

Abstract
Classic galactosemia is a potentially lethal metabolic disorder that results from profound impairment of the enzyme galactose-1-phosphate uridylyltransferase (GALT); despite decades of research, the underlying mechanism of pathophysiology remains unclear. Previous studies of plasma and tissue samples from patients with classic galactosemia have revealed defects of protein and lipid glycosylation, however, the underlying bases for these defects and their clinical significance, if any, has remained unclear. As a step toward addressing these questions we characterized both the N- and O-linked glycomes of plasma proteins from neonates, infants, children, and adults with galactosemia using mass spectrometry and asked (1) whether similar or disparate defects exist for N-linked and O-linked modifications, (2) what factors correlate with the severity of these defects in different patients, and perhaps most important, (3) whether there is any apparent relationship between chronic glycosylation defects and long-term outcome in patients. We found that some but not all of the galactosemic neonates tested exhibited abnormal N- and O-linked glycosylation of plasma proteins. The types of abnormalities seen were similar between N- and O-linked moieties, but the extent of the defects varied between patients. Age, gender, GALT genotype, and predicted residual GALT activity all failed to explain the extent of the glycosylation defect in the samples studied. Dietary galactose restriction markedly normalized both the N- and O-linked glycosylation patterns for all infants tested; however, any remaining glycosylation defects evident in the plasma of older children or adults on galactose-restricted diets showed no correlation with clinical outcome. These data cannot rule out the possibility that subtle or localized glycosylation defects, not detectable by our methods or not reflected in plasma, may contribute to acute or long-term outcome severity.
Keywords: Classic galactosemia, LC-MS/MS, MALDI-TOF, N-glycome, O-glycome, Plasma
1. Introduction
Classic galactosemia (OMIM 230400) is a potentially lethal metabolic disorder that results from profound impairment of galactose-1P uridylyltransferase (GALT), the middle enzyme in the highly conserved Leloir pathway of galactose metabolism (reviewed in ref. [1]). Enzymes of the Leloir pathway: galactokinase (GALK, EC 2.7.1.6), GALT (EC 2.7.7.12), and UDP-galactose 4′-epimerase (GALE, EC 5.1.3.2) mediate the conversion of dietary galactose to glucose-1-phosphate (glc-1P), enable the endogenous biosynthesis of UDP-galactose (UDP-gal) when exogenous sources are lacking, and control the intracellular ratio of UDP-gal to UDP-glucose (UDP-glc) (reviewed in ref. [2]). In humans and other metazoans, GALE also enables the endogenous biosynthesis of UDP-N-acetylgalactosamine (UDP-galNAc) and controls the intracellular ratio of UDP-galNAc to UDP-N-acetylglucosamine (UDP-glcNAc). All four UDP-sugars: UDP-glc, UDP-gal, UDP-glcNAc, and UDP-galNAc are essential substrates for the biosynthesis of glycoproteins and glycolipids.
A literature trail extending back more than 40 years documents glycosylation defects apparent in plasma or tissue samples from patients with classic galactosemia. In the early 1970s, Haberland and colleagues [3,4] demonstrated an abnormal pattern of glycoproteins in the postmortem brain of a galactosemic patient. Twenty years later, Petry and colleagues [5] described a deficiency of glycolipids containing galactose or N-acetylgalactosamine, and an accumulation of the precursors of these moieties in the postmortem brain of a neonate with galactosemia. Also in the early 1990s, Jaeken and colleagues described abnormalities in the glycosylated serum lysosomal enzymes from patients with galactosemia [6], and Dobbie and colleagues [7] and Ornstein and colleagues [8] reported defective galactosylation of complex carbohydrates and glycoproteins in fibroblasts derived from patients with galactosemia.
More recently, three reports from different groups have both corroborated and extended the results of these earlier studies. Stibler and colleagues [9] demonstrated the presence of carbohydrate-deficient isoforms of serum transferrin in the blood of patients with classic galactosemia. In particular, these authors found abnormal asialo- and/or disialo-transferrin in samples derived from untreated patients; these carbohydrate-deficient isoforms were rarely seen in samples from patients on galactose restricted diets. Charlwood and colleagues [10] corroborated those results and went on to apply HPLC analysis of transferrin-derived N-linked glycans to describe the structural basis of the glycosylation defect. Specifically, these authors found that, compared with controls, transferrin from untreated patients contained four major truncated glycans and reduced levels of the di-sialylated biantennary complex type [10]. These authors concluded that the truncated glycans were deficient in sialic acid and galactose and that their structures suggested inadequate galactosyltransferase activity in the biosynthetic tissue, presumed to be liver. Notably, while the serum transferrin glycans largely normalized in patients following dietary restriction of galactose, they did not become completely normal in all patients, even after prolonged treatment. These subtle but chronic glycosylation defects observed in some but not all patients despite treatment raised the intriguing possibility that chronic aberrant glycosylation might contribute to the long-term complications experienced by some but not all treated patients.
Further studies of serum transferrin from untreated patients with classic galactosemia [11,12] again corroborated the earlier conclusions, and also demonstrated that the glycosylation defects fell into two categories representing abnormalities of both N-glycan assembly and processing. Coman and colleagues [13] also explored the N-glycan profiles of whole serum and IgG from 10 treated patients and found deficiencies of di-galactosylated structures with increases of agalactosylated and mono-galactosylated serum glycoproteins. More recently, Coss and colleagues [14] compared N-glycosylation patterns of IgG in serum samples from galactosemic adults on galactose restricted diets versus after varying degrees of diet liberalization. These authors concluded that IgG N-glycan profiles may be a highly sensitive marker of galactose tolerance and ongoing processing defects. Finally, Staubach and colleagues [15] explored the N-linked glycans from epithelial plasma membranes in urine; control samples demonstrated largely high-mannose-type glycans while samples from patients on galactose-restricted diets demonstrated more complex–type glycans. Again, these chronic defects despite treatment raised the question of whether chronic abnormal glycosylation might underlie at least some of the long-term complications experienced by some well-managed patients.
As a first step toward addressing this question, we characterized both the N- and O-linked glycomes of plasma proteins collected in a time-course fashion from newly diagnosed galactosemic neonates and also from unrelated older children and adults whose outcomes were known. Our results confirmed both N- and O-linked glycosylation defects in most but not all of the neonates, with normalization or near-normalization of those defects over time following dietary restriction of galactose. The N- and O-linked glycomes of the older children and adults tested, all of whom reported following galactose restricted diets for years, showed few if any detectable glycosylation defects, and those defects that were seen showed no apparent relationship with outcome severity. However, we cannot rule out the possibility that glycosylation defects early in development, or impacting only specific cells, tissues, or macromolecules, might contribute to outcome severity.
2. Materials and methods
2.1. Chemicals
Iodomethane, dimethyl sulfoxide anhydrous (DMSO), 2, 5-dihydroxybenzoic acid (DHB), sodium hydroxide, trifluoroacetic acid (TFA), raffinose, sodium borohydrate, and sodium acetate were all purchased form Sigma-Aldrich (St. Louis, MO, USA). Peptide N-glycosidase F (PNGase F), including denaturation buffer, digestion buffer, and NP-40 buffer were all purchased from New England Biolabs (Ipswich, MA). Extra-Clean SPE Carbos were purchased from Grace Davison Discovery Science (Deerfield, IL). The Sep-Pak Vac C18 cartridge 3 cc was from Waters (Milford, MA). The P-Lacto-N-hexaose (pLNH) was from V-labs (Covington, LA). Acetonitrile, chloroform, methanol, sodium hydroxide (w/w, 50%), and sodium acetate were all from Fisher Scientific (Fairlawn, NJ, USA).
2.2. Study volunteers, sample collection, and sample storage
Plasma samples from the seven galactosemic infants in this study were collected as clinical lab discards from the Emory Genetics Lab with authorization under a HIPAA waiver granted by the Emory University Institutional Review Board (protocol 618-99, PI: Fridovich-Keil). These infants ranged in age from 8 to 13 days. Plasma samples from the seven galactosemic girls ages 2–9 years old were collected with informed consent, again authorized under Emory IRB Protocol 618-99. The plasma samples from adults with classic or Duarte galactosemia were collected at Children’s Hospital Boston with informed consent under a protocol approved by the Children’s Hospital Boston Institutional Review Board (protocol 09-08-0392, PI: Berry). Except for the initial samples collected from neonates, all samples were collected from patients whose diagnoses had already been confirmed by clinical lab tests, and who were under careful dietary restriction of galactose.
Controls were anonymous samples from our diagnostic sample archives. They were from adult volunteers, and from children who were sibling controls for diagnoses other than primary or secondary glycosylation disorders or galactosemia. Additional reference samples were from asymptomatic infants initially evaluated for borderline positive Georgia state newborn screening results but ultimate determined not to have a metabolic disorder. The control samples in this study represented both children and adults of different ages and both genders. Many samples were from individuals listed as Caucasian, African-American, or Hispanic, though some were of unknown race. A set of 150 control samples was analyzed and the data were used to establish the reference ranges for the N-glycans quantified. Of note, the data obtained from these control samples were in agreement with data published earlier from samples from 10 healthy adults and nine healthy children [16], and also from samples collected from a cross-section of the populations of the Croatian islands of Vis and Korcula [17]. A smaller set of 40 control samples were also analyzed to establish the normal reference ranges for the O-glycans quantified. In all cases, the reference ranges are expressed as mean ± 2SD of the control group, representing at least 95% of the population range. This means that a small number of control samples will show individual glycan moieties that fall outside the defined “reference range.”
