

Abstract
Formation of myelin sheaths by oligodendrocytes (OLs) in the central nervous system (CNS) is essential for rapid nerve impulse conduction. Reciprocal signaling between axons and OLs orchestrates myelinogenesis but remains largely elusive. In this study, we present a novel multi-compartment CNS neuron-glia microfluidic co-culture platform. The platform is capable of conducting parallel localized drug and biomolecule treatments while carrying out multiple co-culture conditions in a single device for studying axon-glia interactions at a higher throughput. The novel “micro-macro hybrid soft-lithography master fabrication” (MMHSM) technique enables large number of precisely replicated PDMS devices incorporating both millimeter and micrometer scale structures to be rapidly fabricated without any manual reservoir punching processes. Axons grown from the neuronal somata were physically and fluidically isolated inside the six satellite axon/glia compartments for localized treatments. Astrocytes, when seeded and co-cultured after the establishment of the isolated axons in the satellite axon/glia compartments, were found to physically damage the established axonal layer as they tend to grow underneath the axons. In contrast, oligodendrocyte progenitor cells (OPCs) could be co-cultured successfully with the isolated axons and differentiated into mature myelin basic protein-expressing OLs with processes aligning to neighboring axons. OPCs inside the six axon/glia compartments were treated with high concentration of ceramide (150 μM) to confirm the fluidic isolation among satellite compartments. In addition, isolated axons were treated with varying concentrations of chondroitin sulfate proteoglycan (CSPG, 0-25 μg/ml) within a single device to demonstrate the parallel localized biomolecular treatment capability of the device. These results indicate that the proposed platform can be used as a powerful tool to study CNS axonal biology and axon-glia interactions with the capacity for localized biomolecular treatments.
Introduction
The proper functioning of vertebrate nervous system depends on rapid nerve impulse conduction achieved by insulating axons with multi-layered myelin sheaths. Myelin sheaths are formed by oligodendrocytes (OLs) in the central nervous system (CNS) and Schwann cells in the peripheral nervous system (PNS). In the CNS, OLs extend their processes, align and spirally wrap around certain axons to form multi-segments of compact myelin layers that maximize the axonal conduction velocity.1, 2 Despite recent progress in understanding the molecular signals in PNS myelination3, little is known about how CNS axons regulate the unique feature of OLs – i.e. forming myelin sheath around axons. OLs are post-mitotic cells that arise from their progenitors, oligodendrocyte progenitor cells (OPCs), which proliferate and migrate throughout the CNS during late embryonic development and subsequently mature into pre-myelinating oligodendrocytes before finally differentiating into myelinating cells in the white matter.4, 5 Regulation of OL development has been studied extensively and the development of an efficient method to grow pure OPCs in culture6 greatly facilitated the identification of many signaling molecules that control OPC proliferation, survival, and differentiation.7-9 The CNS myelination process is highly regulated by reciprocal signaling between myelinating OLs and the axons to be ensheathed10-12. However, the molecular basis of axon-glia signaling in formation of CNS myelin still remains largely unknown. This is in part due to the lack of appropriate in vitro models that are easily accessible for experimental manipulations to unravel the cellular and molecular basis of axon-glia interactions. An in vitro model system that mimics physiological axon-glia interactions with capabilities to easily manipulate the biochemical and physical environment will allow detailed understanding into axon-glia communications.
Conventional cell culture methods, where neurons and glial cells are co-cultured in randomly mixed form, fail to provide means to locally manipulate the physical and biochemical environments in culture, making it difficult to investigate localized interaction of axons and glia in the absence of neuronal somata for detailed mechanistic studies. In order to overcome the limitations of conventional cell culture methods, microfluidic technologies have been applied for cell culture and several neuron culture microsystems were recently demonstrated.13-21 Hur et al. utilized compartmentalized neuron culture device to demonstrate that inhibition of NMII ATPase activity promotes axon regeneration over inhibitory molecules22 and Zhang et al. locally treated isolated DRG axons with nerve growth factor (NGF) for imaging the axonal transport of NGF using a similar scheme.23 We have previously developed a circular microfluidically compartmentalized culture platform composed of one circular soma compartment in the center and one co-centric ring shape axon/glia compartment for the co-culture of OLs with isolated axons.24 Primary cortical neuron cells were cultured inside the device for up to four weeks and axons were successfully isolated inside the axon/glia compartment from neuronal somata. The unique circular design significantly increased the efficiency of axon isolation, and OPCs cultured on isolated axonal layer successfully differentiated into mature OLs after two weeks of co-culture period. However, previously introduced designs allowed only a single treatment to be performed on each cell culture platform and the time consuming manual reservoir punching process made it not suitable for high-throughput testing of drugs or growth factors. Furthermore, the manual punching process often results in inconsistent devices from batch-to-batch since the distance between the two manually punched reservoirs determines the length of the axon-guiding microchannels. This makes it difficult to conduct parallel comparison studies and obtain reproducible results. Hosmane et al. recently introduced a compartmentalized cell culture microsystem where the number of compartments can be modified by punching multiple reservoirs, yet the device still required time-consuming manual punching process.25 In addition, the compartment overhang structure, resulting from the punching procedures, hinders neurons to be positioned close to channel inlets and prevents cells to be co-cultured from being directly loaded on top of the dense isolated axonal layers, significantly limiting axon-glia interactions.
