obrador GT, Mahdavi-Mazdeh M, Collins AJ. Establishing the Global Kidney Disease Prevention Network (KDPN): a position statement from the National Kidney Foundation. Am J Kidney Dis. 2011;57:361–370. doi: 10.1053/j.ajkd.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Collins AJ, Gilbertson DT, Snyder JJ, Chen SC, Foley RN. Chronic kidney disease awareness, screening and prevention: rationale for the design of a public education program. Nephrology (Carlton) 2010;15(Suppl 2):37–42. doi: 10.1111/j.1440-1797.2010.01312.x. [DOI] [PubMed] [Google Scholar]
- 5.Ina K, Kitamura H, Tatsukawa S, Takayama T, Fujikura Y, Shimada T. Transformation of interstitial fibroblasts and tubulointerstitial fibrosis in diabetic nephropathy. Med Electron Microsc. 2002;35:87–95. doi: 10.1007/s007950200011. [DOI] [PubMed] [Google Scholar]
- 6.Wolak T, Kim H, Ren Y, Kim J, Vaziri ND, Nicholas SB. Osteopontin modulates angiotensin II-induced inflammation, oxidative stress, and fibrosis of the kidney. Kidney Int. 2009;76:32–43. doi: 10.1038/ki.2009.90. [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez-Iturbe B, Vaziri ND, Herrera-Acosta J, Johnson RJ. Oxidative stress, renal infiltration of immune cells, and salt-sensitive hypertension: all for one and one for all. Am J Physiol Renal Physiol. 2004;286:F606–616. doi: 10.1152/ajprenal.00269.2003. [DOI] [PubMed] [Google Scholar]
- 8.Alsaad KO, Herzenberg AM. Distinguishing diabetic nephropathy from other causes of glomerulosclerosis: an update. J Clin Pathol. 2007;60:18–26. doi: 10.1136/jcp.2005.035592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yokozawa T, Zheng PD, Oura H, Koizumi F. Animal model of adenine-induced chronic renal failure in rats. Nephron. 1986;44:230–234. doi: 10.1159/000183992. [DOI] [PubMed] [Google Scholar]
- 10.Janaky R, Dohovics R, Hermann A, Oja SS, Saransaari P. Effects of metabotropic glutamate receptor agonists and antagonists on D-aspartate release from mouse cerebral cortical and striatal slices. Neurochem Res. 2001;26:1217–1224. doi: 10.1023/a:1013963222332. [DOI] [PubMed] [Google Scholar]
- 11.Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 12.Pompella A, Visvikis A, Paolicchi A, De Tata V, Casini AF. The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol. 2003;66:1499–1503. doi: 10.1016/s0006-2952(03)00504-5. [DOI] [PubMed] [Google Scholar]
- 13.Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, Krauth M, Ruiz S, Audhya P, Christ-Schmidt H, Wittes J, Warnock DG. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med. 2011;365:327–336. doi: 10.1056/NEJMoa1105351. [DOI] [PubMed] [Google Scholar]
- 14.Adabag AS, Ishani A, Bloomfield HE, Ngo AK, Wilt TJ. Efficacy of N-acetylcysteine in preventing renal injury after heart surgery: a systematic review of randomized trials. Eur Heart J. 2009;30:1910–1917. doi: 10.1093/eurheartj/ehp053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hilmi IA, Peng Z, Planinsic RM, Damian D, Dai F, Tyurina YY, Kagan VE, Kellum JA. N-acetylcysteine does not prevent hepatorenal ischaemia-reperfusion injury in patients undergoing orthotopic liver transplantation. Nephrol Dial Transplant. 2010;25:2328–2333. doi: 10.1093/ndt/gfq077. [DOI] [PubMed] [Google Scholar]