All blood samples were collected into sodium heparin tubes and shipped or stored at 4 °C for less than or equal to 1 day before processing. Heparinized bloods were centrifuged at 1800 rpm for 15 min at RT in a Sorvall 6000B refrigerated centrifuge (Donpont); plasma was collected and aliquots were stored at −85 °C until use.
2.3. Evaluation of ovarian function in pre-pubertal girls
Ovarian status was evaluated by measuring plasma anti-Müllerian Hormone (AMH, also called Müllerian Inhibiting Substance, MIS) as described previously [18,19]. In brief, frozen plasma samples were coded and sent for analysis to the Biomarkers Core Laboratory of the Yerkes Primate Research Center at Emory University, or to the Reproductive Endocrine Unit Reference Laboratory, Massachusetts General Hospital (Boston, MA). Both labs used a kit from Diagnostic Systems Laboratories, Inc. to quantify AMH/ MIS. The minimum reportable concentration of the test was 0.03 ng/mL. Testing was monitored using quality control sera (two levels); the intra-assay coefficient of variability (CV) was <6%, and the inter-assay CV was <12%. Girls designated as having “good” ovarian function had AMH values >1.5 ng/mL and girls designated as having evidence of premature ovarian insufficiency (POI) had AMH values <0.03 ng/mL (Tables 3 and 7).
Table 3.
N-linked glycans released from total plasma glycoprotein from girls with well-managed galactosemia who do and do not demonstrate ovarian dysfunction as evidenced by AMH value. Patient number, gender, age at blood draw, AMH at blood draw, and GALT genotype are listed.
| Glycan structure | Reference range from 150 controls (%) | P8 ♀ | P9 ♀ | P12 ♀ | P10 ♀ | P11 ♀ | P13 ♀ | P14 ♀ |
|---|---|---|---|---|---|---|---|---|
| 2 years | 5 years | 9.5 years | 6 years | 6 years | 9 years | 8.5 years | ||
| AMH 3.55 μg/L | AMH 1.69 μg/L | AMH 3.085 μg/L | AMH 0.02 μg/L | AMH 0.02 μg/L | AMH 0.025 μg/L | AMH 0.015 μg/L | ||
| Q188R/N314D/L218L/D197G | T138M/V157I | K285N/D98N | Q188R/Q188R | Q188R/Q188R | Q188R/Q188R | Q188R/Q188R | ||
|
|
0.27-1.23 | 0.66 | 0.61 | 1.08 | 0.63 | 0.35 | 0.69 | 0.72 |
|
|
0.02-0.19 | 0.00 | 0.07 | 0.11 | 0.06 | 0.08 | 0.09 | 0.00 |
|
|
0.25-0.92 | 0.53 | 0.48 | 0.57 | 0.51 | 0.28 | 0.50 | 0.40 |
|
|
0.00-0.14 | 0.06 | 0.05 | 0.06 | 0.06 | 0.00 | 0.07 | 0.00 |
|
|
0.10-0.38 | 0.13 | 0.17 | 0.30 | 0.21 | 0.15 | 0.15 | 0.22 |
|
|
0.21-0.59 | 0.41 | 0.50 | 0.61 | 0.51 | 0.30 | 0.46 | 0.54 |
|
|
0.07-0.29 | 0.16 | 0.20 | 0.18 | 0.16 | 0.08 | 0.16 | 0.15 |
|
|
0.00-0.14 | 0.06 | 0.03 | 0.00 | 0.06 | 0.00 | 0.04 | 0.10 |
|
|
0.08-0.49 | 0.22 | 0.25 | 0.23 | 0.23 | 0.17 | 0.25 | 0.23 |
|
|
0.47-1.15 | 1.18 | 1.19 | 1.56 | 1.25 | 0.97 | 1.41 | 1.33 |
|
|
0.00-0.11 | 0.11 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
|
|
0.14-0.52 | 0.15 | 0.21 | 0.23 | 0.26 | 0.27 | 0.19 | 0.17 |
|
|
2.71-6.6 | 4.53 | 4.49 | 5.87 | 5.35 | 3.71 | 6.08 | 4.77 |
|
|
0.29-1.98 | 0.15 | 0.32 | 0.52 | 0.44 | 0.33 | 0.44 | 0.48 |
|
|
10.91-20.43 | 20.16 | 20.81* | 15.31 | 19.81 | 16.25 | 22.53** | 18.99 |
|
|
0.09-1.92 | 1.08 | 1.01 | 1.30 | 1.41 | 1.14 | 1.95 | 1.39 |
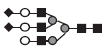
|
0.25-0.99 | 0.83 | 0.91 | 0.65 | 0.88 | 0.73 | 0.68 | 0.67 |
|
|
0.00-0.32 | 0.00 | 0.05 | 0.24 | 0.11 | 0.33** | 0.35** | 0.00 |
|
|
0.97-4.16 | 5.01** | 3.46 | 1.74 | 2.74 | 2.32 | 2.40 | 1.91 |
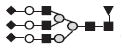
|
0.25-2.14 | 0.87 | 0.64 | 0.73 | 0.92 | 1.80 | 1.25 | 1.12 |
P10, P11, P13, and P14 all showed evidence of ovarian dysfunction; P8, P9, and P12 did not. Glycan symbols, shading, and statistical significance: same as Table 1.
Table 7.
O-linked glycans released from total plasma glycoprotein from girls with well-managed galactosemia who do and do not demonstrate ovarian dysfunction as evidenced by AMH value. Patient number, gender, age at blood draw, AMH at blood draw, and GALT genotype are listed.
| Glycan structure | Reference range from 40 controls | P8 ♀ | P9 ♀ | P12 ♀ | P10 ♀ | P11 ♀ | P13 ♀ | P14 ♀ |
|---|---|---|---|---|---|---|---|---|
| 2 years | 5 years | 9.5 years | 6 years | 6 years | 9 years | 8.5 years | ||
| AMH 3.55 μg/L | AMH 1.69 μg/L | AMH 3.085 μg/L | AMH 0.02 μg/L | AMH 0.02 μg/L | AMH 0.025 μg/L | AMH 0.015 μg/L | ||
| Q188R/ N314D/ L218L/ D197G | T138M/ V157I | K285N/ D98N | Q188R/ Q188R | Q188R/ Q188R | Q188R/ Q188R | Q188R/ Q188R | ||
|
|
0.22 μM -1.14 μM | 0.51 | 0.56 | 2.9** | 0.5 | 0.86 | 3.45** | 1.77** |
|
|
11.7 μM -31.4 μM | 18.8 | 15.5 | 36.3** | 15.3 | 21.7 | 34.9** | 28.4 |
| T/ST | ≤0.06 | 0.03 | 0.04 | 0.08** | 0.03 | 0.0 4 | 0.10** | 0.06 |
P10, P11, P13, and P14 all showed signs of ovarian dysfunction; P8, P9, and P12 did not. Core1Tn-antigen (T), sialyl-T antigen (ST), and the ratio of T/ST are presented. Glycan symbols: □: N-acetylgalactosamine (GalNAc); ○: galactose; ◆ : sialic acid (Neu5Ac). Shading and statistical significance are the same as for Table 1
2.4. Evaluation of neurological function in adult volunteers
Four adults with classic galactosemia and one adult with Duarte galactosemia, all of whom were confirmed by GALT genotype and enzyme activity, participated in this study. Each adult participant received physical and neurological exams, a psychological evaluation, and speech/language assessment performed as part of a separate study at the Children’s Hospital Boston to define the adult galactosemic phenotype [20]. A designation of “Good” vs. “Poor” neurological outcome was utilized. A poor neurological outcome was assigned to those volunteers who had a FSIQ below one standard deviation and neurological findings such as cerebellar ataxia and dysarthria, tremor, and dystonia.
2.5. Analyses of plasma protein N- and O-glycan profiles
2.5.1. Preparation of N-glycans for analysis
An internal standard, P-Lacto-N-hexaose (pLNH, 150 pmol) was added into 20 μL plasma, then mixed with 20 μL of denaturation buffer and 145 μL water for a total volume of 200 μL. Plasma proteins were denatured by incubation at 100 °C for 10 min in a water bath and then cooled to room temperature. 25 μL NP-40 buffer and 25 μL digestion buffer from New England Biolabs were added with 1 μL PNGase F and the reaction was incubated at 37 °C for 16 hours to release the N-linked glycans from glycoproteins. Following digestion, the reaction was clarified of particulates by centrifugation at 15,000 rpm for 4.5 min in an Eppendorf 5424 Centrifuge, and the supernatant was purified through an activated SapPak C18 column followed by an activated carbograph column (Waters). Finally, the purified glycans were lyophilized overnight until completely dry [21].
2.5.2. Preparation of O-glycans for analysis
O-glycans were released from plasma glycoproteins and prepared for analysis essentially as described by Carlson [22], with modifications as described. An internal standard (1250 pmol raffinose in 5 μL) was added to 10 μL of plasma and 65 μl water for a final volume of 100 μL. Next, 100 μL of freshly prepared 2 M sodium borate in 0.1 M sodium hydroxide was added to denature the serum proteins and release the O-glycans; the mixture was incubated at 45 °C for 16 hours to ensure complete reaction. The reaction was neutralized by drop wise addition of 1.6 mL of a 0.25 M acetic acid-methanol solution, and the O-glycans were extracted in methanol. Finally, the extracted glycans were desalted through ion-exchange AG 50W-X8 resin (Bio-Rad, Hercules, CA) following the manufacturer’s instruction and lyophilized overnight. The dried samples were dissolved in DMSO for permethylation (see below).