Here, we present a multi-compartment microfluidic co-culture platform where glial cells can be directly seeded on top of physically and fluidically isolated CNS axons. This multi-compartment configuration enables multiple conditions of treatments to be performed on a single device in parallel for increased throughput. In order to eliminate the alignment errors and device-to-device variation from the manual punching process as well as to reduce the fabrication time, a “Micro-macro Hybrid Soft-lithography Master fabrication” (MMHSM) technique that we have previously developed26 was implemented. This technique allowed macro-scale reservoirs and micro-scale fluidic channels to be fabricated by a single-step poly(dimethylsiloxane) (PDMS) soft-lithography process without any manual punching process. In the previous publication, we have successfully demonstrated the fabrication of a microfluidic device having embedded tubing interfaces and confirmed the technical reliability of the process in replicating micro-scale components by measuring the dimension changes throughout the fabrication process. Embryonic CNS neurons and two different types of glial cells were successfully co-cultured inside the device for up to four weeks. Furthermore, the device was used for investigating the localized effect of chondroitin sulfate proteoglycan (CSPG) on isolated axonal layer. We expect that these results can provide critical guidelines for designing various co-culture compartments to study interactions and signalling between CNS axons and glial cells.
Materials and Methods
Design and fabrication
The proposed multi-compartment microfluidic neuron-glia co-culture platform is composed of one circular soma compartment (38.5 mm2) and six square shaped satellite axon/glia compartments (6.3 mm2 per compartment) that are connected via radially positioned arrays of axon-guiding microchannels (20 μm wide, 3 μm high, and 400 μm long, Figure 1 A). Approximately 30 of these axon-guiding microchannels are connecting the soma compartment with each of the axon/glia compartment. Both the soma compartment and the six axon/glia compartments are designed to be a well-type open compartment, where the compartment itself is a reservoir that can hold 20-100 μl of culture medium. These well-type compartments have several advantages over widely used microfluidic channel-type closed-compartment cell culture platforms. First, cell density inside the compartment can be accurately controlled. In culturing adherent type cells, such as neurons, the areal cell density needs to be carefully managed; however, in channel-type cell culture microsystems where suspended cells are typically loaded from the channel inlet reservoirs, much higher density of cells are often observed near inlet and outlet reservoir areas rather than being uniformly distributed throughout the compartment. Second, the well-type compartment provides better cell culture environments by facilitating CO2 exchange and by minimizing exposure of cells to potential uncured monomers secreted from the PDMS that may negatively impact neuronal cell growth.27 Lastly, the design prevents cells from being exposed to strong fluidic flow during the medium exchange process. Culture medium is typically exchanged every 3-4 days and in the channel-type compartment designs, added medium creates fluidic flow inside the compartment that results in some cells to be washed away to outlet reservoirs.
Fig. 1.
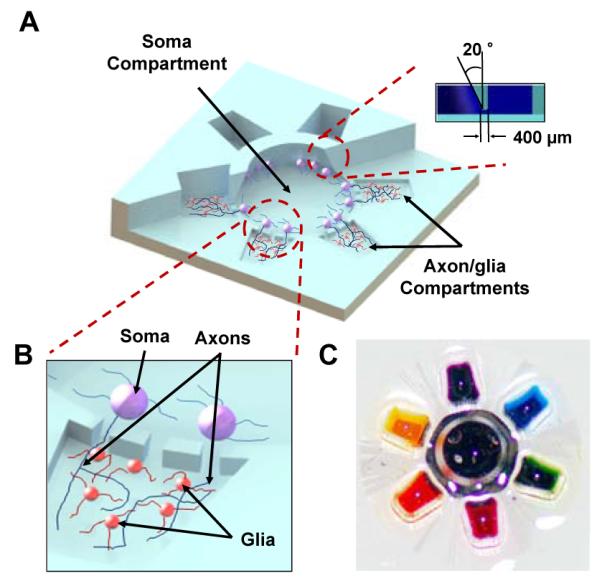
(A) 3D illustration of the multi-compartment neuron-glia co-culture microsystem capable of carrying out multiple localized axon treatments in parallel (Inset: Cross-sectional view of the truncated cone shaped soma compartment). (B) Illustration showing the isolation of axons from neuronal somata for localized axon-glia interaction studies. (C) Photographic image of the neuron-glia co-culture platform (20 × 20 × 4 mm3) filled with seven different color dyes for visualization.
Physical isolation of neuronal somata from axons inside the neighboring axon/glia compartments is achieved by utilizing the significant size difference between the somata and the axons (Figure 1B). Typical size of embryonic day 16 cortical neurons dissected from forebrain of rats are 10 – 20 μm in size, therefore, the shallow height (3 μm) of the axon-guiding microchannels function as a physical barrier and the neuronal somata plated inside the soma compartment are confined within the compartment during the culture period. Axons that differentiate from neuronal somata are much smaller in dimension compared to axon-guiding microchannels and can pass through the shallow microchannel array to form isolated axonal layer inside the neighboring axon/glia compartments (Figure 1B). The six axon/glia compartments surrounding the center soma compartment are fluidically isolated from each other to carry out multiple localized treatments on a single device. Minute fluidic level difference (approximately 500 μm) between the soma compartment and six satellite axon/glia compartments generates slow but sustained flow from the soma compartment toward the six axon/glia compartments during the localized drug treatments. This enables isolated axons inside each axon/glia compartments to be locally treated with drugs or molecular factors while neuronal somata remain unaffected.
