

Abstract
Microglia become activated in humans subsequent to infection with HIV, and uncontrolled brain inflammation plays a key role in neuronal injury and and cognitive dysfunction during HIV infection. Various studies have shown a deleterious role for the HIV regulatory protein Tat in the development and maintenance of HIV-associated neurocognitive disorders (HAND). One cell surface receptor implicated in inhibiting microglial activation is the protein-tyrosine phosphatase (PTP), CD45. It is especially effective at inhibiting microglial activation because its action takes place far upstream from proinflammatory intracellular signaling mediators. To investigate the possible role of CD45 in microglial responsiveness to HIV-1 Tat protein, we treated BV-2 microglia with a tyrosine phosphatase inhibitor [potassium bisperoxo (1, 10-phenanthroline) oxovanadate (phen), 5 μM] and HIV-1 Tat protein (700ng/ml). We found a synergistic pro-inflammatory microglial activation as supported by tumor necrosis factor-alpha (TNF-α) and interleukin 1-beta (IL-1β) release, both of which were dependent on p44/42 mitogen-activated protein kinase (MAPK) activation. Stimulation of microglial CD45 by anti-CD45 antibody markedly inhibited these Tat or Tat/Phen effects via attenuation of p44/42 MAPK, suggesting CD45 negatively regulates microglial activation. As a validation of these findings in vivo, brains from transgenic mice deficient for CD45 through complete genetic ablation, or by CNS delivery of CD45shRNA, demonstrate markedly increased production of TNF-α 24 hours after intracerebroventricular injection of HIV-Tat protein (5μg/mouse) compared to control mice. This increased microglial activation was accompanied by astrogliosis and a significant loss of cortical neurons due to apoptosis in CD45 deficient animals. These results suggest therapeutic agents that activate CD45 PTP signaling may be effective in suppressing microglial activation associated with HAND.
Keywords: HIV, dementia, CD45, microglia, HIV associated neurocognitive disorders
Introduction
Human immunodeficiency virus-1 (HIV-1) infection of the central nervous system (CNS) can result in cognitive, motor, and behavioral deficits, termed collectively HIV-associated neurocognitive disorders (HAND) [1,2] . Soon after infection by the HIV, it rapidly moves into the brain via infected monocytes and lymphocytes [3] and, despite highly active antiretroviral therapy (HAART), persists in parenchymal microglia as well as the perivascular macrophages [4-6]. Importantly, once the virus infects the brain, there is a deleterious immune activation of resident glia. As HIV is unable to productively infect neurons, neuronal cell damage is largely promoted by neurotoxins secreted by these infected and/or activated macrophages, microglia, and astrocytes. In spite of the fact that the clinical severity of HAND has been significantly reduced due to the widespread utilization of HAART, the prevalence and associated morbidity still remain unacceptably high (~50%) [7,8]. The fact that HAND still persists in the current era of HAART, even in patients effectively controlled for systemic viremic load, is incompletely understood. Recent evidence suggests prolonged inflammation in both the brain and periphery may be responsible [9-11].
At the center of this persistence of prolonged CNS inflammation is an increased number of microglia and macrophages in the brain. The presence of these cells positively correlates with the severity of pre-mortem HAND, suggesting the importance of these cell in promoting neuronal damage [4,12-14]. Indeed microglias are key generators of a number of toxic factors, which together impair neuronal function. As such, neurologic deficits in HAND are more closely correlated with the presence of activated macrophage and microglia than with the viral RNA [12,15]. In combination with the neurotoxic secreted factors from microglia are the soluble viral proteins such as Tat and, the glycoprotein, gp120 which can be released from infected microglia and macrophages as well [16]. Circulating levels of HIV-1 Tat have been quantified in patient sera from HIV-1 positive individuals, at levels ranging from 1–40 ng/mL [17,18], although, local extracellular concentrations in the brain may be much higher, especially adjacent to HIV-1 positive perivascular cells [19]. The HIV-1 Tat protein can also exert its proinflammatory activating effect on uninfected cells including other microglia, astrocytes, and neurons. Both infected and activated microglia and astrocytes produce pro-inflammatory cytokines including tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β), which serve to further promote activation of neighboring cells. Infected and activated cells also produce chemokines such as monocyte chemotactic protein-1 (MCP-1), thus attracting more inflammatory monocytes and macrophage in a positive feedback loop [20,21]. Therefore, circulating HIV-1 Tat protein is very likely involved in triggering this self-perpetuating inflammatory loop, ultimately leading to neuron damage and cognitive deficits [17].
One viable target on microglia is the promotion of the CD45 signaling pathway. CD45 is a haemopoietic cell specific protein tyrosine phosphatase (PTP), essential for antigen receptor-mediated signaling in T and B cells [22], as well as microglia [23,24]. It modulates signaling through cytokine receptors as well as cellular adhesion [25,26]. The CD45 protein is encoded by a single gene (PTPRC; protein tyrosine phosphatase, receptor-type C) and different isoforms can be cleaved by alternative splicing of three variable exons: A, B, and C [27].
CD45 modulation may be particularly salient to the clinical features of HAND since microglia in normal human brain express CD45 and upregulation in microglial CD45 expression has been noted in Alzheimer’s disease (AD), graft-versus-host disease (GVHD), multiple sclerosis (MS), and in HIV encephalitis (HIVE) [28-33]. In addition, studies in rodent and human cells indicate CD45 can suppress microglial pro-inflammatory activation. For example, we previously found murine microglia devoid of CD45 expression demonstrate an overactivated phenotype [34,35]. On the other hand, it has concordantly been shown that an agonist antibody (αCD45RO, clone UCHL-1) can stimulate CD45 PTP activity and dampen granulocyte-macrophage colony-stimulating factor (GM-CSF) signal transduction and cell proliferation [36]. In addition, CD45 also mitigates HIV-1 replication in microglia, suggesting there maybe be a potential for targeting this phosphatase as a therapy for HAND [37]. Furthermore, in an animal model of neurodegeneration, upregulation of PTP signaling in activated microglia was found in and around degenerating brain regions [25]. As the phosphorylating enzyme of the related cytoplasmic threonine tyrosine kinase, p44/42 mitogen activated protein kinase (MAPK), is also a response to HIV-1 Tat and other inflammatory molecules including gp120 [38-40]. Thus, it is likely that p44/42 MAPK activation is also critical to the disease CNS inflammatory cascade. Indeed activation of the p38 pathway in microglia or neurons may stimulate the production of inflammatory mediators, thereby contributing to the degeneration or further activation of these cells. Together these data led us to investigate the possible involvement of CD45 PTP signaling as a putative down-regulator of microglial p38 activation in response to HIV-1 Tat protein.