2.5.3. Permethylation
Both N-glycans and O-glycans were permethylated as previously described with minor modification [16]. Briefly, four NaOH pellets (approximately 375 mg) were crushed in 10 mL anhydrous DMSO with 0.5 μL water; 0.5 mL of this slurry and 0.2 mL CH3I were then added to the dried glycans and the mixture was shaken vigorously for 1 hour. The mixture was extracted five times sequentially with a mixture of 200 μL water and 600 μL chloroform. Finally, the combined chloroform phases were dried under nitrogen in the chemical hood (30 min) and the permethylated N- and O-glycans were resuspended in 50 μL of 50% methanol and further purified through a C18 Stage Tip (Thermo Scientific, West Palm Beach, FL) as described previously [16].
2.5.4. Analysis of N-glycan profiles by matrix-assisted laser desorption/ ionization time-of-flight mass spectrometry (MALDI-TOF)
0.5 μL of each permethylated N-glycan sample was spotted onto the MALDI plate and 0.5 μL DHB (11 mg/mL in 50% methanol with 1 mM sodium acetate) was added as a matrix solution. Each spot was then air dried for 10 min. The data were generated using a MALDI-TOF-TOF 4800 plus (Applied Biosystems, Foster City, CA). The method of operation was positive reflector mode with the laser power set at 4880 and the digitizer set at 0.79. Each sample was run 3 times.
Twenty specific N-glycans were selected for semi-quantitative analysis and normalization of their intensities to the sum of all the peaks. The ratio of the most abundant glycan (m/z 2792) and the internal standard (m/z 1357) were recorded to monitor the yield of glycans. The 4000 series explorer software (Applied Biosystems, Foster City, CA) was used to collect the data and integrate the peak intensities.
2.5.5. Quantification of O-linked glycans Core1Tn-antigen and sialyl-T antigen by tandem mass spectrometry coupled with high-performance liquid chromatography (HPLC-MS/MS)
HPLC separation of O-linked glycans was achieved with a Shimadzu Prominence 20 AD LC and a Thermo gold 3-μm C18 column (2 × 100 mm). The binary method used buffer A (acetonitrile:formic acid: water; 1:0.1:99 (v:v:v)) and buffer B (acetonitrile:formic acid: water; 99:0.1:1 (v:v:v)) with a flow rate at 0.25 mL/min under the following gradient conditions: 0–20 min, 50% to 80% buffer B; 20–28 min, 98% buffer B; 28–39 min, 50% buffer B. An injection volume of 10 μl was used for analysis of each sample.
The API-QTRAP 5500 tandem mass spectrometry conditions were as follows: ion source: EPI positive mode; curtain gas: 25; ion source: 5500, EPI positive mode; curtain gas:25; source temperature: 600. MRM transitions for core1 Tn-antigen and sialyl-T-antigen were: m/z 534/298 and m/z 895/520. Calibration curves were constructed with 6 concentrations of Tn-antigen (from 0.0625 to 5 μM). The ST value is based on the ratio of the ST over the T peak area, times the T absolute value.
2.6. GALT activity assays of soluble lysates of yeast expressing hGALT alleles
The presence or absence of residual GALT activity associated with each hGALT allele identified in the patients in this study was ascertained using a null-background yeast expression system for the human enzyme, as described previously [23] with one modification. Rather than using JFy3747 as the host strain we used JFy5555; both W303-derived strains lack endogenous yeast GALT, but unlike JFy3747, JFy5555 also lacks endogenous GALK and GALE. The absence of GALE substantially lowers the background signal in GALT enzymatic assays of lysates from JFy5555 transformants because it prevents the conversion of UDP-glc to UDP-gal by endogenous yeast epimerase. GALT assays performed using JFy5555 are therefore much more sensitive than assays performed using JFy3747, enabling detection of very low levels of residual GALT activity.
3. Results
3.1. N- and O-linked whole glycome defects in plasma proteins from neonates with classic galactosemia
As a first step toward addressing the extent and nature of both N- and O-linked glycosylation defects in classic galactosemia, we characterized and compared the N-and O-linked glycomes of total plasma glycoproteins from neonates with classic galactosemia and controls. Prior reports had investigated the plasma N-glycomes of patients, but to our knowledge, this is the first study of plasma O-glycome changes in classic galactosemia. In an attempt to test samples collected prior to dietary restriction of galactose, we studied plasma samples collected from the initial blood draws from 7 neonates, ages 8–13 days, referred to the Emory Biochemical Genetics Laboratory for diagnostic testing in follow-up to an abnormal newborn screen for galactosemia. Subsequent diagnostic testing for these 7 infants demonstrated that all had classic galactosemia (see Materials and methods). Diet history was available for three of these infants and gal-1P values were available for six; all consistent with the presumption that these infants were consuming milk at the time of their initial blood draw.
N-linked glycans were enzymatically released from glycoproteins in the plasma by (PNGase F and analyzed by MALDI-TOF (see Materials and methods). To avoid overlapping peaks between free glycans and N-linked glycans, only N-glycans with m/z > 1000 were analyzed and semi-quantified. O-linked glycans, including Core1Tn-antigen (T) and mono-sialyl-T antigen (ST), were chemically released from plasma proteins by sodium hydroxide and quantified by HPLC-MS/MS (see Materials and methods).
A reference range for N-linked glycans was established by the Emory Genetics Biochemistry Lab from analyses of 150 control samples representing individuals of different ages, races, and both genders (see Materials and methods). Of note, the glycan values observed for these controls were consistent with control values reported previously by other investigators [16,17]. Samples from age- and gender-matched neonatal controls were also studied in parallel with the cases described here, but the results from these specific control samples were consistent with the reference ranges presented, so these neonatal control data are not shown. Typical plasma N-glycan profiles from control and galactosemic neonates are presented in Fig. 1. Twenty individual N-glycans from 1579.8 to 3776.3 m/z were monitored and semi-quantified from each sample. The structures of the N-glycans corresponding to each of these peaks have been previously confirmed [24-26], and are illustrated in Tables 1-4.
Fig. 1.
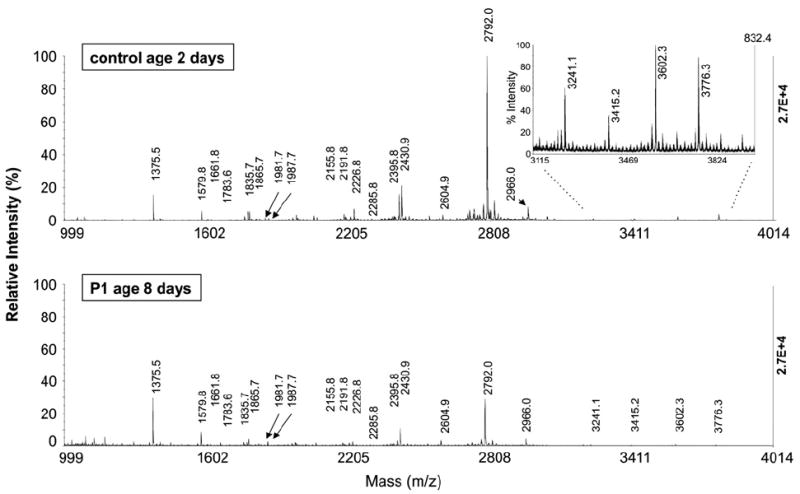
Negative-ion spectra of N-linked glycans from total plasma glycoproteins. (A) Sample from a non-galactosemic neonate, (B) sample from a neonate with classic galactosemia.
Table 1.
N-linked glycans released from total plasma glycoproteins from neonatal patients with galactosemia. Patient number, gender, age at blood draw, GALT genotype, and gal-1P at blood draw are listed. None of these infants had detectable GALT activity in hemolysate, though one (P5) carries a recognized variant allele (S135L).
