

Abstract
(E)-4-Hydroxy-3-methyl-but-2-enyl diphosphate reductase (IspH or LytB) catalyzes the terminal step of the MEP/DOXP pathway where it converts (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate (HMBPP) into the two products isopentenyl diphosphate and dimethylallyl diphosphate. The reaction involves the reductive elimination of the C4 hydroxyl group, using a total of two electrons. Here we show that the active form of IspH contains a [4Fe-4S] cluster and not the [3Fe-4S] form. Our studies show that the cluster is not only the direct electron source for the reaction but that a reaction intermediate is bound directly to the cluster. This active form, has been trapped in a state, dubbed FeSA, that was detected in EPR spectroscopy when one-electron-reduced IspH was incubated with HMBPP. In addition, three mutants of IspH protein have been prepared and studied, His42, His124 and Glu126 (Aquifex aeolicus numbering), with particular attention to the effects on the cluster properties and possible reaction intermediates. None of the mutants affected the properties of the [4Fe-4S]+ cluster significantly, but different effects were observed when one-electron-reduced forms were incubated with HMBPP. Replacing the His42 led to an increased Km value and much lower catalytic efficiency, confirming the role of this residue in substrate binding. Replacing the His124 also resulted in lower catalytic efficiency. In this case, however, enzyme showed the loss of the [4Fe-4S]+ EPR signal upon addition of HMBPP without the subsequent formation of the FeSA signal. Instead, a radical-type signal was observed in some of the samples indicating that this residue plays a role in the correct positioning of the substrate. The incorrect orientation in the mutant leads to the formation of substrate-based radicals instead of the cluster-bound-intermediate complex FeSA. Replacing the Glu126 also resulted in lower catalytic efficiency, with yet a third type of EPR signal being detected upon incubation with HMBPP. 31P- and 2H-ENDOR measurements on the FeSA species incubated with regular and 2H-C4-labeled HMBPP reveal that the substrate binds to the enzyme in close proximity of the active-site cluster with the C4 adjacent to the site of linkage between the FeS cluster and HMBPP. Comparison of the spectroscopic properties of this intermediate to those of intermediates detected in (E)-4-hydroxy-3-methylbut-2-enyl diphosphate synthase and ferredoxin:thioredoxin reductase suggest that HMBPP binds to the FeS cluster via its hydroxyl group instead of a side-on binding as previously proposed for the species detected in the inactive Glu126 variant. Consequences for the IspH reaction mechanism are discussed.
Keywords: (E)-4-Hydroxy-3-methyl-but-2-enyl diphosphate reductase, IspH, EPR, ENDOR, reaction intermediate
The (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate reductase (IspH or LytB) catalyzes the terminal step of the 2-C-methyl-D-erythritol 4-phosphate/1-deoxy-D-xylulose 5-phosphate (MEP/DOXP) pathway, the conversion of (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate (HMBPP) into the two products isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) (Fig. 1).1–3 IspH and other enzymes in the MEP/DOXP pathway are currently in the spotlight since the pathway is one of two that are found in nature for the synthesis of IPP and DMAPP. These are the building blocks for the large group of molecules called isoprenoids or terpenes, which include biologically essential compounds like vitamins, cholesterol, steroid hormones, carotenoids and quinines.4,5 Mammals use the mevalonate pathway to synthesize the isoprene precursors, while eubacteria and apicomplexan parasites use the MEP/DOXP pathway as the sole pathway for isoprene synthesis.4 Several of these microorganisms are pathogens, causing, for example, malaria, tuberculosis, anthrax, plague, cholera and venereal diseases.6 This makes the MEP/DOXP pathway an attractive target for the development of new anti-infective drugs. Since this pathway is not present in humans these inhibitors should demonstrate very low toxicity. Fosmidomycin, an inhibitor of the enzyme DOXP reductoisomerase in the MEP/DOXP pathway, can be used for the treatment of acute uncomplicated Plasmodium falciparum infections (= malaria) but an overall cure rate of 95% in clinical studies was only achieved when fosmidomycin was tested in combination with clindamycin, showing the need for compounds with higher efficacy.7–13 Research efforts focus on finding fosmidomycin analogs that can work as stand-alone drugs14–22 but also on finding inhibitors for the other enzymes in the MEP/DOXP pathway, which could be used in combination with fosmidomycin. Isoprenoids also have biotechnological applications as drugs, flavors, pigments, perfumes or agrochemicals. Detailed knowledge of the mechanisms of the enzymes and regulation of the pathway could or are already benefitting the biotechnological production of commercially interesting isoprenoids, such as carotenoids,23,24 and Taxol.25 The MEP/DOXP pathway is also present in the plastids of plants and targeting this pathway could result in the development of novel herbicides that are less harmful to humans.26,27
Figure 1. IspH Mechanism I.

Reaction mechanism for the conversion of HMBPP by IspH. See text for details.