Reservoirs, the compartment itself in the well-type design, typically need to hold a certain volume of culture medium to supply cells with nutrients for 3-4 day, thus, typically require millimeter scale dimensions in height. This is a challenging size-scale for most microfabrication techniques. Also, manually punching reservoirs is not applicable to the proposed co-culture platform since six axon/glia compartments and the soma compartments are only 400 μm apart, meaning two holes that are 400 μm apart need to be punched out. In order to fabricate the multi-compartment neuron-glia co-culture platform, the “Micro-macro Hybrid Soft-lithography Master fabrication” (MMHSM) technique, recently introduced by the authors, was utilized.26 This process allowed both microscale (height: 3 μm, width: 20 μm) and macroscale (height: 3.5 mm, width: 3-7 mm) structures to co-exist on the PDMS master mold, from which the final PDMS devices could be easily replicated in large quantities. The macroscale reservoir/compartment structures were first cut from a 75 × 75 mm2 sized poly(methyl methacrylate) (PMMA) block (McMaster-Carr, Atlanta, GA) using a bench-top CNC milling machine (MDX 40, Roland, Irvine, CA). Next, an imprint master was prepared by etching a borofloat wafer with hydrofluoric acid to have arrays of radially positioned 3 μm high and 20 μm wide ridge structures. This imprint master was then hot-embossed at 115°C with 1,082 kPa of pressure for 5 min using a temperature controlled hydraulic press (Specac Ltd., London, UK) against the reservoir-machined PMMA block to directly imprint axon-guiding microchannel arrays on the prepared PMMA block. This resulted in a PMMA block having both millimeter scale reservoir structures and micrometer scale channel structures. The PDMS master was then replicated from this PMMA master by pouring PDMS pre-polymer on the master (10:1 mixture, Sylgard® 184, Dow Corning, Inc., Midland, MI), followed by curing at 85°C for 60 min. The 75 × 75 mm2 sized PDMS mater contains four identical 20 × 20 mm2 sized devices so that each replication results in four devices. The final PDMS device having one soma compartment and six axon/glia compartments connected via arrays of shallow microfluidic channels was replicated from the PDMS master by a single soft-lithography process, and was assembled on a poly-d-lysine (PDL) coated 6-well polystyrene culture plates or glass coverslips after oxygen plasma treatment (Plasma cleaner, Harrick Plasma, Ithaca, NY). This PDMS master was then repeatedly used to fabricate large numbers of devices in significantly reduced time. For sterilization, the device was immersed in 70% ethanol for 30 minutes prior to assembly on the PDL coated substrate. Figure 2 shows the overall fabrication steps of the multi-compartment neuron-glia co-culture platform.
Fig. 2.
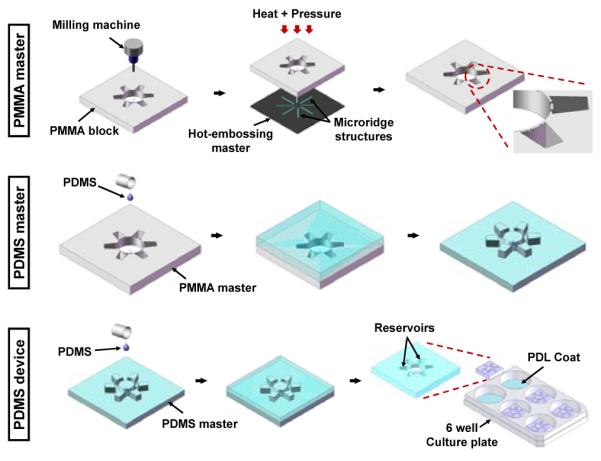
Fabrication and assembly steps for the multi-compartment neuron-glia co-culture microsystem. The PDMS device having both the macroscale reservoirs and the microscale axon-guiding channels is replicated by a single step PDMS soft-lithography process using the ‘MMHSM’ technique. The final PDMS devices are sterilized and assembled on a PDL coated 6-well culture plates for cell culture.
Tissue dissociation and cell culture
Primary CNS neurons were prepared from forebrains of embryonic day 16 Sprague-Dawley rats.28 Briefly, forebrains free of meninges were dissected in ice-cold dissection buffer (Ca2+/Mg2+-free Hank’s Balanced Salt Solution containing 10 mM HEPES), dissociated with L-cysteine activated papain (10 units/ml) in dissection buffer for 5 minutes at 37°C, and resuspended in dissection medium containing trypsin inhibitor (10 mg/ml) for 2-3 minutes. Following two more washes with the trypsin inhibitor solution, the tissue was resuspended in a plating medium (NBB27 + glutamate: neurobasal medium containing 2% B27, 1 mM Glutamine, 25 M glutamic acid, 100 units/ml penicillin, and 100 g/ml streptomycin) and triturated with a fire-polished glass Pasteur pipette until all clumps disappeared. The cells were then passed through a 70 μm cell sieves and live cells were counted using a hemocytometer and trypan blue exclusion assay. The viability of isolated cells was constantly greater than 90-95%.
Primary OLs and astrocytes cultures were prepared from the cerebral hemispheres of Sprague-Dawley rats at postnatal day 1-2 as previously described.29, 30 Forebrains free of meninges were chopped into 1 mm3 blocks and placed into HBSS containing 0.01% trypsin and 10 μg/ml DNase. After digestion, the tissue was collected by centrifugation and triturated with the plating medium DMEM20S (DMEM, 20% fetal bovine serum and 1% penicillin-streptomycin). Cells were plated onto PDL-coated 75 cm2 flasks and were fed with fresh DMEM20S medium every other day for 10-11 days at 37°C in a humidified 5% CO2 incubator. The flasks were pre-shaken for 1 hour at 200 rpm to remove lightly attached microglia followed by overnight shaking to separate OLs from the astrocyte layer. The suspension was plated onto uncoated petri-dishes and incubated for 1 hour to further remove contaminating microglia and astrocytes. Purified OLs were then collected by passing through a 15 μm sieve and centrifuged. OLs isolated in this study were primarily OL progenitors (OPCs) and precursors. Astrocytes were purified (> 95%) from the astrocyte layer in the flask after being exposed to a specific microglia toxin L-leucine methyl ester (1 mM) for 1 hour and were sub-cultured one to two times. Ca2+/Mg2+-free Hank’s balanced salt solution, neurobasal medium, B27, penicillin, streptomycin and goat serum were from Invitrogen (Carlsbad, CA). Poly-D-lysine, papain, trypsin inhibitor, glutamine, glutamic acid, paraformaldehyde and Triton X-100 were from Sigma (St. Louis, MO).
