

Abstract
Betulinic acid derivatives modified at the C28 position are HIV-1entry inhibitors such as compound A43D; however, modified at the C3 position instead of C28 give HIV-1 maturation inhibitor such as bevirimat. Bevirimat exhibited promising pharmacokinetic profiles in clinical trials, but its effectiveness was compromised by the high baseline drug resistance of HIV-1 variants with polymorphism in the putative drug binding site. In an effort to determine whether the viruses with bevirimat resistant polymorphism also altered their sensitivities to the betulinic acid derivatives that inhibit HIV-1 entry, a series of new betulinic acid entry inhibitors were synthesized and tested for their activities against HIV-1 NL4-3 and NL4-3 variants resistant to bevirimat. The results show that the bevirimat resistant viruses were approximately 5- to10-fold more sensitive to three new glutamine ester derivatives (13, 15 and 38) and A43D in an HIV-1 multi-cycle replication assay. In contrast, the wild type NL4-3 and the bevirimat resistant variants were equally sensitive to the HIV-1 RT inhibitor AZT. In addition, these three new compounds markedly improved microsomal stability compared to A43D.
Keywords: HIV-1, Entry inhibitor, Maturation inhibitor, Betulinic acid, Berivimat, Berivimat-resistance
The HIV-1 life cycle is initiated by a multi-step entry of HIV into the target cell that involves interactions of HIV-1 gp120 to the cell surface receptor CD4 and co-receptors CXCR4 or CCR5 (1). Binding of gp120 to the cellular receptors triggers conformational changes in HIV-1 envelope glycoproteins that allow HIV-1 to enter the cells (2). Interference at the various steps of HIV entry has been proved to be a successful strategy for drug development of anti-HIV therapy. Examples of drugs that target HIV entry include the gp41 fusion inhibitor enfuvirtide and the CCR5 receptor antagonist maraviroc (1,3,4). Enfuvirtide has high potency and broad spectrum in HIV inhibition but as a peptide, has limited oral bioavailability. Maraviroc is very effective against HIV-1 R5 viruses that use the CCR5 co-receptor for entry, but not X4 strains that use CXCR4 for entry. In mid 90s, a betulinic acid (BA) derivative was reported with potent anti-HIV-1 activity in the early stage of HIV life cycle (5). It has been shown that BA derivatives with side chain modification at the C28 position inhibited viral entry by blocking the conformational change of HIV gp120 during the process of HIV entry (6,7). Further study suggested that these compounds targeted the V3 region of HIV gp120, preventing the subsequent conformational changes in HIV-1 gp41 in HIV entry (8). Among a series of C28 modified BA derivatives, compound A43D exhibited the most potent anti-HIV-1 entry activity (9).
A43D was effective against a variety of HIV subtypes and displayed the strongest inhibition to the clade C HIV-1 strains (8). However, as shown in the results section, the microsomal stability of A43D was poor when it was compared to bevirimat (BVM). BMV is a BA derivative that targets HIV-1 maturation instead of entry by blocking the processing of Gag precursor protein at a specific step of CA-SP1 cleavage. In clinical trials, BVM displayed good bioavailability and pharmacokinetic profiles (10). However, a high baseline BVM resistance was uncovered during the clinical trials among HIV-1 positive patients due to the polymorphism in the HIV-1 Gag region (11, 12). Therefore, it is important to study the possible drug resistance issue in the synthesis of BA derivatives as potential anti-HIV agent. A43D and BVM share the same BA scaffold (Figure 1). BVM possesses a dimethylsuccinic acid side chain at the C3 position, while A43D has a long side chain modification at the C28 position (13). The BA entry inhibitors, such as A43D, do not have C3 side chain modifications but possess a relatively larger C28 side chain with 7 to 9 carbons to be optimal for their anti-entry activity. Thus, the objectives of this study are to synthesize new BA derivatives with improved microsomal stability and to investigate the effectiveness of the compounds not only to wild type HIV-1 but also the BVM-resistant variants.
Figure 1.

Chemical structures of the BA derivatives A43D and Bevirimat.