The IspH protein is an iron-sulfur-cluster containing enzyme. There is some confusion, however, whether the active-site cluster is a [4Fe-4S] or a [3Fe-4S] cluster. This is mainly due to the crystal structures of IspH proteins from Aquifex aeolicus and Escherichia coli. The A. aeolicus enzyme was crystalized in a relatively ‘open’ form and contained a [3Fe-4S] cluster,28 whereas the E. coli enzyme, which was crystalized under several different conditions, exhibited ‘closed’ structures that either contained a [3Fe-4S] cluster or a [4Fe-4S] cluster.29–31 The structural data places the substrate HMBPP and the iron-sulfur cluster very close to each other. In light of the catalytic reaction, which involves the reductive elimination of the hydroxyl group, the cluster is the most logical direct source of the needed electrons. The redox potential of a 3Fe cluster, however, is too high to fulfill such a role. More importantly the only structure that is obtained with bound HMBPP contains a [4Fe-4S] cluster. The fourth iron in this cluster features a typical Fe-O bond distance of 2.0 Å to the oxygen of the hydroxyl group of HMBPP. This would imply a direct role of the 4Fe cluster in the reaction mechanism, in line with a recent proposal where the binding of HMBPP with its hydroxyl group to the [4Fe-4S] cluster is the starting point of the reaction.32,33 In this hypothetical mechanism (Fig. 1), transfer of the first electron from the cluster to bound HMBPP results in the removal of the hydroxyl group, which leaves as water, and the formation of an allyl anion that could be stabilized by direct or indirect σ/π interaction with the cluster. This type of interaction was proposed based on the spectroscopic properties of a paramagnetic species detected in an IspH mutant (E126Q).32 It was further proposed that the cluster with the bound reaction intermediate receives another electron and the concomitant protonation results in the formation and release of either IPP or DMAPP. The ratio of these products appears to be determined kinetically31 and can be anywhere between 4:1 and 6:1, depending on the origin of the enzyme (A. aeolicus, E. coli, or P. falciparum), or enzyme type (as-isolated or reconstituted).1–3,34 Electrons can be donated by the natural flavodoxin/flavodoxin reductase/NADH system (E. coli)35–37 or ferredoxin (P. falciparum)38, or by artificial donors including photoactivated deazaflavin39 and dithionite.1
The present paper reveals a linear dependency of WT enzyme activity and [4Fe-4S] cluster content, confirming that protein containing this cluster type represents the active form of the enzyme. It further reports that when a one-electron-reduced form of the WT enzyme is incubated with HMBPP a new paramagnetic species is trapped. The properties of this species indicate that it is an intermediate that forms as a stage on the enzyme reaction pathway. The spectroscopic properties of this intermediate formed in WT enzyme, denoted FeSA are different than the species detected in the E126Q mutant, and the behavior of the species formed in the mutant enzyme suggests it may not lie along the reaction pathway. Given the likely significance of the FeSA state, it has been studied by EPR and ENDOR spectroscopies.
A third topic investigated is the effects of site-directed mutagenesis on protein activity and cluster properties. From the crystal structures it can be deduced which amino acids probably play a role in the binding of substrate and in the reaction mechanism. The IspH structure is quite flat and displays a cloverleaf motif built up of α/β domains that surround a central iron-sulfur cluster. In the ‘open’ A. aeolicus structure,28 there is a pronounced cavity, ca. 10 Å × 20 Å, located at the front side. The [3Fe-4S] cluster is found at the bottom of the crevice. Computational studies, with a 4Fe cluster instead of the detected 3Fe cluster, indicated that the diphosphate group of HMBPP is most likely coordinated by His42 and His124 (A. aeolicus numbering) (Fig. 2A).28 The highly conserved Glu126 seemed to be the only candidate to be able to participate in acid-base chemistry. The studies indicated a possible interaction of the hydroxyl group of HMBPP with the unique iron of the active-site cluster. The E. coli structure with bound HMBPP showed a slightly different picture (Fig 2B).30 In this case the enzyme is in the closed conformation and HMBPP is trapped inside. Most of the conserved amino acids do not change position in comparison to the open form, except for His42 (His41 in E. coli) which is shifted about 4 Å.30 In this structure HMBPP has an almost cyclic form and the orientation of the diphosphate group is different from that in the A. aeolicus model by 180°. As mentioned, the hydroxyl group is bound to the fourth iron of the iron-sulfur cluster. The structure also shows a possible relay system where the proton for the last reaction step would come from a water molecule present in most structures that is hydrogen bonded to Glu126, which in turn can pass the proton to Tyr167, which is in a better position to donate a proton to the allyl anion intermediate.
Figure 2. Binding of HMBPP in the active site of IspH.

(A) Modulation of HMBPP in the ‘open’ site of A. aeolicus IspH. (B) HMBPP bound in the crystal structure of E. coli IspH. Note that the numbering is different for the enzymes from both organisms.
Confirmation of the assigned roles of these amino acids in the binding of HMBPP and in the reaction mechanism has to come from site-directed mutagenesis studies. Some of these sites have been probed in this fashion,29,32 but those studies lack detailed information about the kinetic and spectroscopic properties of the produced mutants. Here we present studies of the enzymatic function of IspH mutated at the His42, His124, or Glu126 positions. The characteristics of ‘reaction intermediates’ induced when the one-electron-reduced mutant enzyme is incubated with HMBPP are also discussed.