Materials and methods
Reagents
Monoclonal antibodies (purified rat anti-mouse CD45 and purified rat IgG2bcontrol antibodies) were purchased from PharMingen (San Diego, CA). Antibodies for phospho-p44/42 mitogen-activated protein kinase (MAPK) (Thr-202/Tyr-204) and total p44/42 MAPK were obtained from New England Biolabs (Beverly, MA). PD98059 were obtained from Calbiochem (La Jolla, CA). The phosphatase inhibitor, potassium bisperoxo (1,10-phenanthroline) oxovanadate (phen) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Each of these was dissolved in DMSO before adding to cell culture medium, and DMSO alone was used as a solvent control, which did not differ from the untreated controls presented. Bacterial lipopolysaccharide (LPS) was purchased from Sigma (St. Louis, MO) and dissolved in complete cell culture medium. Anti-mouse and anti-rabbit HRP-conjugated IgG secondary antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Immun-Blot polyvinylidene difluoride (PVDF) membranes were purchased from Bio-Rad (Hercules, CA). Anti-mouse TNF-α polyclonal antibody was obtained from R & D systems (Minneapolis, MN). Anti-neuronal nuclei antibody was obtained from Chemicon (Temecula, CA). Donkey anti-mouse IgG Alexa Fluor 594 and was purchased from Molecular Probes (Eugene, OR). Tris-buffered saline was obtained from Bio-Rad (Hercules, CA) and luminol reagent was obtained from Pierce Biotechnology. Anti-Bcl-xL, and anti-Bax antibodies were purchased from Upstate (Lake Placid, NY). Antiactin antibody was obtained from Roche. Recombinant HIV-1 protein Tat1-86 was obtained from The National Institutes of Health (NIH) AIDS Research and Reference Reagent Program (Rockville, MD).
TNF-α and IL-1 β ELISAs
BV-2 microglial cells were plated in 24-well tissue-culture plates (Costar, Cambridge, MA) at 5 × 104cells per well and stimulated for 12 hr with phen (5 μM), Tat peptides (700 ng/ml), phen/Tat in the presence or absence of anti-CD45 antibody (1:200) or PD98059 (5 μM) pretreatment for 1 hr, or appropriate controls. Cell-free supernatants were collected and assayed by a TNF-α or IL-1β ELISA kit (eBioscience, San Diego, CA) in strict accordance with the manufacturer's instructions. The Bio-Rad (Hercules, CA) protein assay was performed to measure total cellular protein from each of the cell groups under consideration just before quantification of cytokine release by ELISA.
Western immunoblotting
BV-2 microglia were plated in six-well tissue culture plates at a density of 8 × 105 cells per well. These cells were incubated for 30 min with or without phen (5 μM), heat inactive HIV-1 Tat peptide (700 ng/ml) and HIV-1 Tat peptide (700 ng/ml); or phen/Tat in the presence or absence PD98059 (5 μM) pretreatment for 1 hr. Immediately after culturing, microglia were washed in ice-cold PBS three times, and lysed in an ice-cold lysis buffer containing 20 mM Tris, pH 7.5, 150 mMNaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mMβ-glycerolphosphate, 1 mMNa3VO4, 1 μg/ml leupeptin (RIPA buffer), 1 mM PMSF and protease cocktail (Sigma, St. Louis, MO). After incubation for 30 min on ice, samples were centrifuged at the highest speed for 15 min, and supernatants were collected. Total protein content was estimated using the Bio-Rad protein assay. An aliquot corresponding to 50 μg of total protein of each sample was separated by SDS-PAGE and transferred electrophoretically to Immun-Blot PVDF membranes. Nonspecific antibody binding was blocked with 5% nonfat dry milk for 1 hr at room temperature in Tris-buffered saline (20 mM Tris and 500 mM NaCl, pH 7.5). Membranes where first hybridized with a phospho-specific p44/42 MAPK antibody, stripped with β-mercaptoethanol stripping solution (62.5 mM Tris-HCl, pH 6.8, 2% SDS, and 100 mM β-mercaptoethanol), and then reprobed with an antibody that recognizes total p44/42 MAPK. Followed by an anti-rabbit HRP-conjugated IgG secondary antibody as a tracer, the luminol reagent was used to develop the blots. Densitometric analysis was performed for all blots using the Flour-S MultiImager with Quantity One software (Bio-Rad).
For the in vivo studies, Western blot was performed as described previously [39]. Briefly, for the in vivo studies left hemispheres of 3 month old transgenic and nontransgenic mouse brains were lysed by ice-cold RIPA buffer with protease cocktail and an aliquot corresponding to 50 μg of total protein were electrophoretically separated using 10% Tris–glycine gels. Electrophoresed proteins were then transferred to PVDF membranes (Bio-Rad), washed in dH2O, and blocked for 1 h at ambient temperature in Tris-buffered saline containing 5% (w/v) non-fat dry milk. After blocking, membranes were hybridized overnight at 4Ctemperature with various primary antibodies. Membranes were then washed three times (5 min each) in dH2O and incubated for 1 h at ambient temperature with the appropriate HRP-conjugated secondary antibody (1:1000). All antibodies were diluted in TBS containing 5% (w/v) non-fat dry milk. Blots were developed using the luminol reagent. Antibodies used for western blot included: anti-Bcl-xL antibody (Millipore, 1:1000), anti-Bax antibody (Millipore, 1:1000), anti-TNF-α (R & D systems, Minneapolis, 1:1000) and anti-actin antibody (Sigma, 1:10000).
In vivo neurotoxicity analysis
Animals were anesthetized using isoflurane (chamber induction at 4–5% isoflurane, intubation and maintenance at 1–2%). After reflexes were checked to ensure that mice were unconscious, they were positioned on a stereotaxic frame (Stoelting Lab Standard) with ear-bars positioned and jaws fixed to a biting plate. The axis coordinates were taken from a mouse brain atlas, and a 5-mm sterile plastic guide cannula (21 GA; Plastic One, Inc., Roanoke, VA) was implanted into the left lateral ventricle delimited from the stereotaxic coordinates (coordinates relative to bregma: - 0.6 mm anterior/posterior, + 1.2 mm medial/lateral, and -3.0 mm dorsal/ventral) using the stereotaxic device (Stoelting Lab Standard) and an attached probe (cannula) holder. HIV-1 protein Tat (500 ng/mouse) or PBS (10 μl) was administered at the rate of 1 μl/min using a Hamilton syringe (modified with a solder stop to prevent over insertion of the needle) through the implanted cannula. Correctness of the injection was confirmed by trypan blue dye administration and histological examination. The wounds were closed with 1 staple and mice were all observed until anesthesia had cleared. Twenty-four hours after the i.c.v. injections animals were sacrificed with isofluorane and brain tissues collected.
NeuN, GFAP, and IBA-1 immunohistochemistry analysis
Mice were anesthetized with isofluorane and transcardially perfused with ice-cold physiological saline containing heparin (10 U/ml). Brains were rapidly isolated and separated into left and right hemispheres using a mouse brain slicer (Muromachi Kikai, Tokyo, Japan). The left hemispheres were rapidly frozen for western blot. The right hemispheres were used for cryostate sectioning and immunochemistry analysis. NeuN, GFAP, and IBA-1 staining was performed under standard immunofluorescence-labeling procedures according to our previous studies [39]. Briefly, frozen tissue sections were washed in PBS and blocked in 3% bovine serum albumin and 2% normal serum for 2 hr at room temperature, then incubated overnight in primary antibodies, monoclonal mouse antineuronal nuclei antibody NeuN (Millipore, 1:1000), GFAP (Zymed Laboratories, 1:1000), or Iba-1 (Wako Chemicals, USA, 1:1000). The following day, sections were washed in PBS 3 times (10 min each), and then incubated for 1 h in the dark with secondary antibody, donkey anti-mouse IgG Alexa Fluor 594 at 1:100. After another cycle of washing, floating sections were mounted onto slides, dehydrated and coverslipped with Vectashield fluorescence mounting media (Vector Labs., Burlingame, CA). Slides were visualized under dark field using an Olympus BX-51 microscopy.