Dissected primary neuron cells were loaded into the soma compartment of the device at an areal density of 500-1000 cells/mm2 by application and cultured at 37°C in a humidified 5% CO2 incubator. After 14-17 days in culture when dense axonal layer inside the axon/glia compartments had been established, OPCs and astrocytes were loaded on top of isolated axon layer. Areal cell density of co-cultured OPC and astrocyte was adjusted from 500 to 2000 cells/mm2 by the experimental conditions. Neurons were fed with the plating medium (NBB27 + glutamate) for 3 days and then NBB27 medium thereafter until glial cells were added. DMEM/NBB27 medium (NBB27 with DMEM, sodium pyruvate, SATO, and D-Biotin) was used for neuron-glia co-cultures. Culture medium was half changed every 3-4 days.
Parallel localized biomolecular treatment
In order to carry out multiple localized experimental treatments on a single device in parallel, each axon/glia compartment has to be fluidically isolated from each other and from the soma compartment. For the localized biomolecular treatments, 80 μl of culture medium was loaded to the soma compartment, while only 15 μl was applied to each of the surrounding six axon/glia compartments, generating a fluidic level difference of approximately 500 μm between the soma and satellite compartments. The resulting pressure difference prevented biomolecules added to the isolated axons in the axon/glia compartments from diffusing into the soma compartment, thus enabling localized biomolecular treatment. Culture medium level difference was maintained throughout the localized treatment processes over two days to ensure the fluidic isolation. Fluidic isolation was demonstrated by locally treating OLs with high concentrations of ceramide (150 μM) and performing cell viability assay with Calcein-AM (Sigma Aldrich, St. Louis, MO). 1 mM of Calcein-AM was added to OLs and incubated for 20 minutes followed by thorough rinsing with fresh culture medium.
Parallel screening capability of the multi-compartment platform was shown by locally treating isolated axons inside the six axon/glia compartments with varying concentrations of CSPG (0-25 μg/ml) for 72 hours. Effects of localized CSPG treatment at six different concentrations were examined by staining isolated axons inside the six axon/glia compartments with Calcein-AM. Calcein-AM (1mM) was added only to the six axon/glia compartments while maintaining fluidic isolation. axonal layer inside the axon/glia compartments that remained healthy after the localized CSPG treatment was stained with Calcein-AM. Although the soma compartment and axon/glia compartments were fluidically isolated during the CSPG treatment and the Calcein-AM staining process, axons inside the microfluidic channels expressed green fluorescence due to axonal retrograde transport system.
Surface profilometry
Optical profilometry (Veeco NT9100, Veeco, Plainview, NY) and scanning electron microscopy (SEM) (JEOL 6400, JEOL Ltd., Tokyo, Japan) were used to analyze whether the axon-guiding microchannels are clearly defined on the PDMS device with no pattern distortion or significant dimension change occurring throughout the MMHSM process. Surface roughness change throughout the fabrication was also analyzed to ensure that the device bonding is not affected by the process.
Immunocytochemistry
Prior to fixing and immunostaining the cells, PDMS devices were 100 peeled off from the cell culture substrate followed by rinse with PBS. axons tend to attach to the bottom substrate, and are not damaged by this PDMS peeling process. Cells were fixed with 4% paraformaldehyde in phosphate buffered saline (PBS) for 10-20 minutes, washed with PBS, and blocked with TBS-T (50 mM Tris·HCL, pH 7.4, 150 mM NaCl and 0.1 % Triton X-100) containing 5% goat serum. The fixed cells were incubated overnight at 4°C with antibodies against neurofilament-H (NF) at 1:1000 dilution (Chemicon, Temecula, CA), or myelin basic protein (MBP) at 1:1000 (Covance, Berkeley, CA). After 110 washing with TBS-T, secondary antibody conjugated with either Alexa Fluor 488 or Alexa Fluor 594 (1:1000, Molecular Probes, Inc., Eugene, OR) was incubated with the cells for 1 hour at room temperature. Cell images were captured using a fluorescent microscope (Olympus IX71) equipped with a digital camera (Olympus DP70).
Axon growth analysis
Multiple immunofluorescence images of isolated axons from single axon/glia compartment were stitched and converted into black and white images using a commercial software (Photoshop®, Adobe Systems, Inc., San Jose, CA). The percentage of the white area indicating the area covered with axons within the selected region (1.6 × 0.8 mm2) was then measured with NIS-Element 2.30 (Nikon Instruments, Inc., Tokyo, Japan).
Axon-glia co-culture
Neurons (1000 cells/mm2) were co-cultured with high density of OPCs (2000 cells/mm2) for studying OPC development into mature-OLs and pre-myelinating OLs that aligned to neighboring axons. For axon-astrocyte co-culture, initially 500 cells/mm2 of neurons were co-cultured with 500 cells/mm2 of astrocytes. After observing axonal damage by astrocytes, co-culture conditions have been changed to have neurons (500 cells/mm2) co-cultured with 1000 cells/mm2 of OPCs for the axon-OPC co-culture. The same number of neurons (500 cells/mm2) were co-cultured with 500 cells/mm2 of OPCs and 500 cells/mm2 of astrocytes (i.e. same number of glia cells) for the axon-OPC/astrocytes co-culture.