From the comparison of the catalytic properties of WT and mutant IspH and of the spectroscopic studies of the paramagnetic species formed upon incubation of the one-electron-reduced forms of WT and mutant enzyme with HMBPP a clearer picture arises of the role of the three amino acids on cluster properties, substrate binding, and the reaction mechanism of IspH.
MATERIALS AND METHODS
Materials
Elemental 57Fe (95 % enrichment) was from WEB Research Co. 57FeCl3 was prepared by reacting solid 57Fe in 37% HCl. After all iron reacted the pH was adjusted to 4–5 with NaOH. 2H-labelled HMBPP was donated by the group of Dr. Eric Oldfield at the University of Illinois, Urbana-Champaign, IL, USA. Dithionite was from Fisher Scientific. All gases and gas mixtures were from Airgas.
Anaerobic conditions are required for all experiments. This was achieved by performing all purification steps, sample handling and experiments in a glove box (Coy Laboratory Products, Inc.) with an atmosphere of 95% N2 and 5% H2. All buffers and solutions used in the procedures were degassed by boiling them under a nitrogen atmosphere and subsequent cooling down under vacuum for 2 to 12 hours.
Expression and Purification
The expression plasmids for WT IspH from A. aeolicus and P. falciparum were also provided by the group of Dr. Hassan Jomaa from the Justus-Liebig University at Giessen, Germany. The expression plasmid for WT IspH from E. coli was provided by the group of Dr. Michael Groll from the Technical University Munich, Germany. Plasmids with site-directed mutants of IspH from A. aeolicus were provided by the group of Dr. Eric Oldfield. Wild-type IspH protein (from A. aeolicus, E. coli, and P. falciparum) and mutated IspH proteins from A. aeolicus were overexpressed successfully in E. coli XL-1 blue cells. All IspH proteins contained a His6-tag and were purified by immobilized nickel affinity chromatography.
The cell cultures were started with a single colony from an LB-Amp plate that was transferred into SOC medium containing 100 mg/L ampicillin and 300 μM FeCl3. The cultures were incubated at 37 °C under shaking. For 57Fe-isotope-enriched protein, 57e Cl3 was used. For IspH from A. aeolicus (both wild type and mutants) and P. falciparum, anhydrotetracycline was used as inducer. For IspH from E. coli IPTG was used as an inducer. The cells were harvested by centrifugation at 5,500 rpm for 30 minutes (Sorvall RC-5B Refrigerated Superspeed Centrifuge, Sorvall GS-3 Rotor, Du Pont Instrument). The cell pellets were stored at −80 °C until needed.
The purification and subsequent sample handling steps were performed in the Coy box. Buffer containing 50 mM Tris-HCl, 100 mM NaCl, pH 8.0, was used to re-suspend the cell pellets. The cells were disintegrated by sonication, followed by centrifugation at 35,000 rpm for 30 minutes (Beckman XL-70 Ultracentrifuge, YPE 45 Ti Rotor, Beckman Coulter, Inc.). The A. aeolicus cell extract was subsequently incubated at 65 °C in a water bath for 30 minutes, followed by a second centrifugation step after which the supernatant was loaded on the HisTrap Affinity column (Pharmacia, GE Healthcare). The heat treatment was skipped for the P. falciparum and E. coli enzymes. Protein was eluted with increasing amounts of imidazole in the buffer. Enzyme eluted at an imidazole concentration of ±250 mM. The main fractions were collected and used freshly. Unless indicated, all experiments were performed in 50 mM Tris-HCl, 100 mM NaCl, pH 8.0. The protein samples were judged to be pure (> 95%) based on SDS-PAGE (not shown).
Determination of protein and iron
The enzyme concentration was determined by the Bradford method40 or directly based on the absorbance at 280 nm depending on Tyr and Trp content: ε = 26,930 M−1cm−1 for A. aeolicus (32 kDa), ε = 20,190 M−1cm−1 for E. coli (35 kDa), and ε = 41,260 M−1cm−1 for P. falciparum (37 kDa). The iron determination was carried out with a rapid ferrozine-based colorimetric method.41 Adventitiously bound iron was removed by running the protein samples over a chelex 100 column (Biorad).
Reconstitution
Most of the IspH samples showed substoichiometric amounts of the active-site [4Fe-4S] cluster. Reconstitution of the cluster was achieved by incubation over night with dithiothreitol (at least 5 mM), FeCl3 (5x protein concentration), and Na2S (5x protein concentration) in 50 mM TrisHCl, pH 8.0. The samples were centrifuged in an Eppendorf centrifuge (4500 rpm for 5 min) to remove black iron-containing precipitate. The supernatant was desalted by running it over a PD 10 column. The eluted protein was used directly.
Kinetic Studies
The kinetic studies for WT and mutant IspH were performed at RT with the reaction mixture containing dithionite, methyl viologen, and substrate. The activity of IspH was determined by monitoring the oxidation of dithionite-reduced methyl viologen at 732 nm (ε732 = 2,200 M−1cm−1) or at 603 nm (ε603 = 13,600 M−1cm−1). The concentration of substrate HMBPP was varied from 0 μM to 500 μM.