CD45 RNAi knock-down
Four different sequences of CD45 shRNAs, empty vector and scrambled control shRNA were purchased from OriGene (Rockville, MD). To select highest knock-down effects of CD45 shRNAs, four CD45 shRNA victors were transferred into N9 microglial cells first. CD45 protein expression levels were detected by western blot in transferred cell lysates, and the CD45 shRNA with the lowest express levels of CD45 (target CD45 sequencing is TGGCTTCGCGCCCGCACTGAGCTGGAATC) was selected for transfering into mouse brain. HVJ-Envelope vector kit (CosmoBio, Tokyo, Japan), was used a delivery of shRNA plasma into target tissues. Following with manufacturer’ protocols, HVJ-Envelope (HVJ-E) incorporated with CD45 shRNA to yield an HVJ-E CD45 shRNA complex (vector). The victors were introduced into target tissues by membrane-fusion activity of fusion protein. 5 μg of CD45 shRNA, empty vector or scrambled control shRNA are combined with HVJ-E, respectively. The 10 ul of complexes in PBS were delivered by intracerebroventricular (ICV) injection. After 72 hrs, additionally, the mice ICV injected of Tat protein or PBS for control. After 24 hrs, mice were scarified.
Statistical analysis
Data were analyzed using ANOVA followed by post-hoc comparisons of means by Bonferroni's or Dunnett's T3 method, for which Levene's test for homogeneity of variances was used to determine the appropriate method of post hoc comparison. In instances of single-mean comparisons, t test for independent samples was used to assess significance. α levels were set at 0.05 for each analysis. All analyses were performed using SPSS for Windows release 9.0.
Results
CD45 signaling pathway is involved in HIV-1 Tat protein-stimulated microglial activation
It has been shown that a tyrosine phosphorylation cascade plays an important role in HIV-1 Tat-induced microglial activation [41-43]. To test whether promotion of tyrosine phosphorylation could affect Tat-induced microglial activation, we co-incubated BV-2 microglial cells with phen, a specific tyrosine phosphatase inhibitor, and HIV-1 Tat (provided by NIH AIDS Reference and Reagent Program, Germantown, MD) for 12 hr. Microglial activation was measured by TNF-α and IL-1β production. Data showed that phen synergistically enhanced HIV-1 Tat-stimulated microglial activation as evidenced by TNF-α and IL-1β levels (Figure 1A). To further confirm that phen and HIV-1 Tat activated microglia by inhibiting the PTP signaling pathway, we co-cultured BV-2 microglia with HIV-1 Tat and phen (5 μM) and measured TNF-α and IL-1β production. This result led us to focus on stimulating microglial CD45 PTP activity to oppose HIV-1 Tat-induced activation of these cells.
Figure 1.

CD45 signaling pathway is involved in HIV-1 Tat protein-stimulated microglial activation. (A) Co-treatment with phen and HIV-1 Tat protein synergistically increased in Th1 cytokines in microglial cells. BV2 cells were plated at 5 X 104 cells per well in 24-well tissue-culture plates and stimulated with phen (5 μM), Tat protein (700 ng/ml) or phen/Tat combination for 12 hr. Control peptide is heat inactive Tat protein (inact. Tat). Microglial activation was measured by TNF-α (upper panel) and IL-1β (lower panel) productions in culture media (mean ± SD, pg/ml) using ELISA assay. As indicated, phen/Tat co-treatment synergistically increased cytokines in culture media compared by control (PBS), phen, and Tat treatment alone. One-way ANOVA was shown a significant differences between the individual treatment and phen/Tat co-treatment (**P < 0.005). (B) CD45 cross-linking with an antibody specially against CD45 (CD45 Ab) markedly inhibits phen and Tat protein induced microglial activation monitored by TNF-α (upper panel) and IL-1β (lower panel) releasing in culture media (mean ± SD, pg/mL). BV2 cells were pretreated with either rat anti-mouse CD45 antibody (2.5 μg/mL) or rat anti-mouse IgG2b (2.5 μg/ml) as control antibody (Ctrl Ab) for 1 hr and then stimulated with phen and Tat protein (***P < 0.001). (C) PD98059 (PD) as a specific MEK1/2 (the upstream activator of p44/42) inhibitor, significantly mitigated TNF-α (upper panel) and IL-1β (lower panel) release in BV2 culture media induced from phen/Tat. BV2 cells were pre-treated with PD98059 (5 μM) for 1 hr, and then treated with phen and Tat for 12 h. Compared with phen/Tat co-treatment alone, adding PD98059 significantly diminished cytokine production induced by phen/Tat co-administration (*** P < 0.001). Data represented three repeat experiments through A to C.
Therefore, to further characterize the putative role of CD45 in HIV-1 Tat induced microglial activation, we treated BV-2 microglial cells with monoclonal anti-CD45 antibody before stimulation with phen and HIV-1 Tat. Microglial activation, as evidenced by TNF-α and IL-1β release after co-treatment with phen and HIV-1 Tat, was significantly inhibited by cross-linking CD45 (Figure 1B); further substantiating the role of CD45 in negative regulation of microglial activation.
Next, since previous studies have shown activation of p44/42 MAPK is involved in TNF-α production in macrophages, monocytes, and microglia after activation of these cells with a variety of stimuli [39,44] we asked whether the observed effect of CD45 cross-linking on opposing microglial activation might be mediated via activation of the MAPK module. Thus, we first analyzed TNF-α and IL-1β release in microglial cell lysates after co-treatment with PD98059, a selective inhibitor of MEK1/2. We observed that production of TNF-α and IL-1β was markedly decreased compared with appropriate controls within 12 hr after treatment with PD98059 and phen and HIV-1 Tat (Fig. 1C). These data suggest that phen and HIV-1 Tat activation of microglia is subserved by the p44/42 MAPK pathway.
HIV-1 Tat induced microglial activation is mediated by the p44/42 MAPK pathway
Having shown that cross-linking of CD45 opposed HIV-1 Tat induced microglial activation, we next further confirmed whether reduced p44/42 MAPK activity could be responsible for this effect. To investigate this possibility, BV-2 microglial cells were co-incubated with heat inactivated HIV-1 Tat protein (negative control) , HIV-1 Tat protein , and phen. Cell lysates were then analyzed for phosphorylated forms of p44/42 MAPK by Western immunoblotting (WB). Results showed that phen and HIV-1 Tat synergistically enhanced phosphorylation of p44/42 MAPK compared with controls (Figure 2A). Elk1 is a substrate of MAP kinases. In accord, phen and HIV-1 Tat induced phosphorylation of Elk1 as detected by WB probed with phospho-Elk1 antibody (Figure 2A). To determine whether inhibition of p44/42 could oppose this synergistic effect on MAPK activity, the cells were treated with PD98059, a selective inhibitor of MEK1/2. Results showed that addition of PD988059 markedly reduced both p44/42 MAPK and phospho-Elk1 activity in phen- and HIV-1 Tat co-treated cells (Figure 2B).
Figure 2.