| Glycan structure | Theoretical m/z [M - H]- | Observed m/z [M - H]- | Reference range from 150 controls (%) | Initial blood draw | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| P1 ♀ | P2 ♂ | P3 ♀ | P4 ♀ | P6 ♂ | P7 ♀ | P5 ♂ | ||||
| 8 days | 9 days | 8 days | 9 days | 9 days | 13 days | 8 days | ||||
| Q188R/5kbdel | Q188R/Q188R | Q188R/K285N | Q188R/Q188R | Q188R/Y209C | Q206R/Q206R | S135L/K285N | ||||
| gal - 1P 30.3 mg/dL | gal - 1P unk | gal - 1P 15.7 mg/dL | gal - 1P 46.4 mg/dL | gal - 1P 48.1 mg/dL | gal - 1P 52.4 mg/dL | gal - 1P 55.6 mg/dL | ||||
|
|
1579.8 | 1579.8 | 0.27-1.23 | 1.91* | 1.37 | 1.78* | 1.30 | 1.61 | 1.09 | 0.49 |
|
|
1661.8 | 1661.8 | 0.02-0.19 | 0.46 | 0.14 | 0.09 | 0.08 | 0.27 | 0.07 | 0.12 |
|
|
1783.9 | 1783.6 | 0.25-0.92 | 1.06 | 0.90 | 1.20* | 0.46 | 0.79 | 0.55 | 0.27 |
|
|
1835.9 | 1835.7 | 0.00-0.14 | 0.24** | 0.06 | 0.00 | 0.12 | 0.14 | 0.07 | 0.08 |
|
|
1865.9 | 1865.7 | 0.10-0.38 | 0.71** | 0.36 | 0.55 | 0.21 | 0.95** | 0.26 | 0.24 |
|
|
1982.0 | 1981.7 | 0.21-0.59 | 0.61 | 0.40 | 0.70 | 0.72* | 0.83** | 0.49 | 0.43 |
|
|
1988.0 | 1987.7 | 0.07-0.29 | 0.41** | 0.32 | 0.59** | 0.14 | 0.38* | 0.25 | 0.14 |
|
|
2156.1 | 2155.8 | 0.00-0.14 | 0.17 | 0.12 | 0.03 | 0.23 | 0.18 | 0.10 | 0.00 |
|
|
2192.1 | 2191.8 | 0.08-0.49 | 0.34 | 0.27 | 0.38 | 0.11 | 0.36 | 0.19 | 0.04* |
|
|
2227.1 | 2226.8 | 0.47-1.15 | 0.66 | 0.44 | 0.87 | 0.89 | 1.00 | 0.82 | 0.69 |
|
|
2285.2 | 2285.8 | 0.00-0.11 | 0.08 | 0.05 | 0.08 | 0.0 | 0.21** | 0.10 | 0.00 |
|
|
2396.2 | 2395.8 | 0.14-0.52 | 0.28 | 0.14 | 0.44 | 0.13 | 0.30 | 0.20 | 0.06* |
|
|
2431.2 | 2430.9 | 2.71-6.6 | 3.22 | 2.25 | 4.05 | 5.37 | 6.57 | 2.86 | 2.76 |
|
|
2605.3 | 2604.9 | 0.29-1.98 | 1.11 | 0.94 | 0.92 | 1.35 | 1.80 | 0.95 | 0.52 |
|
|
2792.4 | 2792.0 | 10.91-20.43 | 10.82 | 6.73** | 14.85 | 14.49 | 15.18 | 14.12 | 10.12* |
|
|
2966.5 | 2966.0 | 0.09-1.92 | 1.46 | 0.94 | 1.87 | 2.27** | 1.96 | 2.20* | 0.73 |
|
|
3241.6 | 3241.1 | 0.25-0.99 | 0.11* | 0.14* | 0.28 | 0.22 | 0.23 | 0.48 | 0.31 |
|
|
3415.7 | 3415.2 | 0.00-0.32 | 0.13 | 0.08 | 0.35** | 0.12 | 0.11 | 0.32** | 0.10 |
|
|
3602.8 | 3602.3 | 0.97-4.16 | 0.42 | 0.22* | 0.80 | 0.28* | 0.28* | 0.85 | 0.74 |
|
|
3776.9 | 3776.3 | 0.25-2.14 | 0.22 | 0.19 | 0.91 | 0.18 | 0.12 | 0.84 | 0.25 |
Glycan symbols: ■: N-acetylglucosamine (GlcNAc);
 :mannose (Man); ○: galactose (Gal); ◆: sialic acid (Neu5Ac); ▼: fructose (Fuc). Shading: whole cell shaded dark gray indicates the glycan level exceeds the high-end of the reference range; underlined text indicates that the glycan level is below the low-end of the reference range. Statistical significance:
:mannose (Man); ○: galactose (Gal); ◆: sialic acid (Neu5Ac); ▼: fructose (Fuc). Shading: whole cell shaded dark gray indicates the glycan level exceeds the high-end of the reference range; underlined text indicates that the glycan level is below the low-end of the reference range. Statistical significance:
indicates P<0.05% in comparison with controls;
indicates P<0.01% in comparison with controls by two tailed Student’s t test.
Table 4.
N-linked glycans released from total plasma glycoprotein from adults with well-managed galactosemia who do and do not demonstrate neurologic dysfunction. Patient number, gender, age at blood draw, neurological outcome status, and GALT genotype are listed.
| Glycan structure | Reference range from 150 controls (%) | P15 ♀ | P16 ♀ | P17 ♂ | P18 ♀ | P19 ♂ |
|---|---|---|---|---|---|---|
| 37 years | 31 years | 59 years | 54 years | 51 years | ||
| good outcome | poor outcome | poor outcome | good outcome | good outcome | ||
| D/G | Q188R/Q188R | 5kb del/5kb del | 5kb del/5kb del | Q188R/Q188R | ||
|
|
0.27-1.23 | 0.95 | 0.59 | 0.36 | 1.02 | 0.84 |
|
|
0.02-0.19 | 0.07 | 0.14 | 0.10 | 0.18 | 0.30 |
|
|
0.25-0.92 | 0.89 | 0.54 | 0.20 | 0.97 | 0.55 |
|
|
0.00-0.14 | 0.07 | 0.04 | 0.04 | 0.07 | 0.05 |
|
|
0.10-0.38 | 0.19 | 0.39 | 0.21 | 0.36 | 0.44 |
|
|
0.21-0.59 | 0.36 | 0.44 | 0.34 | 0.45 | 0.46 |
|
|
0.07-0.29 | 0.29 | 0.15 | 0.08 | 0.31 | 0.16 |
|
|
0.00-0.14 | 0.06 | 0.05 | 0.04 | 0.05 | 0.09 |
|
|
0.08-0.49 | 0.47 | 0.25 | 0.06 | 0.40 | 0.24 |
|
|
0.47-1.15 | 0.90 | 1.27 | 1.06 | 1.06 | 1.07 |
|
|
0.00-0.11 | 0.00 | 0.00 | 0.06 | 0.06 | 0.00 |
|
|
0.14-0.52 | 0.32 | 0.19 | 0.16 | 0.53 | 0.38 |
|
|
2.71-6.6 | 5.36 | 5.73 | 3.97 | 5.81 | 4.74 |
|
|
0.29-1.98 | 0.43 | 0.39 | 0.49 | 0.45 | 0.69 |
|
|
10.91-20.43 | 22.26** | 20.68* | 20.76* | 20.24 | 15.32 |
|
|
0.09-1.92 | 1.46 | 1.31 | 2.14 | 1.22 | 1.72 |
|
|
0.25-0.99 | 1.17* | 0.96 | 0.68 | 0.86 | 0.34 |
|
|
0.00-0.32 | 0.04 | 0.09 | 0.36** | 0.00 | 0.26 |
|
|
0.97-4.16 | 3.70 | 2.93 | 1.28 | 3.09 | 1.08 |
|
|
0.25-2.14 | 0.61 | 0.61 | 1.76 | 0.39 | 1.02 |
Note: P16, P17, P18, and P19 all have classic galactosemia; P15 has Duarte galactosemia. Further information about these patients is presented in Supplemental Table 1. Glycan symbols, shading, and statistical significance: same as Table 1.
Compared to controls, samples from most neonates with classic galactosemia demonstrated elevated levels of high mannose glycans at m/z of 1579 (Hex5HexNAc2, man5), 1783 (Hex6HexNAc2, man6), 1988 (Hex7HexNAc2, man 7), and also elevated truncated N-glycans with one N-acetyl glucosamine and one galactose, or alternatively one sialic acid at m/z 1865 (Hex4HexNAc4, mono-galactosylated, asialo, agalactobiantennary), 1982 (Neu5Ac1Hex4HexNAc3, mono-sialo, galactosylated, mono N-acetyl glucosamine biantennary), or 2156 (Fuc1Neu5Ac1Hex4HexNAc3, mono-sialo, galactosylated, mono-antennary fucosylated) (Table 1, darkly shaded boxes).
The neonatal samples also demonstrated unusually low levels of sialobiantennary and triantennary N-glycan at m/z values of 2430 (Neu5Ac1Hex5HexNAc4, monosialo, biantennary), 2792 (Neu5Ac2Hex5HexNAc4, disialobiantennary), 3241 (Neu5Ac2Hex6HexNAc5, disialotriantennary), 3602 (Neu5Ac3Hex6HexNAc5, trisialotriantennary), and 3776 (Neu5Ac3Fuc1Hex6HexNAc5, trisialotriantennary fucosylated) (Table 1, underlined text). In short, varying degrees of deficiency of N-glycans containing galactose and sialic acid, and accumulation of truncated and high mannose N-glycans were observed in these samples.
To quantify two specific O-glycans in the plasma, Core1Tn-antigen (T) (HexHexNAcitol, m/z pair 534/298 and monosialyl T-antigen (ST) (Neu5AcHexHexNAcitol, m/z pair 895/520), we applied a novel HPLC-MS/MS method (see Materials and methods). The reference ranges of both T and ST were derived from analyses of 40 control samples from individuals of different ages, races, and both genders, though again we also analyzed samples from age and gender-matched neonatal controls and confirmed that these results indeed fell within the stated reference range. Relative to these controls, samples from neonatal patients demonstrated elevated levels of T and diminished levels of ST so that the ratio of T/ST was increased (Table 5).
Table 5.