UV-vis absorption spectra were obtained under anaerobic conditions using an Ocean Optics USB 2000 miniature fiber optic spectrometer inside the glove box or using stoppered cuvettes in a HP 8451A UV-visible Spectrophotometer or an Agilent 8453 UV-visible Spectrophotometer.
Circular Dichroism (CD) Spectroscopy
CD spectroscopy was carried out to check proper folding of the mutant enzymes. As-isolated enzyme was washed with 5 mM phosphate buffer (pH 8.0) to remove both imidazole and the Tris buffer. An aliquot was transferred into a 0.1 mm cuvette which was capped inside the glove box. All CD data were collected on a J-810 Spectropolarimeter (Jasco).
EPR Spectroscopy
The 4Fe cluster in IspH can be reduced by the addition of dithionite. Incubation with both dithionite and the substrate HMBPP does not result in the detection of an intermediate signal under the conditions used. An ‘intermediate signal’ was induced by incubating the enzyme with an excess of dithionite and subsequently removing the excess by running the sample over a desalting PD10 column. The cluster stays reduced after this procedure and this represents the ‘one-electron-reduced’ form of the enzyme. Addition of HMBPP to this form induced the disappearance of the cluster signal and the formation of a new EPR signal, which for convenience is called FeSA.
CW EPR spectra were measured at X-band (9 GHz) frequency on a Bruker EMX spectrometer, fitted with the ER-4119-HS high sensitivity perpendicular-mode cavity. General EPR conditions were: microwave frequency, 9.385 GHz; field modulation frequency, 100 kHz; field modulation amplitude, 0.6 mT. Sample specific conditions are indicated in the figure legends. The Oxford Instrument ESR 900 flow cryostat in combination with the ITC4 temperature controller was used for measurements in the 4 K to 300 K range using a helium flow. Measurements at 77 K were performed by fitting the cavity with a liquid nitrogen finger Dewar.
A copper perchlorate standard (10 mM CuSO4, 2 mM NaClO4, 10 mM HCl) was used for spin quantifications on spectra measured under non-saturating conditions by comparison of the double integral of the signal from the samples with that from the standard. Signal intensities are presented as amount of spin, which is the fraction of the amount of EPR signal detected over the amount of [4Fe-4S] cluster present in the sample.
Pulsed EPR and ENDOR data (35 GHz and 2 K) were obtained with an instrument described earlier.42,43
RESULTS
Relationship between cluster type and enzyme activity
The purified A. aeolicus enzyme shows the typical 420 nm band characteristic for cuboidal iron-sulfur-cluster-containing proteins (Fig. 3, black line). There is a small shoulder at 320 nm that can be assigned to bound single-iron ions (see below). The 420 nm band can be due to either [4Fe-4S]2+ or [3Fe-4S]+ clusters or a mixture of these cluster types. Typically, these clusters can be reduced to the [4Fe-4S]1+ or [3Fe-4S]0 forms by dithionite. This is also the case here, as detected by the bleaching of the 420 nm band upon addition of dithionite (Fig. 3, gray line). The increase in absorbance at around 330 nm is due to dithionite. Similar spectra of as-isolated and reduced proteins were obtained for WT enzyme from E. coli and P. falciparum (Figs. S1 and S2).
Figure 3. UV-visible absorption of as-isolated (—) and dithionite-reduced (—) IspH protein from A. aeolicus.

The peak at 330 nm is due to dithionite.
Comparison of the as-isolated and the dithionite reduced IspH enzymes in EPR spectroscopy show that only for the A. aeolicus enzyme an EPR signal from a [3Fe-4S]1+ cluster was detected (Fig. S3, trace A). That signal, however, represents less than 1% of the total cluster content. The P. falciparum and the E. coli enzymes do not show 3Fe cluster signals in the as-isolated forms (not shown). Instead, Figure 4, shows strong [4Fe-4S]+ signals from the IspH enzyme of the three different organisms. The data thus show that the main cluster type present in all three enzymes is the 4Fe cluster. The 3Fe cluster content in IspH from all three sources can be increased by exposing any of these enzymes to air (not shown). The 3Fe clusters, however, do not accumulate to significant levels but are an intermediate form on the way to complete cluster breakdown.
Figure 4. EPR spectra of the [4Fe-4S]+ cluster in IspH protein from A. aeolicus (A), P. falciparum (B), and E. coli (C).

The signal intensities have been adjusted for easy comparison. EPR conditions: Microwave power, 0.2 mW.
Figure 5, shows the relationship between cluster content and enzymatic activity for IspH enzyme from P. falciparum. This enzyme was chosen since it displayed the highest (reconstituted) cluster content of all three IspH enzymes studied. Different samples were prepared for this plot. The first type was as-isolated enzyme (±15% cluster content), the second type was enzyme that was reconstituted (60 or 86% cluster content), and the third type was enzyme that was exposed to air for a prolonged time (0% cluster content). All types were run over a 5-ml column containing Chelex 100 resin to remove non-bound and adventitiously bound iron. In the case of WT enzyme, the 420 nm band is not affected to a great extent by the Chelex treatment, but the shoulder at 320 nm is (Fig. S4). Therefore this band is assigned to single iron ions coordinated to one or more of the Cys residues present in the active site. The cluster content was determined after the Chelex treatment. Control EPR measurements showed that either no or very low-intensity 3Fe cluster signals were present in the samples, confirming that all clusters that are present have the 4Fe form. The cluster content was calculated based on the iron determination. Figure 5 shows that there is a linear relationship between [4Fe-4S] cluster content (plotted as %) and enzyme activity (plotted as Vmax/[E]).