HIV-1 Tat induced microglial activation is subserved by the p44/42 MAPK pathway. Microglia were treated with control (PBS), control peptide (heat inactivated HIV-1 Tat, 700 ng/ml), HIV-1 Tat (700 ng/ml), phen (5 μM), or both phen/Tat for 30 min in 6-well culture plates. Cell lysates were analyzed by western blot using specific antibodies that recognize phosphorylation or total p44/42 MAPK. The ratio of band intensities of phospho-p44/42 vs. total-p44/42 is shown below the immunoblots (pp44/42 / total p44/42 MAPK) (n=3 for each condition experiments). Compared with phen or HIV-1 Tat individual treatments, co-treatment with phen and HIV-1 Tat significantly promoted phosphorylation of p44/42 MAPK (p < 0.01). (A) Phosphorylation of Elk 1, as a substrate for MAPK, was significantly increased by co-treatment phen/Tat. The expression levels of phospho- Elk 1 normalized by actin were significantly increased by co-treatment with phen and HIV-1 Tat compared to either treatment individually (p < 0.05). (B) PD98059 dramatically inhibited phosphorylation of p44/42 MAPK (upper panel) and Elk 1 (lower panel) by co-treatment of with phen and Tat (p < 0.01). n=3 for each condition experiments.
Intracerebroventricular (ICV) injection of HIV-1 Tat results in neuronal loss and T-helper-1 (Th1) associated microglial activation in CD45 deficient mice
Next, to test whether CD45 plays a role in the modulation of HIV-1 Tat-induced neuronal injury, we treated C57BL/6 mice, and CD45-deficient mice (B6.129-Ptprctm1Holm/J) (n = 3) with heat inactivated HIV-Tat (500 ng/mouse; negative control) or HIV-1 Tat via the ICV route. Twenty-four hours later, these mice were sacrificed and then brain tissues were collected. Mouse brain sections from cortical regions were stained with NeuN and NeuN/DAPI as well as GFAP and GFAP/DAPI. Results indicated a marked increase in neuronal damage in cortical brain regions from CD45 deficient mice ICV injected with HIV-1 Tat compared to controls (heat inactivated HIV-1 Tat), HIV-1 Tat treatment in CD45 sufficient mice, or PBS treatment in CD45 sufficient mice (Figure 3A). In addition, brain homogenates from these mice were prepared for Western blot analysis of Bcl-XL and Bax protein expression as well as TNF-α expression. Consistently, a significant reduction in the ratio of Bcl-XL to Bax in CD45 deficient/HIV-1 Tat condition was observed (Figure 3B). TNF-α expression was also concordantly significantly increased in CD45 deficient/HIV-1 Tat condition compared with other groups (Figure 3C) (* p < 0.05). One-way ANOVA followed by post hoc comparison revealed significant differences between HIV1 Tat / CD45 deficiency and Tat in wild type for the relative intensity of western blot band ratio of Bcl-XL to Bax. In CD45-/- mice, the ratio of Bcl-XL to Bax trended to be decreased by HIV- Tat treatment but did not reach significance due to the relatively small number of mice. Importantly, the TNF-α expression levels are significantly upregulated by ICV injection of HIV-1 Tat in CD45-/- mice compared to CD45-/- mice treated with heat inactivated HIV-1 Tat injection same type of mice (C) (* p < 0.05).
Figure 3.

HIV-1 Tat intracerebroventricular (ICV) injection promotes neuronal cell loss and Th1 associated microglial activation in CD45 deficient mice. (A) CD45 deficiency promoted HIV-1 Tat induced neuronal damage/death. Half-brain sections from 3-month-old CD45 sufficient and deficient mice (C57BL/6J, CD45-/-) after ICV injection of Tat (500 ng/mouse) or heat inactivate HIV-1 Tat (500 ng/mouse) as a control for 24 hr, were stained by NeuN antibody (a-d), and GFAP (i-l) antibody. Counter staining was performed by DAPI (e–h, m-p) (n=3 for each group). HIV-1 Tat treatment induced neuronal cell damage/loss in CD45 sufficient mice compared with PBS treated mice (b vs. a). Furthermore, HIV-1 Tat treatment in the CD45-/- mice further promoted neuronal death/damage (d vs. a-c). Further, astrocyte activation was shown highest in HIV-1 Tat treated mice compared to the CD45 deficient condition (l vs. i-k). Images are represented 10 × object magnifications. (B) Brain homogenates were prepared from half of brain tissues and probed by western blot using antibodies against Bcl-XL and Bax. In CD45-/- mice, the ratio of Bcl-XL to Bax trended to decrease but did not reach significance due to the relatively small number of animals. (C) TNF-α expression is significantly upregulated in the HIV-1 Tat ICV injected CD45-/- mice compared to heat-inactivated HIV-1 Tat injected mice (* p < 0.05, n=3 for each group).
HIV-1 Tat exacerbates neuronal injury and gliosis in wild type mice with stable brain CD45 knock-down
CD45 knock-down was performed by stable transfection of CD45 short hairpin RNA (shRNA) into 3-month-old C57BL/6L mice brain (CD45 RNA interference [RNAi]) by ICV injection. For controls, empty victor (EV), scrambled shRNA (SCR), and PBS were ICV injected respectively (n=3 for each group). Subsequently, each mouse from four groups was injected with 500 ng of HIV-1 Tat by ICV. Sections from each hemi-brain were stained by immunofluorescence for: NeuN (Figure 4 a-d), DAPI (Figure 4 e-h; 10 ×), GFAP (Figure 4i-l), (DAPI, m-p; 10 ×), Iba1 (Figure 4 q-t) and (DAPI, Fig 4u-x; 20 ×). As expected, HIV-1 Tat exacerbated neuronal injury in CD45 knock- down mice in cortical regions examined (Figure 4 d/h) compared with other controls (Figure 4 a-c/e-g). Since astrogliosis is a common feature of the HAND brain and promotes neuronal loss [45], we examined GFAP as well. As expected we found HIV-1 Tat augmented astrocytosis determined by GFAP staining (Figure 4 l/p) in CD45 knock-down mice compared with control groups (Figure 4 i-k/m-o). Likewise due to the pathological importance of activated microglia, we immunohistochemically stained with Iba-1. Here we found HIV-1 Tat injection was associated increased in microglial expression in CD45 knock down mice (Figure 4 t/x) vs. control groups (Figure 4 q-s/u-w) in hippocampi. As additional confirmation of the role of CD45 in the modulation of HIV-1 Tat-induced neuronal injury in vivo, we prepared brain homogenates from these mice for Western blot analysis of CD45 expression to confirm stable knock-down (Fig 5a) as well as Bcl-XL and Bax protein expression to monitor anti- and proapoptotic signaling respectively. One-way ANOVA followed by post hoc comparison revealed significant differences between CD45 RNAi compared to PBS, EV, or SCR for both TNF-α and IL-β release (** p < 0.001) (Figure 5c) upon HIV-1 Tat ICV injection in CD45 knockdown mice compared to the three control groups (PBS, EV, and SCR) (n=3 for each group of mice). Moreover the relative intensity of western blot band density ratio of Bcl-XL to Bax was significantly decreased in CD45 knock-down mice compared with scrambled mice PBS group or the mice receiving EV (** p < 0.01) (Figure 5b).
Figure 4.