O-linked glycans released from total plasma glycoprotein from neonates with galactosemia. Patient number, gender, age at blood draw, GALT genotype, and gal-1P at blood draw are listed. None of these infants had detectable GALT activity in hemolysate, though P5 carries a recognized variant allele (S135L).
| Glycan structure | Reference range from 40 controls | Initial blood draw | ||||||
|---|---|---|---|---|---|---|---|---|
| P1 ♀ | P2 ♂ | P3 ♀ | P4 ♀ | P6 ♂ | P7 ♀ | P5 ♂ | ||
| 8 days | 9 days | 8 days | 9 days | 9 days | 13 days | 8 days | ||
| Q188R/ 5kbdel | Q188R/ Q188R | Q188R/ K285N | Q188R/ Q188R | Q188R/ Y209C | Q206R/ Q206R | S135L/ K285N | ||
| gal - 1P 30.3 mg/dL | gal - 1P unk | gal - 1P 15.7 mg/dL | gal - 1P 46.4 mg/dL | gal - 1P 48.1 mg/dL | gal - 1P 52.4 mg/dL | gal - 1P 55.6 mg/dL | ||
|
|
0.22 μM -1.14 μM | 3.19** | 0.2 | 0.62 | 0.94 | 0.72 | 0.33 | 0.59 |
|
|
11.7 μM -31.4 μM | 13.9 | 1.9** | 11.8 | 8.8** | 5.6** | 4.5** | 21.8 |
| T/ST | ≤0.06 | 0. 23** | 0. 11** | 0.05 | 0.11** | 0.13** | 0.07 | 0.05 |
Core1Tn-antigen (T), sialyl-T antigen (ST), and the ratio of T/ST are presented. Glycan symbols: □: N-acetylgalactosamine (GalNAc); ○: galactose; ◆: sialic acid (Neu5Ac). Shading and statistical significance are the same as for Table 1.
3.2. Variance of plasma protein N- and O-glycome defects is not explained by gender, age, GALT genotype, or predicted residual GALT activity
Of the galactosemic neonates whose plasma samples we studied, all 7 demonstrated at least some glycosylation abnormality, but the extent of the defect ranged from mild to pronounced. For each patient, the degree of abnormality of N- and O-glycans was consistent though not identical. For example, P1 and P6, who demonstrated severe N-glycome defects, also showed severe O-glycome defects, and P4 and P7, who both demonstrated mild N-glycome defects, also showed mild O-glycome defects (Tables 1, 5).
To explore possible modifiers of this biochemical trait we first stratified the samples by gender and age, but saw no clear patterns; the three most severely impacted patients included two females (P1 and P3) and one male (P6), and the two least severely impacted patients included one of the youngest (P4) at 9 days, and the oldest (P7) at 13 days (Tables 1, 5). We also tried stratifying the samples according to GALT genotype and predicted presence or absence of residual GALT activity associated with that genotype (see Materials and methods); again we saw no clear pattern. Specifically, the 7 patients studied collectively comprised 6 different GALT genotypes: Q188R/Q188R (P2 and P4), Q188R/Y209C (P6), Q188R/K285N (P3), Q188R/5 kb deletion (P1), Q206R/Q206R (P4), and S135L/K285N (P5). All 7 patients were reported from clinical studies of hemolysate to have no detectable GALT activity, however, three carried GALT alleles reported to demonstrate residual activity, at least in some tissues or model systems (K285N (0.6 ± 0.1% of WT activity in a yeast model system), S135L (0.7 ± 0% of WT activity in a yeast model system) and Y209C (13.6 ± 1.3% of WT activity in a yeast model system), see Materials and methods [27,28]. Of the four patients with no detected or predicted residual GALT activity, two (P1 and P2) demonstrated more notable glycosylation defects and two (P4 and P7) demonstrated only mild defects. Of the three patients with some predicted residual GALT activity, two demonstrated a marked glycosylation defect (P3 and P6) and the other one (P5) demonstrated only a mild defect (Table 1). In short, there was no apparent relationship between severity of the glycosylation defect and the presence or absence of predicted residual GALT activity.
Finally, we had limited data on race for these seven infants: P2, P4, and P6 were described as Caucasian, P5 was described as African-American, and P7 was described as Hispanic; P1 and P3 were of unknown race. While it is certainly intriguing to note that the two least severely affected samples derived from the two infants who were not Caucasian, while all three samples from infants listed as Caucasian were more severely affected, this sample size is far too small to draw a general conclusion about race and glycosylation in galactosemia.
3.3. Both N- and O-linked glycosylation defects respond to dietary restriction of galactose in infants
Prior studies have reported that N-linked glycosylation defects associated with classic galactosemia normalize, or almost normalize in infants following dietary restriction of galactose [11] but the question has remained open whether O-linked defects would also normalize, and if so, how the kinetics for normalization of the two glycan populations would compare.
To address this question we obtained follow-up plasma samples from three of the seven neonates initially studied and characterized the N- and O-linked glycomes of these new samples using the same approaches described above. The new samples, collected sequentially after each infant was diagnosed and switched to a galactose restricted diet, revealed a clear improvement in the levels of high mannose N-glycans and truncated N-glycans, and also near normalization of the sialobiantennary and triantennary N-glycans (Fig. 2, Table 2). For O-glycans, Tn-antigen moved to within the reference range as did the monosialyl T-antigen (Table 6). The kinetics of the changes, however, were not the same. Of note, although we do not have formal confirmation of diet history on four of these infants, we do have gal-1P values derived from the same blood samples, and these confirm that for neonatal patients P1, P4, P5, P6, and P7 the first gal-1P values were extremely high (e.g. >30 mg/dL), consistent with what would be expected for a neonate with classic galactosemia consuming milk (Table 1). The initial gal-1P value for P3 was also high (15.7 mg/dL) though not as high as the others. The initial gal-1P value for infant P2 was unavailable, but follow-up samples from this patient showed still elevated values that took months to fall to within the “therapeutic” range, implying that the gal-1P level was substantially elevated at the time of the initial blood draw.
Fig. 2.
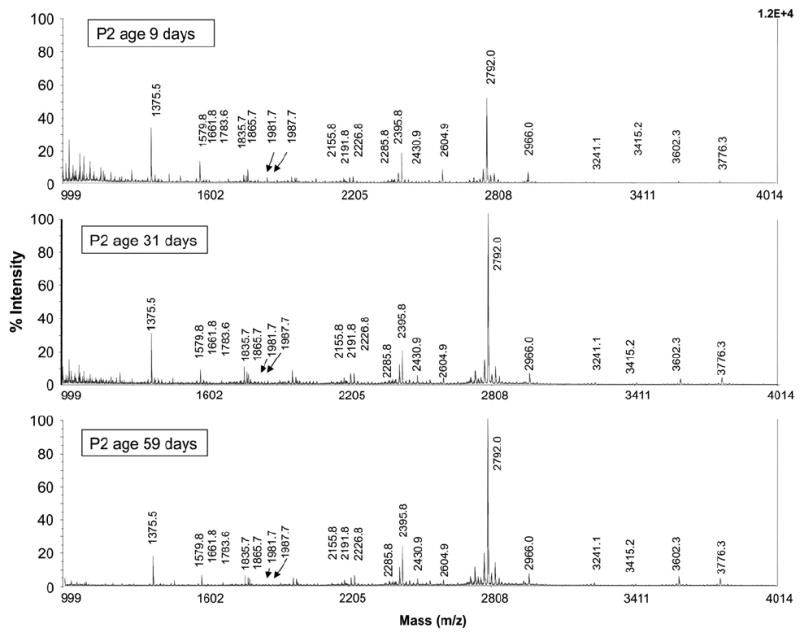
MALDI-TOF spectra of N-linked glycans from total plasma glycoproteins from a single galactosemia patient before and after dietary restriction of galactose. (A) Plasma from the blood sample collected at age 9 days; (B) plasma from blood collected from the same infant at age 31 days; (C) plasma from blood collected from the same infant at age 59 days.
Table 2.
N-linked glycans released from total plasma glycoprotein in longitudinal plasma samples from infants with galactosemia. Patient number, gender, age at blood draw, and gal-1P at blood draw are listed. None of these infants had detectable GALT activity in hemolysate.
| Glycan structure | Reference range from 150 controls (%) | Initial | Follow-up | Initial | Follow-up | Initial | Follow-up | ||
|---|---|---|---|---|---|---|---|---|---|
| P2# ♂ | P2 ♂ | P2 ♂ | P2 ♂ | P3# ♀ | P3 ♀ | P7# ♀ | P7 ♀ | ||
| 9 days (gal - 1P unk) | 31 days (9.5 mg/dL) | 59 days (5.3 mg/dL) | 206 days (1.5 mg/dL) | 8 days (15.7 mg/dL) | 56 days (7.2 mg/dL) | 13 days (52.4 mg/dL) | 109 days (3.1 mg/dL) | ||
|
|
0.27-1.23 | 1.37 | 0.61 | 0.60 | 0.44 | 1.78* | 0.64 | 1.09 | 0.67 |
|
|
0.02-0.19 | 0.14 | 0.08 | 0.04 | 0.00 | 0.09 | 0.00 | 0.07 | 0.31 |
|
|
0.25-0.92 | 0.90 | 0.45 | 0.48 | 0.24 | 1.20* | 0.38 | 0.55 | 0.42 |
|
|
0.00-0.14 | 0.06 | 0.11 | 0.08 | 0.07 | 0.00 | 0.00 | 0.07 | 0.12 |
|
|
0.10-0.38 | 0.36 | 0.11 | 0.11 | 0.14 | 0.55 | 0.13 | 0.26 | 0.14 |
|
|
0.21-0.59 | 0.40 | 0.40 | 0.47 | 0.49 | 0.70 | 0.50 | 0.49 | 0.48 |
|
|
0.07-0.29 | 0.32 | 0.15 | 0.21 | 0.10 | 0.59** | 0.14 | 0.25 | 0.08 |
|
|
0.00-0.14 | 0.12 | 0.10 | 0.09 | 0.07 | 0.03 | 0.00 | 0.10 | 0.09 |
|
|
0.08-0.49 | 0.27 | 0.15 | 0.15 | 0.11 | 0.38 | 0.17 | 0.19 | 0.20 |
|
|
0.47-1.15 | 0.44 | 0.61 | 0.78 | 1.38 | 0.87 | 1.05 | 0.82 | 1.03 |
|
|
0.00-0.11 | 0.05 | 0.08 | 0.13* | 0.08 | 0.08 | 0.07 | 0.10 | 0.06 |
|
|
0.14-0.52 | 0.14 | 0.16 | 0.22 | 0.21 | 0.44 | 0.30 | 0.20 | 0.22 |
|
|
2.71-6.6 | 2.25 | 1.88* | 3.05 | 4.27 | 4.05 | 3.65 | 2.86 | 2.72 |
|
|
0.29-1.98 | 0.94 | 0.42 | 0.54 | 0.27 | 0.92 | 0.44 | 0.95 | 0.48 |
|
|
10.91-20.43 | 6.73** | 11.13 | 16.65 | 20.22 | 14.85 | 24.27** | 14.12 | 16.69 |
|
|
0.09-1.92 | 0.94 | 0.86 | 1.15 | 1.14 | 1.87 | 1.57 | 2.20* | 1.26 |
|
|
0.25-0.99 | 0.14* | 0.10* | 0.27 | 0.83 | 0.28 | 0.40 | 0.48 | 0.34 |
|
|
0.00-0.32 | 0.08 | 0.06 | 0.10 | 0.18 | 0.35** | 0.00 | 0.32** | 0.20 |
|
|
0.97-4.16 | 0.22* | 0.52 | 1.05 | 2.86 | 0.80 | 1.29 | 0.85 | 1.36 |
|
|
0.25-2.14 | 0.19 | 0.54 | 0.87 | 0.77 | 0.91 | 2.07 | 0.84 | 0.91 |
Glycan symbols, shading, and statistical significance: same as Table 1.