Figure 5. Relationship between enzyme activity and [4Fe-4S] cluster content.

The plot was prepared with P. falciparum IspH. The cluster content was calculated from the protein iron content.
For completeness is must be noted that the EPR spectrum of the dithionite-reduced A. aeolicus enzyme shows a feature, at around 100–150 mT that can be attributed to a form of the [4Fe-4S]1+ species with a spin S = 3/2, in addition to the signal from a form with S = ½ between 300 and 350 mT 44 (Fig. S3, trace B). Addition of ethylene glycol enhances the S = 1/2 contribution and sharpens its EPR signal to one with g123 = 2.035, 1.916, and 1.841 (Fig. S3, trace C and Fig. 4, trace A).
Detection and characterization of reaction intermediates in WT enzyme
Based on the fact that the [4Fe-4S] cluster can only donate one electron at-a-time and the reaction requires two electrons it could be expected that there would be a short lived one-electron-reduced HMBPP species during the reaction. The incubation of reduced enzyme in the presence of excess HMBPP and excess dithionite resulted in the disappearance of the [4Fe-4S]1+ signal (Fig. S5, traces A and B) indicating that the enzyme was being oxidized by the addition of substrate. No other signals were detected, however, within a time interval of 6 sec to 5 min (not shown). This indicates that under normal steady-state conditions no intermediate is accumulated to any high extent.
Next it was investigated what would happen if only one electron was available for the reaction. “One-electron-reduced” protein can be obtained by incubating IspH with dithionite and removing the excess dithionite anaerobically with a PD10 desalting column. Optical spectroscopy (Fig. S6, dashed trace) shows that after this treatment the cluster stays reduced since the 420 nm is still absent and nearly all dithionite has been removed since only a small band is still present at 330 nm. (This band can be completely removed when the procedure is repeated.) The cluster was still present as [4Fe-4S]+ as shown by EPR spectroscopy (Fig. S5, trace C). The incubation of the one-electron-reduced A. aeolicus protein with HMBPP resulted in the formation of a new paramagnetic species (Fig. 6, trace A/black line and Fig S5, trace D). This species is not an isolated organic radical, indicated by the large spread in g values: g123 = 2.173, 2.013, and 1.997. EPR samples that were made with 57Fe-enriched WT A. aeolicus IspH showed a significant broadening of the EPR signal (Fig. 6, trace A/gray line). The broadening from the nuclear spin of 57Fe, shows that the signal is iron-sulfur based. For the discussion here we will call this signal FeSA. For completeness, the experiments with one-electron-reduced enzyme were performed for the E. coli and P. falciparum enzymes. In both cases a new EPR signal was detected with properties almost identical to that of A. aeolicus enzyme (Fig. 6, traces B and C). When additional dithionite was added to one-electron reduced IspH incubated with HMBPP the FeSA signal disappeared again showing it is probably not a dead-end product.
Figure 6. EPR signals of the FeSA species detected in IspH from A. aeolicus (A), P. falciparum (B), and E. coli (C).

The spectra for the A. aeolicus IspH are from enzyme from cells grown on natural abundance iron containing medium (—) and from cell grown on 57Fe-enriched medium (—). EPR conditions: Microwave power, 2 mW.
It was noted that several of the samples contained an additional EPR signal due to Mn2+ ions. In a study by Rohdich and coworkers it was found that the activity of the IspH enzyme in E. coli cell extracts was significantly higher in the presence of divalent metal ions like Co2+ and Mn2+.45 None of the crystal structures, however, showed the presence of additional metal ions. The specroscopic behavior of all three enzymes in our studies was identical whether the Mn2+ signal was present or not, suggesting that this ion does not play a direct role in catalysis.
For completeness, the temperature dependence of the EPR signals and their amplitudes were measured for both the [4Fe-4S]1+ and the FeSA species. The [4Fe-4S]1+ state shows representative behavior for such a cluster: above 20 K, the signal starts to broaden (Fig. S7, panel A). Conversely, the FeSA signal can be observed up to 75 K without shifts or extensive broadening (Fig. S7, panel A).
Figure 7, shows 35 GHz CW EPR and 31P-Mims ENDOR spectra from FeSA prepared from P. falciparium IspH and HMBPP. The ENDOR spectra show a doublet centered at the 31P Larmor frequency with a small, highly anisotropic hyperfine splitting with a maximum coupling of Amax = 0.17 MHz. Simulations show the maximum dipolar coupling is 2Tmax ≈ Amax, with a negligible isotropic coupling (not shown). The observation of this signal indicates the presence of a substrate 31P-nucleus in proximity to the cluster spin, while the small coupling means that the phosphate group is not directly bound to the cluster. Detailed information about spin coupling within the cluster would permit a reliable estimate of the distance from 31P to the cluster. However, for heuristic purposes, we may assume the cluster to act as a point spin on a single nearby Fe ion of the FeS cluster, 2Tmax ~ 2gβgnβn/r3 = 0.17 MHz, gives a representative Fe-31P distance of r ~ 7 Å.