Results and Discussions
Fabrication
As shown in Figure 2, the final PDMS neuron-glia co-culture device is replicated from the PDMS master mold obtained from the PMMA master mold by two PDMS soft-lithography processes. The MMHSM technique allowed replicating both the compartments and microchannels at once. However, the soma compartment and the surrounding axon/glia compartments were only 400 μm apart and the wall separating the two compartments (aspect ratio: 9) was initially easily torn during the PDMS replication process. Therefore, the design of the soma compartment was modified from a cylinder shape into a truncated cone shape with sidewalls tilted 20° toward the center (Figure 1A - inset). This modified design fortified the PDMS walls separating the compartments during the PDMS replication process, and no damage to the final PDMS neuron-glia co-culture device was observed. It can be seen from Figure 3 that millimeter scale compartments as well as micrometer scale channels (3 × 20 × 400 μm3) connecting the soma compartment and the axon/glia compartments were successfully transferred to the final PDMS device without any noticeable distortion. In order to further inspect the reliability and pattern replication capability of the fabrication process, the pattern dimensions and the surface roughness were measured throughout the fabrication process (Figure 3C-D). The average width of the axon-guiding microchannels changed from 20.7 ± 1.02 μm (PMMA master, n = 13) to 20.7 ± 1.17 μm (PDMS master, n = 12), and then to 20.2 ± 0.25 μm (PDMS device, n = 6). The depth of the microchannels changed from 2.96 ± 0.02 μm (PMMA master, n = 14) to 3.0 ± 0.03 μm (PDMS master, n = 12), and then to 3.34 ± 0.06 μm (PDMS device, n = 10). The results clearly indicate that no significant changes in dimension occurred throughout the fabrication process. In addition, the surface roughness was measured to be 14.20 ± 2.44 nm (n = 7) for the PMMA master, 17.95 ± 4.36 nm (n = 11) for the PDMS master, and 38.63 ± 7.70 nm (n =18) for the final PDMS device. Although the surface roughness slightly increased through the process, device bonding onto the PDL coated substrates was not affected by this increase in surface roughness, and no leakage between the device and the substrate was observed after the assembly.
Fig. 3.
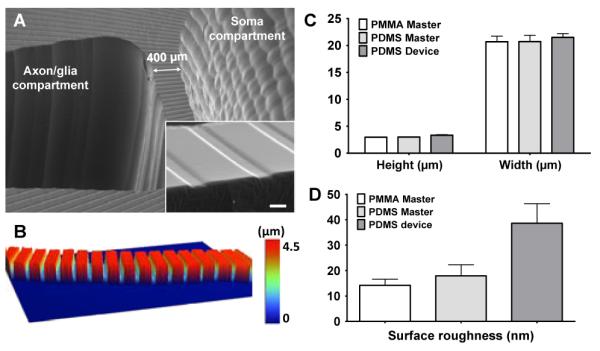
(A) SEM images of the multi-compartment PDMS neuron-glia co-culture device fabricated by the ‘MMHSM’ technique. Scale bar = 20 μm. (B) 3D reconstructed optical profilometry image of the axon-guiding microchannels connecting the soma and the axon/glia compartment. (C) Average height and width of the axon-guiding microchannels on the PMMA master, PDMS master and the PDMS device (mean ± SD). (D) Changes in surface roughness throughout the fabrication process (mean ± SD).
Axon isolation and growth
Primary neurons, loaded into the soma compartment of the co-culture platform at the areal density of 1000 cell/mm2, were cultured successfully for up to four weeks with no toxicity issues from the device. Neuronal somata were uniformly distributed and confined inside the soma compartment due to the shallow height of the axon-guiding microchannels (Figure 4A). In contrast, axons from the confined neuronal somata passed through the axon-guiding microchannels and grew into the surrounding six axon/glia compartments, achieving physical isolation from somata. As previously reported24, the axon isolation efficiency is largely dependent on the distance between the neuronal somata and the axon-guiding microchannel inlets since axons show random directional growth and are not chemically guided toward the neighboring axon/glia compartments. The circular well-type soma compartment fabricated by the MMHSM technique does not have compartment overhangs around axon-guiding channels and enforced neurons plated inside the soma compartment to position close to the axon-guiding microchannel inlets due to surface tension and the radial culture medium flow toward the satellite axon/glia compartments during the cell loading process (Figure 4B). Figure 4C shows the distances of the closest neuron cell from the axon-guiding channel inlets. The average distance of the closest cell from channel inlets was measured to be only 23.8 ± 12.15 μm (mean ± SD, n = 51) and this resulted in high axon isolation efficiency. After two weeks of culture, more than 90% of the axon-guiding microchannels were filled with axons (Figure 4D-E).
Fig. 4.

(A-B) Calcein-AM (green) stained images of neurons confined inside the soma compartment at DIV 1. (C) Histogram showing the distances of the closest neuron cell from the axon-guiding microchannel inlets. (D) Close-up view of axons crossing into the neighboring axon/glia compartment from the soma compartment. More than 90% of channels were filled with axons after two weeks of culture. (E) Reconstructed image of isolated axons in an axon/glia compartment immunostained for NF. White dotted box delineates the area (0.8 × 1.6 mm2) analyzed for ACR. (F) Close-up view of dense axonal layer formed inside the axon/glia compartment at DIV 17. (G) ACR analysis of the multi-compartment neuron-glia co-culture platform showing device-to-device repeatability and axon/glia compartment-to-compartment variations within a single device. (H) Isolated axons inside the six axon/glia compartments of a single device (Stained for NF = red).
The growth of the axons as well as the isolation capability of the multi-compartment neuron-glia co-culture platform was investigated by analyzing the percentage of the area covered with isolated axons, axon coverage ratio (ACR), within the axon/glia compartment (1.6 × 0.8 mm2, white dotted area in Figure 4E). Isolated axons formed dense axonal network layer inside the axon/glia compartments after two weeks of culture (Figure 4F) and were fixed and immunostained for neurofilament (NF-red) at DIV 17 for ACR analysis. Average ACR of the isolated axonsinside the axon/glia compartment of a multi-compartment device was 59 ± 10.8% (mean ± SD, n = 12, Figure 4G), which is comparable to previously reported results obtained from a circular neuron-OPC co-culture platform.24 Figure 4H shows immunostained axons in each of the six axon/glia compartment of a single device. Similar axonal layer densities were observed in all six satellite compartments (coefficient of variation = 0.18), demonstrating reproducibility and consistency of axonal growth and isolation in each axon/glia compartment. This indicates that results from each axon/glia compartment can be directly compared with others, allowing multiple experimental conditions to be carried out in parallel within a single device.