Table 6.
O-linked glycans released from longitudinal samples of total plasma glycoprotein from infants with classic galactosemia. Patient number, gender, age at blood draw, GALT genotype, and gal-1P at blood draw are listed. None of these infants had detectable GALT activity in hemolysate.
| Glycan structure | Reference range from 40 controls | Initial | Follow-up | Initial | Follow-up | Initial | Follow-up | ||
|---|---|---|---|---|---|---|---|---|---|
| P2# ♂ | P2 ♂ | P2 ♂ | P2 ♂ | P3# ♀ | P3 ♀ | P7# ♀ | P7 ♀ | ||
| 9 days (gal - 1P unk) | 31 days (9.5 mg/dL) | 59 days (5.3 mg/dL) | 206 days (1.5 mg/dL) | 8 days (15.7 mg/dL) | 56 days (7.2 mg/dL) | 13 days (52.4 mg/dL) | 109 days (3.1 mg/dL) | ||
|
|
0.22 μM -1.14 μM | 0.2 | 0.37 | 0.31 | 0.50 | 0.62 | 0.47 | 0.33 | 0.36 |
|
|
11.7 μM -31.4 μM | 1.9** | 5.9** | 7.1** | 14.3 | 11.8 | 13.5 | 4.5** | 43.1** |
| T/ST | ≤0.06 | 0. 11** | 0.06 | 0.03 | 0.05 | 0.05 | 0.07 | 0.07 | 0.01 |
Core1Tn-antigen (T), sialyl-T antigen (ST), and the ratio of T/ST are presented. Glycan symbols: □: N-acetylgalactosamine (GalNAc); ○: galactose; ◆: sialic acid (Neu5Ac). Shading and statistical significance are the same as for Table 1.
Follow-up plasma samples from P2 were analyzed at ages 31 days, 59 days, and 206 days, which corresponded to 22 days, 50 days, and 197 days after the initial sample was collected and the diet was ostensibly switched to soy formula (Table 2). Analysis of the first of these follow-up samples showed normalized high mannose N-glycans and truncated N-glycans, but still abnormally low levels of N-glycan at m/z 2431, 3241, and 3602. For O-linked glycans, the first follow-up sample showed a normalized level of Tn-antigen, but still abnormally low monosialyl T-antigen. The two later samples from this infant both showed normal or near-normal N-glycans containing galactose and sialic acid (Fig. 2, Table 2) and O-glycan monosialyl T-antigen (Table 6). The glycan profiles of sequential samples from this patient clearly showed that truncated N-glycans, high-mannose-N-glycans, and asialo O-glycans normalized first, followed by the sialo biantennary and tri-antennary N-glycans and sialo O-glycans, which normalized more slowly.
Follow-up samples from infants P3 and P7 also both showed near-normalization of N- and O-linked glycans, though it is interesting to note that in some instances, e.g. P3 in Table 6, the later samples showed minor abnormalities that were not evident in the initial sample(s).
3.4. Plasma glycosylation defects evident in treated children and adults with galactosemia fail to correlate with long-term outcome severity
Despite neonatal or even prenatal diagnosis and life-long dietary restriction of galactose, a majority of patients with classic galactosemia grow to experience profound long-term complications ([29], reviewed in [1]). To test whether chronic, global defects of protein glycosylation might underlie these outcomes we characterized the N- and O-linked glycomes of plasma proteins from 11 patients with classic galactosemia and one with Duarte galactosemia, each of whom reported adhering to a carefully galactose restricted diet, and for each of whom positive or negative outcome information was known. Of these 12, 7 (P8 to P14) were girls ages 2 to 9 years who had been previously characterized with regard to ovarian function by measuring plasma anti-Müllerian hormone levels. Previous studies indicate that AMH is a sensitive biomarker of ovarian status in girls and women [18,19,30,31], so that low to undetectable AMH in pre-pubertal girls and reproductive age women is an indicator of premature ovarian insufficiency (POI). Of the seven 2–9.5 year old girls studied here, 3 had normal AMH levels and 4 had low to undetectable AMH levels, yet no corresponding patterns were evident in profiles of N- and O-linked glycans released from plasma proteins (Tables 3 and 7).
We also studied the N- and O-linked plasma protein glycomes from 4 adults with classic galactosemia and one adult with the mild variant Duarte galactosemia. All 5 of these adult volunteers had received careful physical, neurological, psychological, cognitive, and speech/language assessment [20] allowing them to be stratified into two groups, one group demonstrating severe central nervous system disabilities, and the other demonstrating essentially normal neurological function (Supplemental Table 1). Despite these outcome differences, those few glycosylation abnormalities that were detected (Tables 4 and 8) appeared scattered almost randomly among the samples, so that no relationship with outcome severity was evident.
Table 8.
O-linked glycans released from total plasma glycoprotein from adults with well-managed galactosemia who do and do not demonstrate neurologic dysfunction. Patient number, gender, age at blood draw, neurological outcome status, and GALT genotype are listed.
| Glycan structure | Reference range from 40 controls | P15 ♀ | P16 ♀ | P17 ♂ | P18 ♀ | P19 ♂ |
|---|---|---|---|---|---|---|
| 37 years | 31 years | 59 years | 54 years | 51 years | ||
| good outcome | poor outcome | poor outcome | good outcome | good outcome | ||
| D/G | Q188R/ Q188R | 5kb del/ 5kb del | 5kb del/ 5kb del | Q188R/ Q188R | ||
|
|
0.22 μM -1.14 μM | 0.5 | 0.75 | 0.59 | 0.66 | 2.45** |
|
|
11.7 μM -31.4 μM | 15.3 | 14.6 | 12.7 | 17.8 | 19.9 |
| T/ST | ≤0.06 | 0.06 | 0.05 | 0.05 | 0.04 | 0.12** |
Note: P16, P17, P18, and P19 all have classic galactosemia; P15 has Duarte galactosemia. Further information about these patients is presented in Supplemental Table 1. Core1Tn-antigen (T), sialyl-T antigen (ST), and the ratio of T/ST are presented. Glycan symbols: □: N-acetylgalactosamine (GalNAc); ○: galactose; ◆ : sialic acid (Neu5Ac). Shading and statistical significance are the same as for Table 1.
4. Discussion
The studies presented here were designed to answer three questions: (1) how similar or disparate are the N- and O-linked glycosylation defects seen in plasma proteins from untreated galactosemic neonates, (2) what intrinsic or environmental factors might explain differences in the extent of glycosylation defect seen in individual neonates, and perhaps most important, (3) is there any apparent relationship between chronic glycosylation defect and long-term outcome in ostensibly well-managed older children and adults with classic galactosemia?
To address these questions we applied mass spectrometry to characterize the plasma N- and O-linked protein glycomes from three groups of patients: galactosemic neonates studied both before and after dietary restriction of galactose, galactosemic girls whose ovarian functional status was known, and galactosemic adults whose neurological status was known. The 7 galactosemic neonates studied ranged in age from 8 to 13 days; each had been flagged by an abnormal newborn screen for galactosemia and subsequently diagnosed with classic galactosemia by a combination of biochemical and molecular testing (Materials and methods). Three of these infants also provided follow-up plasma samples collected weeks to months after dietary restriction of galactose was initiated. The 7 galactosemic girls studied ranged in age from 2 to 9 years; these girls were selected for study because we had plasma AMH levels for all 7 demonstrating that 3 had essentially normal ovarian function and 4 had premature ovarian insufficiency. Finally, of the 5 adults studied, 4 had classic galactosemia and one had the mild variant Duarte galactosemia (DG), however all 5 reported adhering to galactose restricted diets. These adults were assessed for neurological outcome as part of a prior study [20]; three, including the DG patient, had good neurological outcomes and two had poor neurological outcomes.