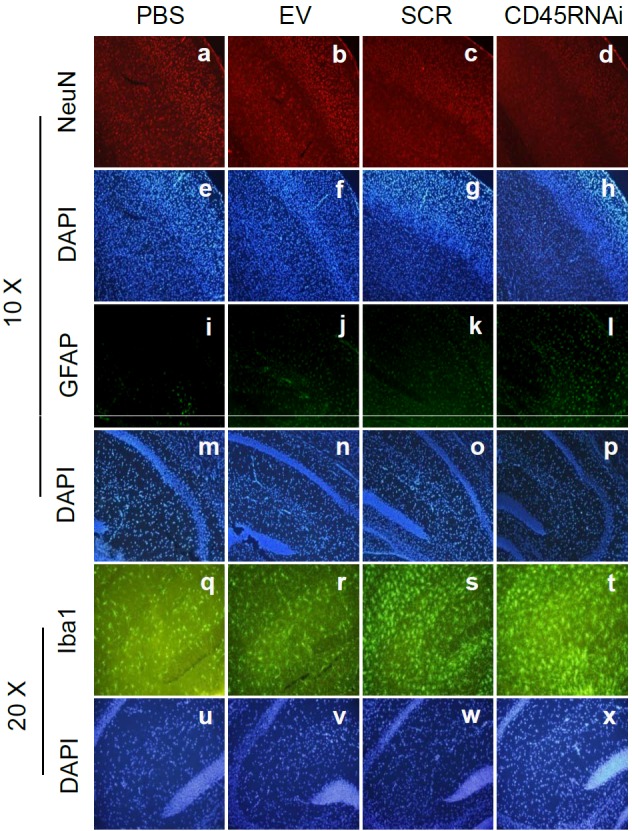
HIV-1 Tat was promoted neuronal death/damage and gliosis in stable knock down CD45 mouse brain detected by immunofluorescence. CD45 knock-down was performed by stable transfection of CD45shRNA into 3-month-old C57BL/6L mice brain (CD45 RNAi) by ICV injection; as controls, empty victor (EV), scrambled shRNA (SCR), and PBS were ICV injected to mouse brain, respectively (n=3 for each group). Subsequently, each mouse from four groups was injected 500 ng of HIV-1 Tat by ICV. Sections from a half-brain were stained by immunofluorescence of NeuN (a-d; for DAPI, e-h; 10 ×), GFAP (i-l; for DAPI, m-p; 10 ×) and Iba1 (q-t; for DAPI, u-x; 20 ×). HIV-1 Tat exacerbated neuronal loss in CD45 knock-down mouse brain sections in cortex (d/h) compared with other controls (a-c/e-g). Further, HIV-1 Tat injection augmented astrocytosis as determined by GFAP staining (i/p) compared with control groups (i-k/m-o). Finally, HIV-Tat injection was increased in microglial expression visualized by Iba1 staining in CD45 knock down mice (t/x) vs. control groups (q-s/u-w) in hippocampus.
Discussion
Inflammation caused by microglial cells drives, at least in part, many neurodegenerative diseases such as MS, PD , HAND and AD; suggesting that therapeutics targeting microglial activation might be efficacious in treating such diseases [2,34,46-53].
Upon challenging, microglia undergo dramatic phenotypic, immunochemical, and functional changes, collectively referred to as ‘activation’ and the activated microglia produce a variety of bioactive molecules with potential toxicity to neurons. Ample evidence indicates that immune and inflammatory responses mediated by activated microglia have a pivotal role in the pathogenesis of HAND [52,53].
Further, microglia modulate both mature and neural stem-cell proliferation, survival, and differentiation [54-57]. For example, stimulating microglia with interleukin- 4 (IL-4) results in the release of factors that promote neurogenesis [58,59]. On the other hand similar stimulaton of CD40 ligand has the opposite effect (reviewed in [60,61]). Thus, microglia represent a double- edged sword which can positively or negatively impact CNS function [60]. Nevertheless, the molecular regulation of microglial behavior is not well understood and attempts to date at reducing neuroinflammation caused by microglial activation have only been partially efficacious [62], possibly because of the fact that such strategies are more general inhibitors of inflammation than specific inhibitors of microglial-associated neuroinflammation.
Studies have also shown that HIV-1-infected, and immune-activated microglia, release a number of soluble substances including proinflammatory cytokines, chemokines, excitatory amino acids, nitric oxide (NO), and reactive oxygen species (ROS), viral proteins, which can diffuse and injure surrounding and distant neurons; contributing to HAND pathogenesis [63-65]. Therefore, it is important to identify potential target(s) to regulate microglia activation and their resultant production of neurotoxins in order to control microglia-associated neurotoxicity.
Following from this idea, pharmacotherapeutics specifically aimed at blocking microglial activation may well be more efficient at ameliorating microglial-associated neuropathology in HAND. In this study, we focused on identifying a specific cell surface receptor target, which, when activated, could inhibit microglial activation far upstream of intracellular proinflammatory mediators such as the MAPK pathway. Our rationale for such investigation was that, if we could inhibit microglial activation by HIV-1 Tat protein very early on, the amplification of the inflammatory response associated with activation of proinflammatory intracellular signal transduction cascades could be abated. Our data show that microglia can be activated after treatment with HIV-1 Tat proteins and, the PTP inhibitor, phen. This result led us to investigate stimulation of this membrane-bound PTP as a negative regulator of microglial activation. Data showed that cross-linking CD45 markedly reduced microglial activation resulting from HIV-1 Tat and phen co-treatment. Furthermore, we observed decreased activation of p44/42 MAPK under these conditions, suggesting that CD45 crosslinking stimulates the CD45-associated PTP pathway, and that stimulation of this pathway negatively controls p44/42 MAPK activation. In accord, co-treatment of HIV-1 Tat and phen-activated microglia with PD98059, an inhibitor of MEK1/2 (the upstream activator of p44/42 MAPK), resulted in statistically interactive blockade of microglial pro-inflammatory activation. We found that microglia deficient for CD45 could be directly activated by HIV-1 Tat proteins in vitro, and brains from wild type mice deficient for CD45 (via ICV CD45 knock-down or by complete genetic ablation [CD45-/- mice]) demonstrated markedly increased TNF-α and IL-1β levels upon HIV-1 Tat treatment compared CD45-sufficient mouse brains. These results suggest that stimulation of CD45 is a viable approach for mitigating HIV-1 Tat-induced microglial activation.
More specifically, in our original experiment we showed that that co-treatment with the PTP inhibitor phen and HIV-Tat resulted in microglial activation as evidenced by increased IL-β and TNF-α release (Figure 1). However, the question arose of whether this effect was dependent on PTP inhibition as opposed to inhibition of other phosphatases. Thus, we co-treated wild-type primary culture microglia with HIV-1 Tat and either sodium orthovanadate, another PTP inhibitor, or okadaic acid, an inhibitor of protein phosphatase 2A, and measured IL-1β and TNF-α release. We observed that sodium orthovanadate treatment in conjunction with HIV-1 Tat produced results similar to those of phen and Aβ peptide co-treatment. However, IL-1β and TNF-α were not detectable in the media of okadaic acid- and Aβ-co-treated microglia; suggesting that treatment of microglia with specific inhibitors of PTPs, as opposed to general phosphatase inhibitors, along with HIV-1 Tat, promotes microglial activation; the specific effect of PTP stimulation via CD45 in opposing microglial activation induced by phen and HIV-1 Tat.
Aside from its importance to microglia, past studies have shown that HIV- 1 induced inhibition of CD45 PTP activity positively correlates with disease progression and apoptosis, and negatively correlates with anti-CD3-induced T lymphocyte proliferation. Indeed CD45 opposes HIV-1-induced T cell hyporesponsiveness and apoptosis [66]. Giovanni and colleagues [66] found the proliferative response to anti-CD3 as well as the CD45-associated PTP activity were significantly reduced in HIV progressors [66].