Parallel localized biomolecular treatment
Fluidic isolation among the six axon/glia compartments for parallel localized treatments was first demonstrated by treating OLs inside the three axon/glia compartments of a single device with a high concentration of ceramide (150 μM) to induce cell death.31 As expected, OLs treated with ceramide were completely dead and were detached from the surface. However, OLs inside the neighboring axon/glia compartments that were not directly exposed to ceramide remained unaffected and were viable as demonstrated by Calcein-AM viability assay (Figure 5).
Fig. 5.
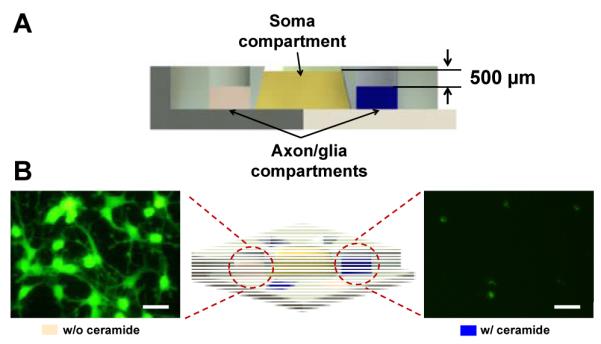
(A) Schematic illustration showing fluidic level difference between the soma compartment and the axon/glia compartments for fluidic isolation. Minute flow from the soma compartment toward the axon/glia compartments prevent localized treatments to isolated axons from diffusing into the soma compartment. (B) Calcein-AM stained images of OLs inside the axon/glia compartments without ceramide treatment and with ceramide treatment (150 μM) after 48 hours. Scale bars = 20 μm.
To further investigate the parallel localized biomolecular treatment capability of the multi-compartment neuron-glia co-culture platform, isolated axons were locally exposed to chondroitin sulfate proteoglycan (CSPG), a proteoglycan known to negatively regulate axon growth and cause retraction of the established axons.32 Pre-established isolated axonal layer inside the six axon/glia compartments of a single device was treated with six different concentrations of CSPG (0-25μg/ml) for 72 hours to find the effective dosage that causes axon retraction. After staining the CSPG treated axons inside the six axon/glia compartments with Calcein-AM, we found that CSPG at concentrations lower than 250 ng/ml was not sufficient to cause the retraction of pre-established axons. However, at concentrations higher than 2.5 μg/ml, CSPG induced robust axonal retraction (Figure 6). This result is in agreement with previously reported effective dosage of CSPG (3 μg/ml) obtained from conventional cell culture method.33 It should be noted that axons inside axon-guiding microchannels were not significantly affected even when CSPG caused complete axon retraction in the compartment (Figure 6 bottom), suggesting that ceramide loaded into the axon/glia compartments was properly confined within the compartment during the localized treatment process.
Fig. 6.
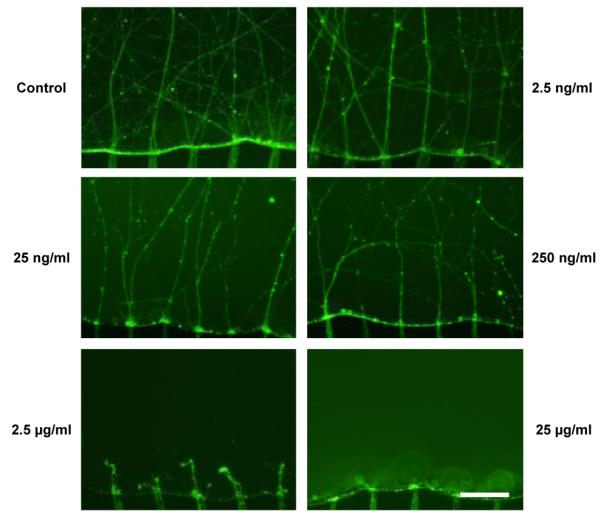
Isolated axons inside the six axon/glia compartments screened with six different concentrations of CSPG. Scale bar = 100 μm.
Co-culture of CNS neuron and glia
When seeded into the axon/glia compartment, OPCs distributed uniformly on top of the established axonal layer due to the non-flow characteristic of the well-type compartment (Figure 7A), which overcomes the limitation of previously reported channel-type compartments where the number of cells cannot be precisely controlled due to the fluidic flow within the compartment. The co-cultured OPCs interacted extensively with the axons (marked with white arrowheads in Figure 7B) and differentiated along the oligodendrocyte lineage stages. Mature OLs, as identified by immunostaining of myelin basic protein (MBP), were abundant two weeks after axon-OPC co-culturing (Figure 8A) with many processes contacting and aligning neighboring axons (Figure 8B, white arrow heads). Although robust formation of compact myelin, which normally is readily identifiable as smooth tubing structures, was not observed in the neuron-OPC co-culture platform, OL alignment to axons is a pre-stage required for myelination. These results demonstrate that the device can be a unique tool for studying CNS axon-glia interactions and OL alignment toward mechanistic studies of CNS myelination.
Fig. 7.
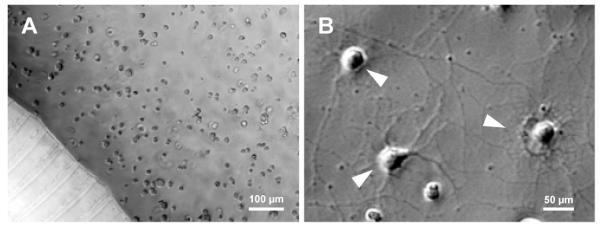
(A) OPCs uniformly distributed inside an axon/glia compartment. (B) Close-up view of differentiating OPCs inside the axon/glia compartment after two days of co-culture.
Fig. 8.

Immunostained images of axons and OLs inside the axon/glia compartment at DIV 29. (A) OPCs co-cultured on top of isolated axonal layer successfully differentiated into mature MBP-expressing OLs. (B) OL processes aligning with axons inside the axon/glia compartment to form myelin-like sheaths. (axon: NF-red, mature OL: MBP-green). Scale bars = 20 μm.