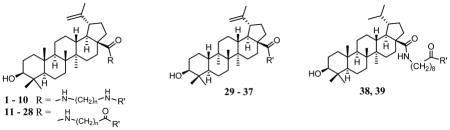
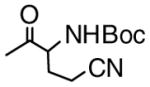
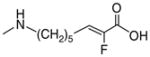
It is clear that the HIV-1 Gag polymorphism greatly reduces the effectiveness of BVM. In light of the structural similarity between BVM and BA derivatives that inhibit HIV-1 entry, it is possible that HIV-1 variants with BVM resistant polymorphism could have an altered sensitivity to the BA entry inhibitors as well. Therefore, in addition to identifying new HIV-1 entry inhibitors with improved metabolic stability, their effectiveness against BVM resistant HIV-1 strains was also determined. As a result, 39 new BA derivatives with a variety of modifications at the C28 position were synthesized. Among them, compounds 1 – 10 have a BA scaffold with C28 modified with a 1,ω-diamino alkane spacer and terminal Boc-masked amino acid (R′). Compounds 11 – 28 are similar to the first ten compounds except that the spacers are ω-aminoalkanoic acid that terminate with an ester of amino acid or aminoalkyl cyano moiety. The spacer length varied from 6 to 9 methylene groups as suggested by previous structure-activity relationship studies (5,9,14,15). Compounds 29 – 37 contain new spacers such as a double bond, fluoride substituent, or oxygen atoms in contrast to those with methylene groups. Compounds 38 and 39 have the dihydrobetulinic acid scaffold with the same or similar C28 modification of 13.
The C-28 modified BA or dihydro-BA derivatives were synthesized using previously described methods (15). As shown in Scheme 1, BA 3-O-acetate was treated with oxalyl chloride and subsequently reacted with an alkyl di-amine or amino alkanoic acid ester in the presence of triethylamine to form an intermediate of BA with a C28 amide linker (2a, 4a, 8a, 11a – 13a, 24a). The intermediate was hydrolyzed to remove the ester group(s) and then coupled with a boc-amino acid, amino acid ester, or aminoalkyl cyano reagent in the presence of N,N′-dicyclohexylcarbodimide/hydroxybenzotriazole/triethylamine to form the final products 2 – 28. Compounds 29 – 37, which contain some unusual linkers at their C28 position such as ethylene glycol (PEG), fluoride substituted or unsaturated hydrocarbons, were also synthesized using a protocol similar to that described in Scheme 1. Compounds with a dihydro-BA scaffold, such as 38 and 39, were synthesized by the same methods applied for compounds 13 and 31 except dihydro-BA 3-O-acetate was used as the starting material.
Scheme 1. Synthesis of betulinic acid derivatives.

Anti-HIV activity of BA derivatives in single cycle infectivity assay
The anti-HIV activities of the BA derivatives were initially evaluated in a single cycle HIV-1 infection assay with NL4-3 virus (Table 1) (15). This assay detects HIV-1 tat-mediated luciferase production in TZM-bl cells after HIV-1 entry. Therefore, it is a convenient and effective assay for HIV-1 entry, but not maturation. The purpose of using this assay was to evaluate and identify new BA derivatives with potent anti-HIV entry activity. The results indicated that most of these new compounds displayed anti-HIV activities except for those compounds with PEG linkers (33 – 37). The results indicated that saturated hydrocarbon linkers are better than less saturated side chains (29 – 32) for anti-HIV-1 activity of these compounds. Previous studies have suggested that the optimal linker length of the C28 side chain is 8 methylene groups for BA derivatives to exhibit maximal activity (5,15). The data of this study indicate that the optimal linker length to be 7 or 8 methylene groups for compounds with a C28 –(CH2)n-NH-COR side chain (1 – 10), and 8 or 9 methylene groups for compounds with a C28 –(CH2)n-CO-NHR side chain (11 – 28). A terminal glutamine group (2, 5, 13, 15, 24, 38 and 39) is favored among a variety of amino acid residues (aa) investigated for the most effective anti-HIV-1 activity. None of the tested compounds were toxic to TZM-bl cells in the single-cycle assay at 4 μM.
Table 1.
Inhibition of HIV-1 NL4-3 by BA entry inhibitors.