To examine whether increasing CD45 activity could block microglial activation resulting from co-treatment with phen and HIV-1 Tat, we activated wild-type microglia with phen and HIV-1 Tat, added CD45 recombinant protein (20 U/ml) to these cells, and measured IL-1β and TNF-α release. We observed marked reduction of IL-β and TNF-α after addition of CD45 recombinant protein to activated microglia compared with appropriate controls. In accord, treatment of activated microglia with CD45 recombinant protein blocked IL-1β and TNF-α release to an extent similar to that resulting from cross-linking CD45, further substantiating that CD45 crosslinking stimulates the CD45 PTP pathway.
To further confirm CD45-mediated downregulation of microglial activation induced by co-treatment with phen and HIV-1 Tat protein, we performed shRNA knockdown via ICV injection of specific CD45shRNA. Data showed that CD45 knock-down markedly attenuated microglial activation as evidenced by IL-1β and TNF-α release (Figure 5). These data raise the possibility that stimulation of the CD45 pathway negatively controls microglial activation induced by HIV-1 Tat proinflammatory stimuli in vitro and in vivo and suggest that therapeutics targeting stimulation of CD45 may be beneficial in suppressing microglial activation, a central pathogenic component of HAND.
Figure 5.
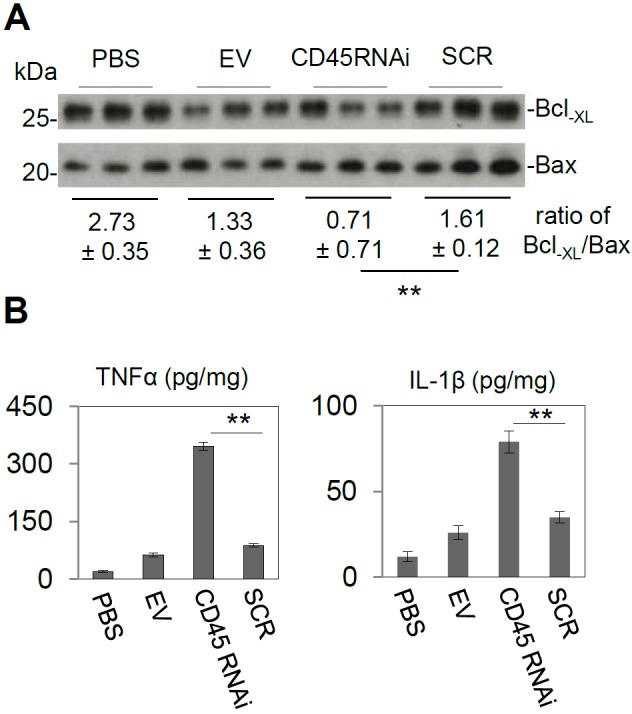
ICV injection of HIV-1 Tat promotes gliosis in stable knock-down CD45 mice subjected to stable CNS CD45 knockdown (A) Confirmation by western blot of CD45 knock-down mice (CD45 RNAi) and three control groups (PBS, EV, and SCR) (n=3 for each group of mice). (B) Brain homogenates were prepared from half of brain tissues and probed by western blot using antibodies against Bcl-XL (neuronal survival) and Bax (apoptosis). The relative intensity of the western blot band ratio of Bcl-XL to Bax was significantly decreased in CD45 knock-down mice compared with scrambled mice (** p < 0.01) and other control groups. (C) TNF-α (left panel) and IL-1β (right panel) ELISA showed significantly increased CD45 knock-down mice compared with scrambled mice (SCR) and other control groups (PBS and EV) (** p < 0.001).
Acknowledgements
BG is supported by NIMH/NIH grant (1K08 MH082642-01A1) (PI). JT is supported by NIH grants (1R41AG031586-01), (1R43AG033417-01), and 1R43AT004871-01 as well as a Veterans Administration grant (MH080168). We extend thanks to Dr. Paula Bickford and Dr. Allison Willing for the use of their confocol microscopy system. In addition we thank Dr. Linella Gemma and Dr. Josh Morganti for providing BV-2 microglia. HIV-1 Tat protein was provided by the NIH AIDS Reference and Reagent Program, Germantown, MD.
References
- 1.McArthur JC, Steiner J, Sacktor N, Nath A. Human immunodeficiency virus-associated neurocognitive disorders: Mind the gap. Ann Neurol. 67:699–714. doi: 10.1002/ana.22053. [DOI] [PubMed] [Google Scholar]
- 2.Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69:1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunfee R, Thomas ER, Gorry PR, Wang J, Ancuta P, Gabuzda D. Mechanisms of HIV-1 neurotropism. Curr HIV Res. 2006;4:267–278. doi: 10.2174/157016206777709500. [DOI] [PubMed] [Google Scholar]
- 4.Petito CK, Cho ES, Lemann W, Navia BA, Price RW. Neuropathology of acquired immunodeficiency syndrome (AIDS): an autopsy review. J Neuropathol Exp Neurol. 1986;45:635–646. doi: 10.1097/00005072-198611000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Ho DD, Rota TR, Schooley RT, Kaplan JC, Allan JD, Groopman JE, Resnick L, Felsenstein D, Andrews CA, Hirsch MS. Isolation of HTLV-III from cerebrospinal fluid and neural tissues of patients with neurologic syndromes related to the acquired immunodeficiency syndrome. N Engl J Med. 1985;313:1493–1497. doi: 10.1056/NEJM198512123132401. [DOI] [PubMed] [Google Scholar]
- 6.Koenig S, Gendelman HE, Orenstein JM, Dal Canto MC, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986;233:1089–1093. doi: 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- 7.Robertson KR, Smurzynski M, Parsons TD, Wu K, Bosch RJ, Wu J, McArthur JC, Collier AC, Evans SR, Ellis RJ. The prevalence and incidence of neurocognitive impairment in the HAART era. Aids. 2007;21:1915–1921. doi: 10.1097/QAD.0b013e32828e4e27. [DOI] [PubMed] [Google Scholar]
- 8.Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, Stern Y, Albert S, Palumbo D, Kieburtz K, De Marcaida JA, Cohen B, Epstein L. HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirol. 2002;8:136–142. doi: 10.1080/13550280290049615. [DOI] [PubMed] [Google Scholar]
- 9.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 10.Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, Zaman T, Stone D, Mefford M, Morgello S, Singer EJ, Wolinsky SM, Gabuzda D. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One. 2008;3:e2516. doi: 10.1371/journal.pone.0002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eden A, Price RW, Spudich S, Fuchs D, Hagberg L, Gisslen M. Immune activation of the central nervous system is still present after >4 years of effective highly active antiretroviral therapy. J Infect Dis. 2007;196:1779–1783. doi: 10.1086/523648. [DOI] [PubMed] [Google Scholar]
- 12.Glass JD, Fedor H, Wesselingh SL, McArthur JC. Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol. 1995;38:755–762. doi: 10.1002/ana.410380510. [DOI] [PubMed] [Google Scholar]
- 13.Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol. 2005;64:529–536. doi: 10.1093/jnen/64.6.529. [DOI] [PubMed] [Google Scholar]
- 14.Cross SA, Cook DR, Chi AW, Vance PJ, Kolson LL, Wong BJ, Jordan-Sciutto KL, Kolson DL. Dimethyl fumarate, an immune modulator and inducer of the antioxidant response, suppresses HIV replication and macrophage-mediated neurotoxicity: a novel candidate for HIV neuroprotection. J Immunol. 187:5015–5025. doi: 10.4049/jimmunol.1101868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Avison MJ, Nath A, Greene-Avison R, Schmitt FA, Bales RA, Ethisham A, Greenberg RN, Berger JR. Inflammatory changes and breakdown of microvascular integrity in early human immunodeficiency virus dementia. J Neurovirol. 2004;10:223–232. doi: 10.1080/13550280490463532. [DOI] [PubMed] [Google Scholar]