4.1. Glycosylation defects in galactosemia
4.1.1. Neonates
Our results demonstrated a range of N- and O-linked glycosylation defects in a majority of the galactosemic neonates studied; these included abnormally high levels of high mannose N-glycans and truncated N-glycans and abnormally low levels of complex N-glycans containing galactose and sialic acid. We also saw elevated levels of the O-glycan Core1 Tn-antigen (T) and diminished levels of the monosialyl T-antigen (ST). Those samples that showed the most significant defects in N-linked glycosylation (P1, P3, P6) overlapped with but were not identical to those that showed the most significant defects in O-linked glycosylation (P1, P2, P4, P6, P7).
Age, gender, GALT genotype, and predicted residual GALT activity all failed to explain the apparent differences between those neonates who exhibited the highest and lowest levels of glycosylation defect. Race did show an interesting pattern, namely that two subjects listed as African-American or Hispanic both showed milder glycosylation defects than three subjects listed as Caucasian; however, this sample size is too small to establish a significant correlation.
It is important to note that although we were not able to secure a confirmed diet history for four of the infants at the time of their first blood draw, from their elevated gal-1P values measured using these same blood samples, or subsequent blood samples, we can surmise that all seven were still on milk diets, or had switched off of milk only recently. The three diet histories that were available (P4 switched to soy at about 8 days, P5 switched to soy at about 8 days, P7 switched to soy at about two weeks) were fully consistent with this conclusion. Of note, for those samples that had corresponding gal-1P values measured, there was no apparent correlation between gal-1P level and the degree of glycosylation defect seen (Tables 1 and 5). At face value, this observation suggests that elevated gal-1P may not underlie the glycosylation defect, at least in plasma, although clearly the situation may be complex and gal-1P may still play a contributing role.
The apparent failure of predicted residual GALT activity to explain severity of the glycosylation defect might also reflect an oversimplification. Our analyses of residual activity associated with each allele were performed using a null-background yeast expression system for the human enzyme [32,33]; this system predicted non-zero residual GALT activity associated with each of the following amino acid substitutions: K285N (0.6 ± 0.1% wild-type activity), S135L (0.7 ± 0% wild-type activity), and Y209C (13.6 ± 1.3% wild-type activity). Plasma from the patient who carries two of these alleles in combination (K285N/ S135L, P5) indeed showed mild if any N- or O-linked glycosylation defects, while P3, who carries one of these alleles (K285N) in combination with a null allele (Q188R, 0 ± 0% wild-type activity) showed significant N-linked defects but no clear O-linked defects. The mystery was P6, who carries a Y209C allele that is predicted from yeast studies to have significant residual activity, yet this sample demonstrated pronounced N- and O-linked glycosylation defects. There are two possible explanations for this disparity—either residual GALT activity is irrelevant to the glycosylation defects observed, or alternatively, the Y209C allele may encode residual activity in a yeast model system, but in its natural context (human) it may not have residual activity. For example, the nucleic acid change that predicts a missense substitution acting at the level of protein might, in fact, alter the function or stability of the RNA, which cannot be adequately modeled in the yeast system.
Blood samples collected from three of the infants following dietary restriction of galactose demonstrated a striking and sequential resolution of both the N- and O-linked defects (Tables 2 and 6) such that the truncated N-glycans, high-mannose-N-glycans, and asialo O-glycans normalized first, followed by the sialo biantennary and triantennary N-glycans and sialo O-glycans, which normalized more slowly. The significance of this order is unclear; it could reflect differential turn-over rates in different subcellular compartments, or alternatively the truncated N-glycans, high-mannose-N-glycans, and asialo O-glycans may normalize first because until they do the sialo biantennary and tri-antennary N-glycans and sialo O-glycans cannot accumulate to normal levels.
4.1.2. Implications for mechanism
N- and O-linked glycosylation are complex biosynthetic processes that require the coordinated activities and appropriate subcellular localization of numerous gene products and metabolites, including glycosyltransferases and glycosidases, nucleotide sugar transporters, UDP-sugars, and more (Supplemental Fig. 1). The glycosylation defects seen by ourselves and others [10,11] in samples from untreated neonates with classic galactosemia are suggestive of errors in both glycan assembly (or transfer) and processing, implicating defects in both the ER and Golgi.
4.2. Implications for outcome
One of the principle motivations for studying glycosylation in galactosemia has been the intriguing possibility that subtle abnormalities in glycosylation might underlie the long-term complications experienced by patients. Considering the essential roles of glycosylation in myriad processes of development and homeostasis, the involvement of Leloir enzymes in biosynthesis and maintenance of UDP-sugar substrate pools, and the clear demonstrations that galactosemic patients experience acute and perhaps also chronic defects of glycosylation despite treatment (e.g. [10]), this hypothesis was not a stretch.
Nonetheless, our data presented here do not support this hypothesis. Part of the problem may be that the glycosylation abnormalities detected in treated patients are subtle, and given that each reference range is defined as the window in which 95% but not 100% of all control values fall, by definition finding occasional mild “abnormalities” in the glycans of patients may not be entirely meaningful. Given that hemolysate gal-1P is not a reliable marker of low-level galactose ingestion in older patients [14], it is also possible that some of the volunteers in this study were consuming dietary galactose, either knowingly or unknowingly. Relevant technologies have also changed substantially over the past decades, influencing what is seen and how it is interpreted. For example, in 1997 Prestoz and colleagues [34] used gel electrophoresis and western blotting to detect what they interpreted to be abnormally glycosylated isoforms of follicle stimulating hormone (FSH) in blood samples from galactosemic women. In 2011, Gubbels and colleagues [35] asked the same question using FPLC chromato-focusing with measurement of FSH concentration per fraction and concluded that they could not find evidence of abnormally glycosylated FSH isoforms in blood samples from galactosemic women. Of course, both studies used small cohorts so it also remains possible that both conclusions were internally correct.
Our study also used a single sample type—plasma—derived from relatively small cohorts of cases representing specific age groups. Our study therefore cannot rule out the possibility that glycosylation defects early in development, or specific to tissues other than plasma, or specific to individual glycoproteins undetectable in a whole glycome study, may yet contribute to clinical outcome. Future studies in different tissues, or at earlier times in development—for example from amniocytes or cord blood—might help to resolve this question.
4.3. Conclusions
We conclude that many infants with classic galactosemia demonstrate galactose-dependent aberrant N- and O-linked protein glycosylation but that chronic, global protein glycosylation defects detected in older children and adults with classic galactosemia do not correlate with the long-term complications experienced by these patients.
Supplementary Material
Acknowledgments
We are especially grateful to all the volunteers and their families who participated in this study. We also thank colleagues in the Emory Genetics Laboratory and Children’s Hospital Boston for their contributions to this project. The work was supported in part by grant NIH DK059904 (to JLFK) and in part by funds from the Manton Center for Orphan Disease Research (to GTB) and the Galactosemia Foundation (to GTB). MH was supported in part by grant U54HD061939 from the Sterol and Isoprenoid Diseases (STAIR) consortium, a part of the NIH Rare Diseases clinical Research Network (RDCRN), funded by the NICHD and the NIH Office of Rare Diseases Research (ORDR).
Abbreviations
- AMH
anti-Müllerian hormone
- DHB
2, 5-dihydroxybenzoic acid
- DMSO
dimethyl sulfoxide
- GALE
UDP-galactose 4′-epimerase
- GALT
galactose-1-phosphate uridylyltransferase
- UDP-Gal
uridine diphosphate galactose
- UDP-GalNAc
uridine diphosphate-N-acetyl-d-galactosamine
- UDP-Glc
uridine diphosphate glucose
- UDP-GlcNAc
uridine diphosphate-N-acetyl-d-glucosamine
- HPLC-MS/MS
high-performance liquid chromatography coupled with tandem spectrometry
- MALDI-TOF
matrix-assisted laser desorption/ionization time-of-flight mass spectrometry
- PNGase F
peptide N-Glycosidase F
- pLNH
P-Lacto-N-hexaose
- ST
monosialyl T-antigen
- T
Core1 Tn-antigen
Footnotes
Supplementary related to this article can be found online at http://dx:doi.org/10.1016/j.ymgme.2012.05.025.
Conflict of interest
The authors have no conflicts of interest to report.