| |||
|---|---|---|---|
| Cpd | n | R′ | EC50 (μM)a |
| 1 | 7 | -COCH3 | 0.08 |
| 2 | 7 | -Gln-NHBoc | 0.04 |
| 3 | 7 | -Ala-NHBoc | 0.11 |
| 4 | 8 | -Ala-NHBoc | 0.09 |
| 5 | 8 | -Gln-NHBoc | 0.06 |
| 6 | 8 | -Val-NHBoc | 2.43* |
| 7 | 8 | -Pro-NH-Boc | 0.16 |
| 8 | 9 | -Gln-NHBoc | 0.12 |
| 9 | 9 | -Ala-NHBoc | >4* |
| 10 | 8 |
|
0.08 |
| 11 | 6 | -Gln-OMe | >4* |
| 12 | 7 | -Gln-OMe | 0.14 |
| 13 | 8 | -Gln-OMe | 0.09 |
| 14 | 8 | -Gln-O-tBu | 0.14 |
| 15 | 8 | -Gln-OBn | 0.12 |
| 16 | 8 | -Asn-OMe | 0.11 |
| 17 | 8 | -Glu-Di-OMe | 0.06 |
| 18 | 8 | -Ala-OMe | 0.07 |
| 19 | 8 | -Ile-OMe | 0.14 |
| 20 | 8 | -Leu-OMe | 0.16 |
| 21 | 8 | -Phe-OMe | 0.72* |
| 22 | 8 | -Pro-OMe | 0.16* |
| 23 | 8 | -(O-tBu)-Thr-OMe | 3.49* |
| 24 | 9 | -Gln-OMe | 0.06 |
| 25 | 9 | -Asn-OMe | 0.08 |
| 26 | 8 | -NHCH2CN | 0.44* |
| 27 | 8 | -NHC2H4CN | 0.34* |
| 28 | 8 | -N(Me)CH2CN | 0.81* |
| 29 | - |
|
>4* |
| 30 | - |
|
>4* |
| 31 | - |
|
0.15 |
| 32 | - |
|
0.41* |
| 33 | - |
|
>4* |
| 34 | - |
|
>4* |
| 35 | - |
|
>4* |
| 36 | - |
|
>4* |
| 37 | - |
|
>4* |
| 38 | - | -Gln-OMe | 0.05 |
| 39 | - | -Gln-OH | 0.04 |
Data is average of two independent experiments.
Data from one experiment.
Anti-HIV activity of BA derivatives against BVM-resistant variant (V370A)
In contrast to the discussed C28 modified BA derivatives with anti-HIV entry activities, it is known that the C3 modified BA derivative BVM [3-O-(3′,3′-dimethylsuccinyl)-betulinic acid] does not inhibit HIV-1 entry. However, it is a potent HIV-1 maturation inhibitor. Phase II clinical trials revealed a high baseline drug resistance due to pre-existing polymorphisms at the QVT-motif (amino acid residues 369–371) within HIV-1 Gag SP1 (12,16,17). Variation such as V370A, V370M, or V370 deletion resulted in high resistance to BVM, while deletion of T371 resulted in medium resistance to BVM (18,19). The V370A polymorphism was found to be the most prevalent among HIV-1 positive patients (18,19). We have constructed a panel of NL4-3 variants with BVM resistant genotypes (Figure 2).
Figure 2. Genotype of three BVM-resistant variants.
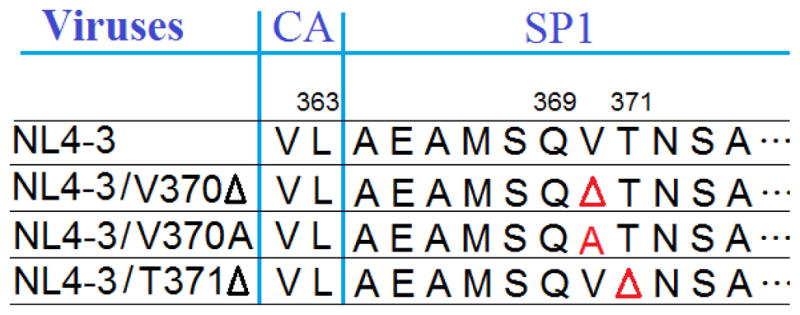
BVM-resistant variants (V370, V370A, and T371Δ) were constructed from HIV-1 NL4-3 with mutations in the QVT motif of HIV-1 Gag (17). The symbol Δ denotes deletion of an amino acid.
The V370A variant was tested extensively in our study to evaluate anti-HIV activity of new BA derivatives against the BVM-resistant strain. We observed that BA derivatives 1 (A43D), 13 and 15 were 2- to 3-fold more potent against the resistant variant, while AZT was 2 times less potent against the resistant variant in a single cycle infection assay (Figure 3).
Figure 3. BVM-resistant HIV-1 variants were more sensitive to BA entry inhibitors.

Inhibition of HIV-1 infection was measured as the reduction in luciferase gene expression in TZM-bl cells after a single round of virus infection as described previously (15). The bevirimat sensitive virus HIV-1 NL4-3 (solid lines) and resistant virus NL4-3 V370A (dashed lines) were used in this assay. Control: HIV-1 NL4-3 or HIV-1 V370A infection, expressed as relative fluorescence units, in the absence of the compounds.