Figure 7. 35 GHz CW EPR and pulsed ENDOR spectra of the FeSA species.

A: Pulsed 31P ENDOR spectra. Spectra were collected at the positions indicated on the pulse-echo detected EPR spectra shown on the right of the panel. ENDOR spectra are normalized to a fixed intensity for clarity. Sample contained 1.7 mM one-electron-reduced IspH from P. falciparum and 33 mM HMBPP. Sample was incubated for 25 s at RT before freezing. Conditions: Mims pulse sequence: microwave pulse length, 30 ns; RF pulse length, 20 μs; repetition rate, 20 ms; τ = 800 n; microwave frequency, 34.871 GHz; temperature, 2 K.
Figure 8, shows a 2D field-frequency 2H ENDOR spectra comprised of spectra collected across the EPR envelope of FeSA prepared by incubating reduced enzyme with HMBPP labeled with deuterium at the C4 position. Labeled compound was synthesized as a racemate mixture. From the perspective of an ENDOR measurement an intermediate prepared with such a mixture is equivalent to a preparation with 50% of the di-deutero compound, and will be discussed as such. The spectra exhibit ν+/ν− branches centered at the 2H Larmor frequency and separated by the 2H hyperfine coupling; each branch is further broadened and/or split by 2H quadrupole interactions. The hyperfine splitting is considerably anisotropic, with maximum coupling of Amax ~ 1.4 MHz at g2. The presence of two magnetically inequivalent deuterons whose hyperfine couplings may differ and whose hyperfine and quadrupole tensor orientations in general will differ, leads to the broad and poorly defined ν+/ν− branches other than in the vicinity of g3, where the spectrum is most highly resolved. At g3, a set of four lines can be seen which almost certainly correspond to the quadrupole-split doublet of a single deuteron 2H1 with an observed quadrupole coupling 3P = 0.13 MHz, and hyperfine coupling A = 0.54 MHz. Two weaker peaks observed in the g3 spectrum are then assigned to 2H2, with a smaller hyperfine coupling at g3.
Figure 8. Pulsed 2H-ENDOR spectra of the FeSA species.

Spectra were collected at the field positions indicated. Sample contained 1.2 mM one-electron-reduced IspH from P. falciparum and 33 mM 2H-HMBPP. Sample was incubated for 25 s at RT before freezing. Conditions: Mims pulse sequence: microwave pulse length, 30 ns; RF pulse length, 20 μs; repetition rate, 20 ms; τ = 800 n; microwave frequency, 34.871 GHz; temperature, 2 K.
Although ENDOR transitions arising from 2H2 undoubtedly contribute across the entire pattern, the majority of the 2D pattern can be reasonably well described in terms of a dominant contribution from 2H1. One begins by assigning Amax to 2H1, and recognizing that the value of the quadrupole splitting for 3P(2H1) at g3 suggests that this is likely to be associated with the ‘perpendicular’ component (3Pζ) of the axial C-H quadrupole interaction. A series of simulations (not shown) then allows an estimate of the hyperfine tensor components for 2H1, A ~ [0.5, 1.4, 0.5] MHz, with an isotropic contribution of aiso(2H1) ~ 0.8 MHz, corresponding to aiso(1H1) ~ 5 MHz. A heuristic distance calculation, similar to that performed for the 31P above, furnishes an Fe-D distance to C4-deuterium of 3.4 Å, undoubtedly an underestimate because it ignores any local anisotropic contribution. The presence of such a large isotropic interaction requires that FeSA contains HMBPP or a product of its reaction that is bound to the paramagnetic cluster. The size of aiso further suggests that C4 is adjacent to the site coordinated to the metal ion, consistent with binding of the hydroxyl group to the unique Fe of the cluster, as seen in the E. coli crystal structure showing that substrate binds to the oxidized cluster.
To further explore substrate binding to A. aeolicus IspH, circular dichroism spectroscopy was utilized. The oxidized cluster present in the reconstituted protein only shows a broad feature at around 350 nm (Fig. S8, black trace). The spectrum changes significantly in the presence of one equivalent of HMBPP (Fig. S8, gray trace). Additional equivalents of HMBPP caused no further change. This finding is consistent with binding of the HMBPP hydroxyl to the [4Fe-4S]2+ cluster. Analogous results were obtained by monitoring changes in the Mössbauer spectra of the [4Fe-4S]2+ induced by treatment with HMBPP.46
Studies with mutant IspH
Based on computer modeling studies with A. aeolicus enzyme and the E. coli structure with bound HMBPP, residues His42, His124, and Glu126 (A. aeolicus numbering) are predicted to have important roles in the binding of HMBPP and/or the reaction itself. To test this, two mutations were made at each of the three positions, 42, 124, and 126: H42A and H42F; H124A and H124F; E126A and E126Q.