References
- 1.Fridovich-Keil JL, Walter JH. Galactosemia. In: Valle D, Beaudet A, Vogelstein B, Kinzler K, Antonarakis S, et al., editors. The Online Metabolic & Molecular Bases of Inherited Disease. McGraw Hill; 2008. http://www.ommbid.com/ [Google Scholar]
- 2.Holden HM, Rayment I, Thoden JB. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J Biol Chem. 2003;278:43885–43888. doi: 10.1074/jbc.R300025200. [DOI] [PubMed] [Google Scholar]
- 3.Haberland C, Perou M, Brunngraber EG, Hof H. The neuropathology of galactosemia. A histopathological and biochemical study. J Neuropathol Exp Neurol. 1971;30:431–447. doi: 10.1097/00005072-197107000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Witting L, Haberland C, Brunngraber E. Ganglioside patterns in galactosemia. Clin Chim Acta. 1972;37:387–389. doi: 10.1016/0009-8981(72)90459-7. [DOI] [PubMed] [Google Scholar]
- 5.Petry K, Greinix HT, Nudelman E, Eisen H, Hakomori S, et al. Characterization of a novel biochemical abnormality in galactosemia: deficiency of glycolipids containing galactose or N-acetylgalactosamine and accumulation of precursors in brain and lymphocytes. Biochem Med Metab Biol. 1991;46:93–104. doi: 10.1016/0885-4505(91)90054-o. [DOI] [PubMed] [Google Scholar]
- 6.Jaeken J, Kint J, Spaapen L. Serum lysosomal enzyme abnormalities in galactosaemia. Lancet. 1992;340:1472–1473. doi: 10.1016/0140-6736(92)92664-2. [DOI] [PubMed] [Google Scholar]
- 7.Dobbie JA, Holton JB, Clamp JR. Defective galactosylation of proteins in cultured skin fibroblasts from galactosaemic patients. Ann Clin Biochem. 1990;27:274–275. doi: 10.1177/000456329002700317. [DOI] [PubMed] [Google Scholar]
- 8.Ornstein KS, McGuire EJ, Berry GT, Roth S, Segal S. Abnormal galactosylation of complex carbohydrates in cultured fibroblasts fom patients with galactose-1-phosphate uridyltransferase deficiency. Pediatr Res. 1992;31:508–511. doi: 10.1203/00006450-199205000-00020. [DOI] [PubMed] [Google Scholar]
- 9.Stibler H, von Dobeln U, Kristiansson B, Guthenberg C. Carbohydrate-deficient transferrin in galactosaemia. Acta Paediatr. 1997;86:1377–1378. doi: 10.1111/j.1651-2227.1997.tb14917.x. [DOI] [PubMed] [Google Scholar]
- 10.Charlwood J, Clayton P, Keir G, Mian N, Winchester B. Defective galactosylation of serum transferrin in galactosemia. Glycobiology. 1998;8:351–357. doi: 10.1093/glycob/8.4.351. [DOI] [PubMed] [Google Scholar]
- 11.Sturiale L, Barone R, Fiumara A, Perez M, Zaffanello M, et al. Hypoglycosylation with increased fucosylation and branching of serum transferrin N-glycans in untreated galactosemia. Glycobiology. 2005;15:1268–1276. doi: 10.1093/glycob/cwj021. [DOI] [PubMed] [Google Scholar]
- 12.Quintana E, Navarro-Sastre A, Hernández-Pérez J, García-Villoria J, Montero R, et al. Screening for congenital disorders of glycosylation (CDG): transferrin HPLC versus isoelectric focusing (IEF) Clin Biochem. 2009;42:408–415. doi: 10.1016/j.clinbiochem.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 13.Coman D, Murray D, Byrne J, Rudd P, Bagaglia P, et al. Galactosemia, a single gene disorder with epigenetic consequences. Pediatr Res. 2010;67:286–292. doi: 10.1203/PDR.0b013e3181cbd542. [DOI] [PubMed] [Google Scholar]
- 14.Coss K, Byrne J, Coman D, Adamczyk B, Abrahams J, et al. IgG N-glycans as potential biomarkers for determining galactose tolerance in Classical Galactosaemia. Mol Genet Metab. 2012;105:212–220. doi: 10.1016/j.ymgme.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 15.Staubach S, Schadewaldt P, Wendel U, Nohroudi K, Hanisch F-G. Differential glycomics of epithelial membrane glycoproteins from urinary exovesicles reveals shifts toward complex-type N-glycosylation in classical galactosemia. J Proteome Res. 2012;11:906–916. doi: 10.1021/pr200711w. [DOI] [PubMed] [Google Scholar]
- 16.Guillard M, Morava E, van Delft F, Hague R, Körner C, et al. Plasma N-glycan profiling by mass spectrometry for congenital disorders of glycosylation type II. Clin Chem. 2011;57:593–602. doi: 10.1373/clinchem.2010.153635. [DOI] [PubMed] [Google Scholar]
- 17.Knezevic A, Gornik O, Polasek O, Pucic M, Redzic I, et al. Effects of aging, body mass index, plasma lipid profiles, and smoking on human plasma N-glycans. Glycobiology. 2010;20:959–969. doi: 10.1093/glycob/cwq051. [DOI] [PubMed] [Google Scholar]
- 18.Fridovich-Keil J, Gubbels C, Spencer J, Sanders R, Land J, et al. Ovarian function in girls and women with GALT-deficiency galactosemia. J Inherit Metab Dis. 2011;34:357–366. doi: 10.1007/s10545-010-9221-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanders R, Spencer J, Epstein M, Pollak S, Vardhana P, et al. Biomarkers of ovarian function in girls and women with classic galactosemia. Fertil Steril. 2009;92:344–351. doi: 10.1016/j.fertnstert.2008.04.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waisbren S, Potter N, Gordon C, Green R, Greenstein P, et al. The adult galactosemic phenotype. J Inherit Metab Dis. 2012;35:279–286. doi: 10.1007/s10545-011-9372-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morelle W, Flahaut C, Michalski JC, Louvet A, Mathurin P, et al. Mass spectrometric approach for screening modifications of total serum N-glycome in human diseases: application to cirrhosis. Glycobiology. 2006;16:281–293. doi: 10.1093/glycob/cwj067. [DOI] [PubMed] [Google Scholar]
- 22.Carlson DM. Structures and immunochemical properties of oligosaccharides isolated from pig submaxillary mucins. J Biol Chem. 1968;243:616–626. [PubMed] [Google Scholar]
- 23.Chhay J, Openo K, Eaton J, Gentile M, Fridovich-Keil J. A yeast model reveals biochemical severity associated with each of three variant alleles of galactose-1P uridylyltransferase segregating in a single family. J Inherit Metab Dis. 2008;31:97–107. doi: 10.1007/s10545-007-0786-5. [DOI] [PubMed] [Google Scholar]
- 24.Morelle W, Slomianny MC, Diemer H, Schaeffer C, van Dorsselaer A, et al. Fragmentation characteristics of permethylated oligosaccharides using a matrix-assisted laser desorption/ionization two-stage time-of-flight (TOF/TOF) tandem mass spectrometer. Rapid Commun Mass Spectrom. 2004;18:2637–2649. doi: 10.1002/rcm.1668. [DOI] [PubMed] [Google Scholar]
- 25.Faid V, Chirat F, Seta N, Foulquier F, Morelle W. A rapid mass spectrometric strategy for the characterization of N- and O-glycan chains in the diagnosis of defects in glycan biosynthesis. Proteomics. 2007;7:1800–1813. doi: 10.1002/pmic.200600977. [DOI] [PubMed] [Google Scholar]
- 26.Morelle W, Faid V, Chirat F, Michalski JC. Analysis of N- and O-linked glycans from glycoproteins using MALDI-TOF mass spectrometry. Methods Mol Biol. 2009;534:5–21. doi: 10.1007/978-1-59745-022-5_1. [DOI] [PubMed] [Google Scholar]
- 27.Fridovich-Keil JL, Langley SD, Mazur LA, Lennon JC, Dembure PP, et al. Identification and functional analysis of three distinct mutations in the human galactose-1-phosphate uridyltransferase gene associated with galactosemia in a single family. Am J Hum Genet. 1995;56:640–646. [PMC free article] [PubMed] [Google Scholar]
- 28.Reichardt JKV, Levy HL, Woo SL. Molecular characterization of two galactosemia mutations and one polymorphism: implications for structure-function analysis of human galactose-1-phosphate uridyltransferase. Biochemistry. 1992;31:5430–5433. doi: 10.1021/bi00139a002. [DOI] [PubMed] [Google Scholar]
- 29.Hughes J, Ryan S, Lambert D, Geoghegan O, Clark A, et al. Outcomes of siblings with classical galactosemia. J Pediatr. 2009;154:721–726. doi: 10.1016/j.jpeds.2008.11.052. [DOI] [PubMed] [Google Scholar]
- 30.Hagen CP, Aksglaede L, Sorensen K, Main KM, Boas M, et al. Serum levels of anti-Mullerian hormone as a marker of ovarian function in 926 healthy females from birth to adulthood and in 172 Turner syndrome patients. J Clin Endocrinol Metab. 2010;95:5003–5010. doi: 10.1210/jc.2010-0930. [DOI] [PubMed] [Google Scholar]
- 31.La Marca A, Sighinolfi G, Radi D, Argento C, Baraldi E, et al. Anti-’Mullerian hormone (AMH) as a predictive marker in assisted reproductive technology (ART) Hum Reprod Update. 2010;16:113–130. doi: 10.1093/humupd/dmp036. [DOI] [PubMed] [Google Scholar]
- 32.Fridovich-Keil JL, Jinks-Robertson S. A yeast expression system for human galactose-1-phosphate uridylyltransferase. Proc Natl Acad Sci U S A. 1993;90:398–402. doi: 10.1073/pnas.90.2.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riehman K, Crews C, Fridovich-Keil JL. Relationship between genotype, activity, and galactose sensitivity in yeast expressing patient alleles of human galactose-1-phosphate uridylyltransferase. J Biol Chem. 2001;276:10634–10640. doi: 10.1074/jbc.M009583200. [DOI] [PubMed] [Google Scholar]
- 34.Prestoz L, Couto A, Shin Y, Petry K. Altered follicle stimulating hormone isoforms in female galactosaemia patients. Eur J Pediatr. 1997;156:116–120. doi: 10.1007/s004310050568. [DOI] [PubMed] [Google Scholar]
- 35.Gubbels C, Thomas C, Wodzig W, Olthaar A, Jaeken J, et al. FSH isoform pattern in classic galactosemia. J Inherit Metab Dis. 2011;34:387–390. doi: 10.1007/s10545-010-9180-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.