- 16.Rumbaugh JA, Nath A. Developments in HIV neuropathogenesis. Curr Pharm Des. 2006;12:1023–1044. doi: 10.2174/138161206776055877. [DOI] [PubMed] [Google Scholar]
- 17.Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature. 1995;375:497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- 18.Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, Jeang KT. Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc Natl Acad Sci USA. 2000;97:11466–11471. doi: 10.1073/pnas.97.21.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayashi K, Pu H, Andras IE, Eum SY, Yamauchi A, Hennig B, Toborek M. HIV-TAT protein upregulates expression of multidrug resistance protein 1 in the blood-brain barrier. J Cereb Blood Flow Metab. 2006;26:1052–1065. doi: 10.1038/sj.jcbfm.9600254. [DOI] [PubMed] [Google Scholar]
- 20.D'Aversa TG, Yu KO, Berman JW. Expression of chemokines by human fetal microglia after treatment with the human immunodeficiency virus type 1 protein Tat. J Neurovirol. 2004;10:86–97. doi: 10.1080/13550280490279807. [DOI] [PubMed] [Google Scholar]
- 21.Eugenin EA, Dyer G, Calderon TM, Berman JW. HIV-1 tat protein induces a migratory phenotype in human fetal microglia by a CCL2 (MCP-1)-dependent mechanism: possible role in NeuroAIDS. Glia. 2005;49:501–510. doi: 10.1002/glia.20137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hermiston ML, Xu Z, Weiss A. CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev Immunol. 2003;21:107–137. doi: 10.1146/annurev.immunol.21.120601.140946. [DOI] [PubMed] [Google Scholar]
- 23.Cosenza-Nashat MA, Kim MO, Zhao ML, Suh HS, Lee SC. CD45 isoform expression in microglia and inflammatory cells in HIV-1 encephalitis. Brain Pathol. 2006;16:256–265. doi: 10.1111/j.1750-3639.2006.00027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stein VM, Baumgartner W, Schroder S, Zurbriggen A, Vandevelde M, Tipold A. Differential expression of CD45 on canine microglial cells. J Vet Med A Physiol Pathol Clin Med. 2007;54:314–320. doi: 10.1111/j.1439-0442.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 25.Irie-Sasaki J, Sasaki T, Matsumoto W, Opavsky A, Cheng M, Welstead G, Griffiths E, Krawczyk C, Richardson CD, Aitken K, Iscove N, Koretzky G, Johnson P, Liu P, Rothstein DM, Penninger JM. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature. 2001;409:349–354. doi: 10.1038/35053086. [DOI] [PubMed] [Google Scholar]
- 26.Irie-Sasaki J, Sasaki T, Penninger JM. CD45 regulated signaling pathways. Curr Top Med Chem. 2003;3:783–796. doi: 10.2174/1568026033452339. [DOI] [PubMed] [Google Scholar]
- 27.Streuli M, Hall LR, Saga Y, Schlossman SF, Saito H. Differential usage of three exons generates at least five different mRNAs encoding human leukocyte common antigens. J Exp Med. 1987;166:1548–1566. doi: 10.1084/jem.166.5.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akiyama H, Ikeda K, Katoh M, McGeer EG, McGeer PL. Expression of MRP14, 27E10, interferon-alpha and leukocyte common antigen by reactive microglia in postmortem human brain tissue. J Neuroimmunol. 1994;50:195–201. doi: 10.1016/0165-5728(94)90046-9. [DOI] [PubMed] [Google Scholar]
- 29.Cosenza MA, Zhao ML, Si Q, Lee SC. Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis. Brain Pathol. 2002;12:442–455. doi: 10.1111/j.1750-3639.2002.tb00461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnston JB, Silva C, Gonzalez G, Holden J, Warren KG, Metz LM, Power C. Diminished adenosine A1 receptor expression on macrophages in brain and blood of patients with multiple sclerosis. Ann Neurol. 2001;49:650–658. [PubMed] [Google Scholar]
- 31.Licastro F, Mallory M, Hansen LA, Masliah E. Increased levels of alpha-1-antichymotrypsin in brains of patients with Alzheimer's disease correlate with activated astrocytes and are affected by APOE 4 genotype. J Neuroimmunol. 1998;88:105–110. doi: 10.1016/s0165-5728(98)00096-4. [DOI] [PubMed] [Google Scholar]
- 32.Masliah E, Mallory M, Hansen L, Alford M, Albright T, Terry R, Shapiro P, Sundsmo M, Saitoh T. Immunoreactivity of CD45, a protein phosphotyrosine phosphatase, in Alzheimer's disease. Acta Neuropathol. 1991;83:12–20. doi: 10.1007/BF00294425. [DOI] [PubMed] [Google Scholar]
- 33.Sedgwick JD, Ford AL, Foulcher E, Airriess R. Central nervous system microglial cell activation and proliferation follows direct interaction with tissue-infiltrating T cell blasts. J Immunol. 1998;160:5320–5330. [PubMed] [Google Scholar]
- 34.Tan J, Town T, Mori T, Wu Y, Saxe M, Crawford F, Mullan M. CD45 opposes beta-amyloid peptide-induced microglial activation via inhibition of p44/42 mitogen-activated protein kinase. J Neurosci. 2000;20:7587–7594. doi: 10.1523/JNEUROSCI.20-20-07587.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan J, Town T, Mullan M. CD45 inhibits CD40L-induced microglial activation via negative regulation of the Src/p44/42 MAPK pathway. J Biol Chem. 2000;275:37224–37231. doi: 10.1074/jbc.M002006200. [DOI] [PubMed] [Google Scholar]
- 36.Suh HS, Kim MO, Lee SC. Inhibition of granulocyte-macrophage colony-stimulating factor signaling and microglial proliferation by anti-CD45RO: role of Hck tyrosine kinase and phosphatidylinositol 3-kinase/Akt. J Immunol. 2005;174:2712–2719. doi: 10.4049/jimmunol.174.5.2712. [DOI] [PubMed] [Google Scholar]
- 37.Kim MO, Suh HS, Si Q, Terman BI, Lee SC. Anti-CD45RO suppresses human immunodeficiency virus type 1 replication in microglia: role of Hck tyrosine kinase and implications for AIDS dementia. J Virol. 2006;80:62–72. doi: 10.1128/JVI.80.1.62-72.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Y, Wu J, Lu Y. Mechanism of HIV-1-TAT induction of interleukin-1beta from human monocytes: Involvement of the phospholipase C/protein kinase C signaling cascade. J Med Virol. 82:735–746. doi: 10.1002/jmv.21720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giunta B, Ehrhart J, Townsend K, Sun N, Vendrame M, Shytle D, Tan J, Fernandez F. Galantamine and nicotine have a synergistic effect on inhibition of microglial activation induced by HIV-1 gp120. Brain Res Bull. 2004;64:165–170. doi: 10.1016/j.brainresbull.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 40.Park IW, Wang JF, Groopman JE. HIV-1 Tat promotes monocyte chemoattractant protein-1 secretion followed by transmigration of monocytes. Blood. 2001;97:352–358. doi: 10.1182/blood.v97.2.352. [DOI] [PubMed] [Google Scholar]