All IspH mutants were expressed at similar levels as seen for WT IspH. Circular dichroism spectra measured for WT and mutant proteins all are very similar, indicating that the proteins are folded properly (Fig. S9). Absorption spectroscopy (not shown) showed that all mutants contained 4Fe clusters. The cluster content was in the range of 10% to 20%. Several of the mutants also displayed a more intense 320 nm band in absorption spectroscopy due to bound iron. Running the samples over a Chelex column greatly reduced the intensity of the 320 nm band (not shown). The cluster content could be increased to 40 to 50% for the different mutants by the cluster reconstitution procedure (Table 1). Activity assays were performed for all six protein variants, as well as the wild-type enzyme (Table 1). The values in Table 1 are similar to those reported in the literature.32 Comparison of the specific activities shows that all mutants, except the H42F mutant, have greatly diminished activity in the range of 5–10% of that of the WT enzyme. The H42F has about 50% WT activity left. Due to the low activity present in most of the mutants it was difficult to obtain the data and small changes in Km and catalytic efficiency should not be over interpreted. A clear difference in catalytic efficiency, however, is found for the H42F and H124F mutants. The Phe residue in the H42F mutant seems to be able to rescue the loss of activity observed in the H42A mutant, but at the same time the Km value increases to 144 μM causing a large decrease in catalytic efficiency. A similar effect is found for the H124F mutant. This indicates that the N-atoms in the His imidazole ring play an important role in the binding of HMBPP.
Table 1.
Comparison of the properties of Wt and mutant A. aeolicus IspH.
| Reconstituted Enzyme | Cluster Content (%) | Specific Activity after Correction for Cluster Content (μmol·min−1·mg−1) | KM (μM) | kcat/KM |
|---|---|---|---|---|
| WT | 42.9 | 1.95 | 6.4 | 0.30 |
| H42A | 29.9 | 0.20 | 1.7 | 0.12 |
| H42F | 45.9 | 1.09 | 144 | 0.008 |
| H124A | 33.2 | 0.14 | 1.2 | 0.12 |
| H124F | 35.6 | 0.20 | 33.6 | 0.006 |
| E126A | 41 | 0.19 | 2.4 | 0.08 |
| E126Q | 29.6 | 0.20 | 0.4 | 0.50 |
EPR spectra collected from as-isolated and cluster-reconstituted mutant proteins are equivalent, except that signal-to-noise ratios were much better in the reconstituted enzyme. Representative samples of each type were used to make the figures. As-isolated mutated proteins show at most only small amounts of [3Fe-4S]1+ cluster; WT and H42A show only radical-type species of unknown origin, not to 3Fe clusters (Fig. S10, panel A). After addition of dithionite to the mutant proteins, the [4Fe-4S]1+ cluster EPR signal was detected in all mutants (Fig. S10, panel B), with an intensity much higher than the intensity of the [3Fe-4S] signals (if any). Hence, it can be concluded that for that mutants the main cluster type present again is the [4Fe-4S] cluster, and the similar g-values show the clusters are no more than minimally perturbed. However, small differences with the WT enzyme can be observed upon addition of 20% EG. This results in different ratios of S = 1/2 and S = 3/2 species for the different mutants. In case of the Glu126 mutants no conversion to the S = 1/2 form was observed. When 33–40% EG was added, however, all mutants showed some S = 1/2 signal in the g = 2 region (not shown).
When one-electron-reduced mutant enzymes were prepared and incubated with HMBPP, several new signals were detected (Fig. 9). Some were similar to the WT FeSA species, but showed differences in temperature behavior. Consider first the H42 mutants. Figure 9, panel A, shows the temperature variation of spectra from the H42A mutant. At 10–20 K an EPR signal due to the [4Fe-4S]1+ can be detected. At higher temperatures the FeSA species can be detected but the signal intensity is very low (0.03 spin). As this mutant has low activity, it can be concluded that not much reaction is taking place. Different results were obtained for the H42F mutant (Fig. 9, panel B). At lower temperatures (from 5 K to 20 K) a mixture of the [4Fe-4S]+ signal and the FeSA signal is present. At 50 K only the FeSA signal is detectable. Whereas the H42A mutant shows low activity and its 4Fe cluster stays reduced upon addition of HMBPP, when HMBPP is added to the H42F mutant a partial oxidation of the 4Fe cluster was detected and the FeSA species was formed. This is in line with the activity data that show that this mutant is still active, although lower activity is observed. When the temperature is increased from 50 K to 70 K, the intensity of the H42F-FeSA signal decreases due to relaxation broadening. This differs from the very wide temperature range over which the WT-FeSA signal can be detected.
Figure 9. Temperature behavior studies of FeSA with reconstituted mutant enzymes from A. aeolicus.

Spectra are presented as an overlay using normalized spectra. All spectra are corrected for differences in gain, temperature and power. A, D and E: Data for enzyme as-isolated. B, C, and F: Data for the reconstituted form of the enzyme.
Next consider the H124 mutants. Neither of the H124A or H124F mutants shows significant activity (Table 1), so it might be expected that they would show the same behavior as the H42 mutants. This is not true, however. Addition of substrate to the H124A or H124F mutants causes the loss of the EPR signal due to the [4Fe-4S]+ cluster (Figure 9, panel C) which indicates that the cluster becomes oxidized upon addition of substrate. This oxidation, however, is accompanied by the formation of only very low levels of the FeSA species (< 0.01 spin). The FeSA species found in these mutants also showed temperature broadening at 70 K, unlike the WT signal.