Unlike the axon-OPC co-cultures, our preliminary results showed that astrocytes appeared to disrupt the axons in the satellite axon/glia compartments when astrocytes were added on pre-established isolated axonal layer. As can be seen by the area marked by the white arrows in Supplementary Figure 1A, damaged axons can be easily observed as compared to a normal neuron culture without the astrocytes (Supplementary Figure 1B). Astrocytes stretch out and form a layer directly on culture substrate in 2D in vitro culture34 and we hypothesized that astrocytes grow underneath the pre-established isolated axonal layer and probably physically pushed away the previously established axons. In order to verify this hypothesis, three axon/glia compartments of a single device were loaded with astrocytes and OPCs while the other three were loaded only with OPCs (all at DIV 17) when dense axonal layers have already formed. To control the loading density of the cells, same number of total glia cells (OPC + astrocytes = 1000 cells/mm2) were co-cultured with axons for both experimental conditions (axon-OPC vs. axon-OPC/astrocyte) in order to rule out the effect of cell density on the established axonal layer. In the case of axon-OPC co-culture, 500 cells/mm2 of neurons were co-cultured with 1000 cells/mm2 of OPCs, while 500 cells/mm2 of neurons were co-cultured with 500 cells/mm2 of OPCs mIXed with 500 cells/mm2 of astrocytes for the axon-OPC/astrocytes co-culture. axons co-cultured only with OPCs remained uniformly distributed inside the axon/glia compartment without any noticeable damage even after two weeks of co-culture period (Figure 9A-D). In contrast, axons co-cultured with astrocytes and OPCs appeared to be damaged. The phase contrast images of the axon/glia compartments, where axons are not clearly visible due to the dense astrocytes, showed nice growth of astrocytes forming layer (black dotted circle in Figure 9G); however, immunolabling of axons revealed that most of the isolated axons inside the axon/glia compartments were damaged and the remaining ones exhibited a morphology as if they were physically pushed to the axon-guiding channel inlet area as indicated by the white arrow heads (Figure 9E-F). This experimental result confirms that the damages to established axons are indeed caused by the co-cultured astrocytes. Despite the axonal damage by astrocytes, differentiation of OPCs was promoted by the co-cultured astrocytes as more MBP-expressing OLs were found in the presence of astrocytes (Figure 9D vs. H, Figure 10). Although the number of OPCs in the axon-OPC co-culture (1000 cells/mm2) was twice as many as that of the axon-OPC/astrocyte co-culture (500 cells/mm2), approximately five times more MBP expressing OLs were observed when OPCs were co-cultured with astrocytes (Figure 10).
Fig. 9.

Images showing co-cultured axons and glial cells at DIV 27. Isolated axons co-cultured with (A-D) OPCs and (E-H) OPCs and astrocytes. Co-cultured astrocytes physically damaged pre-established axonal layer while forming a layer on the substrate. Axons were stained for NF (red) and mature OLs were stained for MBP (green). Scale bars = 100 μm.
Fig. 10.
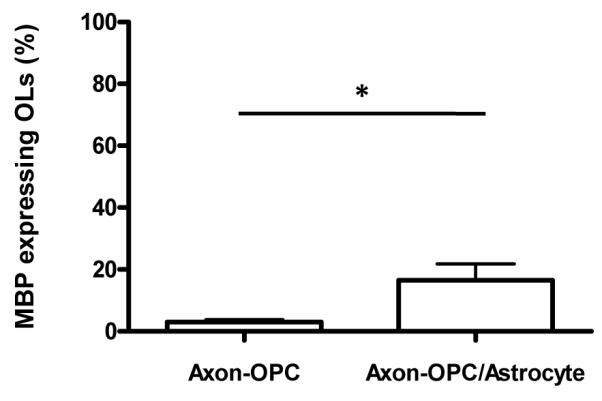
Astrocytes promoted OPC differentiation. Percentage of MBP expressing OLs from total number of OPCs, based on initial seeding density, was analyzed by immunocytochemistry and cell counting. More OPCs developed into mature-OLs when cultured in the presence of astrocytes (mean ± SD, n = 5, p < 0.05).
Conclusions
We have developed a multi-compartment neuron-glia co-culture platform where multiple experimental co-culture conditions as well as six different localized biomolecular treatments can be carried out on a single device in parallel for studying axon-glia interaction, OPC development as well as for investigating axonal response to various stimuli. The multi-compartment device fabricated by the MMHSM technique not only enabled fabrication of the device having multiple compartments that are only 400 μm apart for higher-throughput performance with great reproducibility but also significantly reduced the device fabrication time, approximately 50% compared to previously introduced devices that involve manual reservoir punching process, for its routine use for neuroscience applications. The well-type soma compartment enhanced the axon isolation efficiency with minimum compartment-to-compartment variation for parallel analysis and the multiple localized biomolecular treatment capability of the platform, demonstrated by the CSPG screening, showed its prospective application for studying axonal response to various localized chemical stimuli. Although we did not see robust myelin formation with co-culture of OPCs with isolated axons, differentiation of co-cultured OPCs into mature OLs was evident and perfect alignment of some OL processes with neighboring axons to form myelin sheaths could be clearly seen. Promotion of OPCs development into MBP expressing mature-OLs in the presence of astrocytes was also confirmed using the proposed platform. Astrocytes, however, have been found to physically damage the axonal network when added to pre-established axonal layer. We believe the multiple localized biomolecular treatment capability that enables screening of biomolecules that promote OPC differentiation in the presence of axons along with feature to carry out various co-culture conditions in a single device at increased throughput will make this platform very powerful for studying axon-glia interactions as well as OPC developments in vitro.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. Sunja Kim and Jeffery Thompson for helping with the dissection and the cell culture. This work was supported by the National Institutes of Health / National Institute of Mental Health grant #1R21MH085267 and by the National Institutes of Health / National Institute of Neurological Disorders and Stroke grant #NS060017.