- 41.Milani D, Mazzoni M, Zauli G, Mischiati C, Gibellini D, Giacca M, Capitani S. HIV-1 Tat induces tyrosine phosphorylation of p125FAK and its association with phosphoinositide 3-kinase in PC12 cells. Aids. 1998;12:1275–1284. doi: 10.1097/00002030-199811000-00008. [DOI] [PubMed] [Google Scholar]
- 42.Oshima T, Flores SC, Vaitaitis G, Coe LL, Joh T, Park JH, Zhu Y, Alexander B, Alexander JS. HIV-1 Tat increases endothelial solute permeability through tyrosine kinase and mitogen-activated protein kinase-dependent pathways. Aids. 2000;14:475–482. doi: 10.1097/00002030-200003310-00002. [DOI] [PubMed] [Google Scholar]
- 43.Avraham HK, Jiang S, Lee TH, Prakash O, Avraham S. HIV-1 Tat-mediated effects on focal adhesion assembly and permeability in brain microvascular endothelial cells. J Immunol. 2004;173:6228–6233. doi: 10.4049/jimmunol.173.10.6228. [DOI] [PubMed] [Google Scholar]
- 44.Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, Ehrhart J, Silver AA, Sanberg PR, Tan J. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem. 2004;89:337–343. doi: 10.1046/j.1471-4159.2004.02347.x. [DOI] [PubMed] [Google Scholar]
- 45.Rrapo E, Zhu Y, Tian J, Hou H, Smith A, Fernandez F, Tan J, Giunta B. Green Tea-EGCG reduces GFAP associated neuronal loss in HIV-1 Tat transgenic mice. Am J Transl Res. 2009;1:72–79. [PMC free article] [PubMed] [Google Scholar]
- 46.Jayadev S, Nesser NK, Hopkins S, Myers SJ, Case A, Lee RJ, Seaburg LA, Uo T, Murphy SP, Morrison RS, Garden GA. Transcription factor p53 influences microglial activation phenotype. Glia. 59:1402–1413. doi: 10.1002/glia.21178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henkel JS, Engelhardt JI, Siklos L, Simpson EP, Kim SH, Pan T, Goodman JC, Siddique T, Beers DR, Appel SH. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol. 2004;55:221–235. doi: 10.1002/ana.10805. [DOI] [PubMed] [Google Scholar]
- 48.Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson's disease brains. Acta Neuropathol. 2003;106:518–526. doi: 10.1007/s00401-003-0766-2. [DOI] [PubMed] [Google Scholar]
- 49.Moreau C, Devos D, Brunaud-Danel V, Defebvre L, Perez T, Destee A, Tonnel AB, Lassalle P, Just N. Elevated IL-6 and TNF-alpha levels in patients with ALS: inflammation or hypoxia? Neurology. 2005;65:1958–1960. doi: 10.1212/01.wnl.0000188907.97339.76. [DOI] [PubMed] [Google Scholar]
- 50.Nagatsu T, Sawada M. Inflammatory process in Parkinson's disease: role for cytokines. Curr Pharm Des. 2005;11:999–1016. doi: 10.2174/1381612053381620. [DOI] [PubMed] [Google Scholar]
- 51.Tarkowski E, Rosengren L, Blomstrand C, Wikkelso C, Jensen C, Ekholm S, Tarkowski A. Intrathecal release of pro- and anti-inflammatory cytokines during stroke. Clin Exp Immunol. 1997;110:492–499. doi: 10.1046/j.1365-2249.1997.4621483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Glass JD, Wesselingh SL. Microglia in HIV-associated neurological diseases. Microsc Res Tech. 2001;54:95–105. doi: 10.1002/jemt.1124. [DOI] [PubMed] [Google Scholar]
- 53.Dheen ST, Kaur C, Ling EA. Microglial activation and its implications in the brain diseases. Curr Med Chem. 2007;14:1189–1197. doi: 10.2174/092986707780597961. [DOI] [PubMed] [Google Scholar]
- 54.Barish ME, Mansdorf NB, Raissdana SS. Gamma-interferon promotes differentiation of cultured cortical and hippocampal neurons. Dev Biol. 1991;144:412–423. doi: 10.1016/0012-1606(91)90433-4. [DOI] [PubMed] [Google Scholar]
- 55.Bernardino L, Agasse F, Silva B, Ferreira R, Grade S, Malva JO. Tumor necrosis factor-alpha modulates survival, proliferation, and neuronal differentiation in neonatal subventricular zone cell cultures. Stem Cells. 2008;26:2361–2371. doi: 10.1634/stemcells.2007-0914. [DOI] [PubMed] [Google Scholar]
- 56.Chiu IM, Chen A, Zheng Y, Kosaras B, Tsiftsoglou SA, Vartanian TK, Brown RH Jr, Carroll MC. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci USA. 2008;105:17913–17918. doi: 10.1073/pnas.0804610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clarke RM, Lyons A, O'Connell F, Deighan BF, Barry CE, Anyakoha NG, Nicolaou A, Lynch MA. A pivotal role for interleukin-4 in atorvastatin-associated neuroprotection in rat brain. J Biol Chem. 2008;283:1808–1817. doi: 10.1074/jbc.M707442200. [DOI] [PubMed] [Google Scholar]
- 58.Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, Martino G, Schwartz M. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2006;31:149–160. doi: 10.1016/j.mcn.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 59.Choi SH, Veeraraghavalu K, Lazarov O, Marler S, Ransohoff RM, Ramirez JM, Sisodia SS. Non-cell-autonomous effects of presenilin 1 variants on enrichment-mediated hippocampal progenitor cell proliferation and differentiation. Neuron. 2008;59:568–580. doi: 10.1016/j.neuron.2008.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salemi J, Obregon DF, Cobb A, Reed S, Sadic E, Jin J, Fernandez F, Tan J, Giunta B. Flipping the switches: CD40 and CD45 modulation of microglial activation states in HIV associated dementia (HAD) Mol Neurodegener. 6:3. doi: 10.1186/1750-1326-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, Mattson MP, Flavell RA, Mullan M. Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science. 1999;286:2352–2355. doi: 10.1126/science.286.5448.2352. [DOI] [PubMed] [Google Scholar]
- 62.Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J. Nonsteroidal anti-inflammatory drugs in Alzheimer's disease. Neurology. 1995;45:51–55. doi: 10.1212/wnl.45.1.51. [DOI] [PubMed] [Google Scholar]
- 63.Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, D'Emilia DM, Friedlander RM, Yuan J, Masliah E, Lipton SA. Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J Neurosci. 2002;22:4015–4024. doi: 10.1523/JNEUROSCI.22-10-04015.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- 65.Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci USA. 1999;96:8212–8216. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giovannetti A, Pierdominici M, Mazzetta F, Mazzone AM, Ricci G, Prozzo A, Pandolfi F, Paganelli R, Aiuti F. HIV type 1-induced inhibition of CD45 tyrosine phosphatase activity correlates with disease progression and apoptosis, but not with anti-CD3-induced T cell proliferation. AIDS Res Hum Retroviruses. 2000;16:211–219. doi: 10.1089/088922200309304. [DOI] [PubMed] [Google Scholar]