Inhibition of HIV-1 and BVM-resistant variants by BA entry inhibitors in multiple-cycle replication assay
To further evaluate the BA entry inhibitors on BVM-resistant HIV variants, the new BA derivatives were tested against a panel of HIV-1 variants with the V370A, ΔV370, or ΔT371 genotype using the multi-cycle viral replication assay in MT4 cells. These resistant strains showed markedly decreased sensitivity to BVM when compared with the wild type NL4-3 (Table 2). Five synthesized compounds (2, 13, 15, 38 and 39) were tested against HIV-1 NL4-3 and the resistant variants in the multi-cycle viral replication assay. The majority of the tested compounds were found to be at least 4-fold more potent against BVM-resistant variants than the wild type NL4-3 (Table 2). The ΔV370 and ΔT371 variants were particularly sensitive to these entry inhibitors when compared to the V370A variant and were at least 5- to 10-fold more sensitive to the tested BA entry inhibitors when compared with the wild type NL4-3 virus. NL4-3 and the V370A variant were equally sensitive to the non-nucleoside HIV-1 RT inhibitor, TMC-278.
Table 2.
Effect of BA derivatives on HIV BVM-resistant viruses.
| Compound | EC50 (μM) | Toxicityb TD50 (μM) |
|||
|---|---|---|---|---|---|
| NL4-3a | V370Aa | ΔT371b,c | ΔV370b,c | ||
| BVM | 0.076 | >4 | >4 | >4 | >10, <20 |
| 1 (A43D) | 0.12±0.08 | 0.19±0.35 | 0.027 | 0.006 | 12.6 |
| 2 | 0.36±0.09 | 0.09±0.11 | NDc | ND | 7.4 |
| 13 | 1.72±0.83 | 0.02±0.02 | ND | ND | 4.8 |
| 15 | 0.19±0.14 | 0.05±0.03 | 0.026 | 0.023 | 15.0 |
| 38 | 0.58±0.74 | 0.02±0.01 | ND | ND | 4.6 |
| 39 | 0.83±0.37 | 0.24±0.28 | ND | ND | 18.6 |
| TMC-278 | 0.00055 ±0.00014 | 0.00052 ±0.00011 | ND | ND | >0.1 |
Data is the mean of two or more independent experiments and standard deviation.
Data is the average of two independent experiments.
ND = not determined.
In vitro metabolic stability of BA derivatives
The five selected best entry inhibitors derived from BA (2, 13, 15, 38 and 39) were investigated for their microsomal stability using human liver microsomal preparations under oxidative conditions with a reference compound terfenadine. Terfenadine has a moderate-to-fast half-life in vivo of around 3.5 h. The results in Table 3 show that glutamine residue of the C28 side chain with Boc protected amine terminus (2) exhibited the fastest metabolism with t1/2 of 18.92 min. Leaving the glutamine terminus unprotected (39) did not significantly change its microsomal stability (t1/2: 24.75 min) when compared to A43D. The Benzyl ester protection of the glutamine terminus significantly improved microsomal stability of 15 with a t1/2 of 41.01 min when compared to that of A43D (t1/2: 25.48 min) and terfenadine (t1/2: 29.87 min). In addition to the improved microsomal stability, compound 15 exhibited similar antiviral activity against NL4-3 and better activity against the BVM-resistant V370A variant when compared with A43D.
Table 3.
Microsomal stability of BA derivatives.
| Compound | Terfenadine | A43D | 2 | 13 | 15 | 38 | 39 |
|---|---|---|---|---|---|---|---|
| in vitro t1/2 (min)a | 29.87 | 25.48 | 18.93 | 30.13 | 41.01 | 31.64 | 24.75 |
| CLint (ml/min/mg)a | 0.232 | 0.272 | 0.366 | 0.230 | 0.169 | 0.219 | 0.280 |
Average of two separated experiments with linear regression R2 > 0.95.
In summary, we have designed and synthesized 39 new BA derivatives with different C28 side chains. Among them, compounds 2, 13, 15, 38 and 39, carrying a glutamine terminus, showed potent anti-HIV activity with EC50 ranging from 0.04 to 0.12 μM in a single cycle HIV-1 NL4-3 infection assay (Table 1). These BA entry inhibitors were in general more potent against the BVM-resistant variants than the wild type NL4-3 (Figure 2, Table 2). In addition, compounds 13, 15 and 38 showed improved metabolic stability in vitro when compared to the lead compound A43D. These results suggest that the new BA derivatives may serve as promising leads for further drug development against HIV-1.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID) Grant AI-65310 awarded to C. H. Chen, The D. P. Bolognesi Award (Surgical Science, Surgery Department, Duke University) to L. Huang, and in part by Grant AI-077417 from NIAID awarded to K. H. Lee.