- 16.Rehman T, Fought J, Solomon R. N-acetylcysteine effect on serum creatinine and cystatin C levels in CKD patients. Clin J Am Soc Nephrol. 2008;3:1610–1614. doi: 10.2215/CJN.01560408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shavit L, Korenfeld R, Lifschitz M, Butnaru A, Slotki I. Sodium bicarbonate versus sodium chloride and oral N-acetylcysteine for the prevention of contrast-induced nephropathy in advanced chronic kidney disease. J Interv Cardiol. 2009;22:556–563. doi: 10.1111/j.1540-8183.2009.00500.x. [DOI] [PubMed] [Google Scholar]
- 18.Kamgar M, Zaldivar F, Vaziri ND, Pahl MV. Antioxidant therapy does not ameliorate oxidative stress and inflammation in patients with end-stage renal disease. J Natl Med Assoc. 2009;101:336–344. doi: 10.1016/s0027-9684(15)30881-6. [DOI] [PubMed] [Google Scholar]
- 19.Witschi A, Reddy S, Stofer B, Lauterburg BH. The systemic availability of oral glutathione. Eur J Clin Pharmacol. 1992;43:667–669. doi: 10.1007/BF02284971. [DOI] [PubMed] [Google Scholar]
- 20.Sinha-Hikim I, Shen R, Kovacheva E, Crum A, Vaziri ND, Norris KC. Inhibition of apoptotic signalling in spermine-treated vascular smooth muscle cells by a novel glutathione precursor. Cell Biol Int. 2010;34:503–511. doi: 10.1042/CBI20090349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sinha-Hikim I, Sinha-Hikim AP, Shen R, Kim H, French SW, Vaziri ND, Crum A, Rajavashisth TB, Norris KC. A novel cystine based antioxidant attenuates oxidative stress and hepatic steatosis in diet-induced obese mice. Exp Mol Pathol. 2011;91:419–428. doi: 10.1016/j.yexmp.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crum A. Nutritional or therapeutic compositions to increases bodily glutathione levels. US. RE39734E. 2007. [Google Scholar]
- 23.Arenzana-Seisdedos F, Thompson J, Rodriguez MS, Bachelerie F, Thomas D, Hay RT. Inducible nuclear expression of newly synthesized I kappa B alpha negatively regulates DNA-binding and transcriptional activities of NF-kappa B. Mol Cell Biol. 1995;15:2689–2696. doi: 10.1128/mcb.15.5.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raij L, Azar S, Keane W. Mesangial immune injury, hypertension, and progressive glomerular damage in Dahl rats. Kidney Int. 1984;26:137–143. doi: 10.1038/ki.1984.147. [DOI] [PubMed] [Google Scholar]
- 25.Alvarez V, Quiroz Y, Nava M, Pons H, Rodriguez-Iturbe B. Overload proteinuria is followed by salt-sensitive hypertension caused by renal infiltration of immune cells. Am J Physiol Renal Physiol. 2002;283:F1132–1141. doi: 10.1152/ajprenal.00199.2002. [DOI] [PubMed] [Google Scholar]
- 26.Quiroz Y, Pons H, Gordon KL, Rincon J, Chavez M, Parra G, Herrera-Acosta J, Gomez-Garre D, Largo R, Egido J, Johnson RJ, Rodriguez-Iturbe B. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from nitric oxide synthesis inhibition. Am J Physiol Renal Physiol. 2001;281:F38–47. doi: 10.1152/ajprenal.2001.281.1.F38. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez-Iturbe B, Quiroz Y, Nava M, Bonet L, Chavez M, Herrera-Acosta J, Johnson RJ, Pons HA. Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol Renal Physiol. 2002;282:F191–201. doi: 10.1152/ajprenal.0197.2001. [DOI] [PubMed] [Google Scholar]
- 28.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 29.Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci USA. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Souza JM, Peluffo G, Radi R. Protein tyrosine nitration--functional alteration or just a biomarker? Free Radic Biol Med. 2008;45:357–366. doi: 10.1016/j.freeradbiomed.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 31.Quiroz Y, Ferrebuz A, Romero F, Vaziri ND, Rodriguez-Iturbe B. Melatonin ameliorates oxidative stress, inflammation, proteinuria, and progression of renal damage in rats with renal mass reduction. Am J Physiol Renal Physiol. 2008;294:F336–344. doi: 10.1152/ajprenal.00500.2007. [DOI] [PubMed] [Google Scholar]