References
- 1.Baumann N, Pham-Dinh D. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- 2.Cohen MS, Orth CB, Kim HJ, Jeon NL, Jaffrey SR. Proc. Natl. Acad. Sci. U. S. A. 2011;108:11246. doi: 10.1073/pnas.1012401108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nave K-A, Salzer JL. Curr. Opin. Neurobiol. 2006;16:492–500. doi: 10.1016/j.conb.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 4.Pfeiffer SE, Warrington AE, Bansal R. Trends Cell Biol. 1993;3:191–197. doi: 10.1016/0962-8924(93)90213-k. [DOI] [PubMed] [Google Scholar]
- 5.Colognato H, ffrench-Constant C. Curr. Opin. Neurobiol. 2004;14:37–44. doi: 10.1016/j.conb.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 6.McCarthy KD, de Vellis J. J. Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barres BA, Raff MC. Neuron. 1994;12:935–942. doi: 10.1016/0896-6273(94)90305-0. [DOI] [PubMed] [Google Scholar]
- 8.Miller RH. Prog. Neurobiol. 2002;67:451–467. doi: 10.1016/s0301-0082(02)00058-8. [DOI] [PubMed] [Google Scholar]
- 9.McMorris FA, McKinnon RD. Brain Pathol. 1996;6:313–329. doi: 10.1111/j.1750-3639.1996.tb00858.x. [DOI] [PubMed] [Google Scholar]
- 10.Simons M, Trajkovic K. J. Cell Sci. 2006;119:4381–4389. doi: 10.1242/jcs.03242. [DOI] [PubMed] [Google Scholar]
- 11.Waxman SG. Curr. Biol. 1997;7:R406–410. doi: 10.1016/s0960-9822(06)00203-x. [DOI] [PubMed] [Google Scholar]
- 12.Sherman DL, Brophy PJ. Nature Reviews. 2005;6:683–690. doi: 10.1038/nrn1743. [DOI] [PubMed] [Google Scholar]
- 13.Gabriele S, Versaevel M, Preira P, Théodoly O. Lab Chip. 2010;10:1459–1467. doi: 10.1039/c002257h. [DOI] [PubMed] [Google Scholar]
- 14.Gu W, Zhu X, Futai N, Cho BS, Takayama S. Proc. Natl. Acad. Sci. U. S. A. 2004;101:15861–15866. doi: 10.1073/pnas.0404353101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Millet L, Stewart M, Nuzzo R, Gillette M. Lab Chip. 2010;10:1525–1535. doi: 10.1039/c001552k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor A, Dieterich D, Ito H, Kim S, Schuman E. Neuron. 2010;66:57–68. doi: 10.1016/j.neuron.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frimat J-P, Becker M, Chiang Y-Y, Marggraf U, Janasek D, Hengstler JG, Franzke J, West J. Lab Chip. 2010 doi: 10.1039/c0lc00172d. [DOI] [PubMed] [Google Scholar]
- 18.Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. Nat. Methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Millet L, Stewart M, Sweedler J, Nuzzo R, Gillette M. Lab Chip. 2007;7:987–994. doi: 10.1039/b705266a. [DOI] [PubMed] [Google Scholar]
- 20.Ravula SK, McClain MA, Wang MS, Glass JD, Frazier AB. Lab Chip. 2006;6:1530–1536. doi: 10.1039/b612684g. [DOI] [PubMed] [Google Scholar]
- 21.Erickson J, Tooker A, Tai YC, Pine J. J. Neurosci. Methods. 2008;175:1–16. doi: 10.1016/j.jneumeth.2008.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hur EM, Yang IH, Kim DH, Byun J, Levchenko A, Thakor N, Zhou F-Q. Proc. Natl. Acad. Sci. U. S. A. 2011;108:5057. doi: 10.1073/pnas.1011258108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang K, Osakada Y, Vrljic M, Chen L, Mudrakola H, Cui B. Lab Chip. 2010;10:2566–2573. doi: 10.1039/c003385e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park J, Koito H, Li J, Han A. Biomed. Microdevices. 2009;11:1145–1153. doi: 10.1007/s10544-009-9331-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hosmane S, Yang IH, Ruffin A, Thakor N, Venkatesan A. Lab Chip. 2010;10:741–747. doi: 10.1039/b918640a. [DOI] [PubMed] [Google Scholar]
- 26.Park J, Li J, Han A. Biomed. Microdevices. 2010;12:345–351. doi: 10.1007/s10544-009-9390-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Regehr K, Domenech M, Koepsel J, Carver K, Ellison-Zelski S, Murphy W, Schuler L, Alarid E, Beebe D. Lab Chip. 2009;9:2132–2139. doi: 10.1039/b903043c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koito H, Li J. J. Vis. EXp. 2009:31. doi: 10.3791/1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA. Proc. Natl. Acad. Sci. U. S. A. 2005;102:9936–9941. doi: 10.1073/pnas.0502552102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, Ramenaden ER, Peng J, Koito H, Volpe JJ, Rosenberg PA. The Journal of Neuroscience. 2008;28:5321. doi: 10.1523/JNEUROSCI.3995-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larocca J, Farooq M, Norton W. Neurochem. Res. 1997;22:529–534. doi: 10.1023/a:1027332415877. [DOI] [PubMed] [Google Scholar]
- 32.Wang H, Katagiri Y, McCann TE, Unsworth E, Goldsmith P, Yu ZX, Tan F, Santiago L, Mills EM, Wang Y. J. Cell Sci. 2008;121:3083. doi: 10.1242/jcs.032649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lingor P, Teusch N, Schwarz K, Mueller R, Mack H, Bähr M, Mueller BK. J. Neurochem. 2007;103:181–189. doi: 10.1111/j.1471-4159.2007.04756.x. [DOI] [PubMed] [Google Scholar]
- 34.Gilad GM, Gilad VH. Int. J. Dev. Neurosci. 1987;5:79–89. doi: 10.1016/0736-5748(87)90053-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.