Abbreviations
- BA
betulinic acid
- LA
BVM, bevirimat
- PEG
polyethylene glycol
- Boc
tert-butoxycarbonyl
- t-Bu
tert-butyl
- Bn
benzyl
- Cpd
compound
- AZT
3′-azido-3′-deoxythymidine
- EDC
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride
- THF
tetrahydrofuran
- DCM
dichloromethane
Footnotes
Supplementary data: Methods of organic synthesis, anti-HIV assay, and metabolic stability study as well as spectroscopic data of synthesized compounds were included. The supplementary data are available in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Tilton JC, Doms RW. Antiviral Res. 2010;85:91. doi: 10.1016/j.antiviral.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 2.Sattentau QJ, Moore P. J Exp Med. 1991;174:407. doi: 10.1084/jem.174.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS. Nat Med. 1998;4:1302. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 4.Lieberman-Blum SS, Fung HB, Bandres JC. Clin Ther. 2008;30:1228. doi: 10.1016/s0149-2918(08)80048-3. [DOI] [PubMed] [Google Scholar]
- 5.Soler F, Poujade C, Evers M, Carry JC, Hénin Y, Bousseau A, Huet T, Pauwels R, De Clercq E, Mayaux JF, Le Pecq JB, Dereu N. J Med Chem. 1996;39:1069. doi: 10.1021/jm950669u. [DOI] [PubMed] [Google Scholar]
- 6.Holz-Smith SL, Sun IC, Jin L, Matthews TJ, Lee KH, Chen CH. Antimicrob Agents Chemother. 2001;45:60. doi: 10.1128/AAC.45.1.60-66.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang L, Lai W, Ho P, Chen CH. AIDS Res and Human Retroviruses. 2007;23:28. doi: 10.1089/aid.2006.0137. [DOI] [PubMed] [Google Scholar]
- 8.Lai W, Huang L, Ho P, Li ZJ, Montefiori D, Chen CH. Antimicrob Agents Chemother. 2008;52:128. doi: 10.1128/AAC.00737-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang L, Ho P, Lee KH, Chen CH. Bioorg Med Chem. 2006;14:2279. doi: 10.1016/j.bmc.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 10.Smith PF, Ogundele A, Forrest A, Wilton J, Salzwedel K, Doto J, Allaway GP, Martin DE. Antimicrob Agents Chemother. 2007;51:3574. doi: 10.1128/AAC.00152-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baelen KV, Salzwedel K, Rondelez E, Van Eygen V, De Vos S, Verheyen A, Steegen K, Verlinden Y, Allaway GP, Stuyver LJ. Antimicrob Agents Chemother. 2009;53:2185. doi: 10.1128/AAC.01650-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCallister S, Lalezari J, Richmond G, Thompson M, Harrigan R, Martin D, Salzwedel K, Allaway G. Antivir Ther. 2008;13:A10. [Google Scholar]
- 13.Kashiwada Y, Hashimoto F, Cosentino LM, Chen CH, Lee KHJ. Med Chem. 1996;39:1016. doi: 10.1021/jm950922q. [DOI] [PubMed] [Google Scholar]
- 14.Huang L, Yuan X, Aiken C, Chen CH. AntimicrobAgents Chemother. 2004;48:663. doi: 10.1128/AAC.48.2.663-665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dang Z, Lai W, Qian K, Ho P, Lee KH, Chen CH, Huang LJ. Med Chem. 2009;52:7887. doi: 10.1021/jm9004253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adamson CS, Waki K, Ablan SD, Salzwedel K, Freed EO. J Virol. 2009;83:4884. doi: 10.1128/JVI.02659-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Baelen K, Salzwedel K, Rondelez E, Van Eygen V, De Vos S, Verheyen A, Steegen K, Verlinden Y, Allaway GP, Stuyver LJ. Antimicrob Agents Chemother. 2009;53:2185. doi: 10.1128/AAC.01650-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Margot NA, Gibbs CS, Miller MD. Antimicrob Agents Chemother. 2010;54:2345. doi: 10.1128/AAC.01784-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adamson CS, Sakalian M, Salzwedel K, Freed EO. Retrovirology. 2010;7:36. doi: 10.1186/1742-4690-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.