- 32.Quiroz Y, Ferrebuz A, Vaziri ND, Rodriguez-Iturbe B. Effect of chronic antioxidant therapy with superoxide dismutase-mimetic drug, tempol, on progression of renal disease in rats with renal mass reduction. Nephron Exp Nephrol. 2009;112:e31–42. doi: 10.1159/000210577. [DOI] [PubMed] [Google Scholar]
- 33.Ali BH, Al-Salam S, Al Husseni I, Kayed RR, Al-Masroori N, Al-Harthi T, Al Zaabi M, Nemmar A. Effects of Gum Arabic in rats with adenine-induced chronic renal failure. Exp Biol Med (Maywood) 2010;235:373–382. doi: 10.1258/ebm.2009.009214. [DOI] [PubMed] [Google Scholar]
- 34.Witschi A, Reddy S, Stofer B, Lauterburg BH. The systemic availability of oral glutathione. Eur J Clin Pharmacol. 1992;43:667–669. doi: 10.1007/BF02284971. [DOI] [PubMed] [Google Scholar]
- 35.Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. Possible new role for NF-kappaB in the resolution of inflammation. Nat Med. 2001;7:1291–1297. doi: 10.1038/nm1201-1291. [DOI] [PubMed] [Google Scholar]
- 36.Lawrence T, Fong C. The resolution of inflammation: anti-inflammatory roles for NF-kappaB. Int J Biochem Cell Biol. 2010;42:519–523. doi: 10.1016/j.biocel.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 37.Meyer S, Kohler NG, Joly A. Cyclosporine A is an uncompetitive inhibitor of proteasome activity and prevents NF-kappaB activation. FEBS Lett. 1997;413:354–358. doi: 10.1016/s0014-5793(97)00930-7. [DOI] [PubMed] [Google Scholar]
- 38.Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- 39.Guijarro C, Egido J. Transcription factor-kappa B (NF-kappa B) and renal disease. Kidney Int. 2001;59:415–424. doi: 10.1046/j.1523-1755.2001.059002415.x. [DOI] [PubMed] [Google Scholar]
- 40.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 41.Zhu J, Li S, Marshall ZM, Whorton AR. A cystine-cysteine shuttle mediated by xCT facilitates cellular responses to S-nitrosoalbumin. Am J Physiol Cell Physiol. 2008;294:C1012–1020. doi: 10.1152/ajpcell.00411.2007. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Bardag-Gorce F, Dedes J, French BA, Amidi F, Oliva J, French SW. S-adenosylmethionine prevents Mallory Denk body formation in drug-primed mice by inhibiting the epigenetic memory. Hepatology. 2008;47:613–624. doi: 10.1002/hep.22029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin CC, Yin MC, Hsu CC, Lin MP. Effect of five cysteine-containing compounds on three lipogenic enzymes in Balb/cA mice consuming a high saturated fat diet. Lipids. 2004;39:843–848. doi: 10.1007/s11745-004-1305-4. [DOI] [PubMed] [Google Scholar]
- 44.Sinha-Hikim I, Shen R, Paul Lee WN, Crum A, Vaziri ND, Norris KC. Effects of a novel cystine-based glutathione precursor on oxidative stress in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2010;299:C638–642. doi: 10.1152/ajpcell.00434.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyata T, Takizawa S, van Ypersele de Strihou C. Hypoxia. 1. Intracellular sensors for oxygen and oxidative stress: novel therapeutic targets. Am J Physiol Cell Physiol. 2011;300:C226–231. doi: 10.1152/ajpcell.00430.2010. [DOI] [PubMed] [Google Scholar]