There could be several reasons that the cluster in the H124A/F mutants seems to get oxidized, with the formation of little or no FeSA. In some samples other radical-like signals were observed with g123 = 2.041, 2.034, and 2.015 (Fig. 10, panel A). This suggests that the binding of HMBPP is affected in such a way that electron transfer results in an alternative radical species that is not stabilized by interaction with the cluster. Several samples also showed broad EPR signals (Fig. 10, panel B). From the g values it can be concluded that the signal is due to a paramagnet with S = 3/2. This signal is different than that detected in the reduced enzyme (see Figs. 4 and S3). The H124 spectra have peaks at g = 4.69 and 3.33 which corresponds to an E/D value of 0.11. Since the latter signal is detected in the presence of HMBPP it could also be due to a cluster-HMBPP complex. The binding, however, appears to be unproductive, not resulting in the formation of the FeSA species.
Figure 10. Additional paramagnetic species detected in some of the mutant IspH enzymes from A. aeolicus.

(A) Possible organic radical observed in one-electron reduced H124A-IspH after addition of HMBPP and incubation for 20 sec. (B) S = 3/2 EPR signal observed in one-electron reduced H124F-IspH after addition of HMBPP and incubation for 20 sec. The signal in the 300–350 mT region is the FeSA EPR signal.
Finally, consider the E126A and Q variants, Figure 9, panels E and F. When the two one-electron-reduced enzymes are treated with substrate, both show a signal, with g123 = 2.120, 2.002, and 1.965. These values are identical to those for the species described by Wang et al.32 The signal remains sharp up to 10 K and 20 K, but broadens at higher temperatures. Studies with excess dithionite (not shown) showed that this species, in opposition to the FeSA-like species detected in the other mutants, is not an intermediate whose signal is lost upon further reaction, but accumulated over time. The E126Q mutant also shows an additional species at around 332 mT. It was not possible to obtain a clean EPR signal for this species by subtraction because the main species in these samples showed additional temperature broadening in each spectrum, which induced additional features in the difference spectra.
DISCUSSION
In this paper we have shown that the active form of the IspH enzyme contains a [4Fe-4S] cluster. The EPR signal of this cluster disappears when the enzyme undergoes catalytic turnover with the substrate HMBPP in the presence of excess dithionite, and no new EPR signals appear. The conversion of HMBPP into IPP or DMAPP requires two electrons. Therefore the one-electron-reduced form of the enzyme was prepared with the aim of trapping a reaction intermediate. This strategy was successful, and incubation of the reduced enzyme with HMBPP resulted in the disappearance of the [4Fe-4S]+ signal and the appearance of a new signal. Labeling studies with 57Fe isotope showed that the signal is associated with the [4Fe-4S] cluster, so it was dubbed FeSA. The EPR properties of the FeSA signal, in particular its temperature response, are very different than those of regular 4Fe clusters of proteins involved in electron transport.47 They are, however, very similar to those of intermediates detected in two other [4Fe-4S]-cluster containing enzymes, (E)-4-hydroxy-3-methylbut-2-enyl diphosphate synthase (IspG/GcpE), the penultimate enzyme in the DOXP/MEP pathway, and ferredoxin:thioredoxin reductase (FTR), an enzyme present in oxygenic photosynthetic organisms.48–52 In those two cases there is evidence that the reaction mechanisms involve the attachment of a substrate (IspG) or an additional amino acid (FTR) to the 4Fe cluster. In the recently proposed IspH mechanism I,32,33 HMBPP first coordinates to the cluster through the oxygen of the hydroxyl group at the C4 position (Fig. 1). Subsequent electron transfer results in the formation of a π (or π/σ) complex. Before discussing the properties of the cluster-bound reaction intermediate in more detail, a discussion of the mutagenesis studies is important because the model in Figure 1 is based on a signal detected in the E126A mutant that differs from the FeSA signal detected in WT IspH.
IspH Mutants
Examination of the crystal structures of IspH protein from A. aeolicus and E. coli mutants led to the construction of pairs of mutants at three sites: H42A and H42F; H124A and H124F, E126A and E126Q. Absorption, CD, and EPR measurements show that the mutant enzymes are properly folded and contain 4Fe clusters. As the cluster content was low, reconstitution of the clusters was performed. Analysis of the enzymatic activity of the mutants thus prepared showed that only the H42F mutant shows relatively high activity (Table 1).
His42 was chosen as a mutation site because it was proposed to participate in the interaction with one of the diphosphate oxygens of HMBPP. Replacement of His42 with Ala caused a 90 % decrease in activity. The incubation of one-electron-reduced H42A with substrate does not result in oxidation of the 4Fe cluster or the formation of the FeSA species in significant amounts. The mutation of His42 into Phe resulted in an enzyme that still showed partial activity and formed the FeSA species. The observed Vmax is about half of that of WT enzyme at comparable cluster occupancies. Since the His42 residue is too far away from the HMBPP hydroxyl group to be directly involved in the reaction and it is not involved in coordination of one of the ‘conserved’ water molecules detected in the crystal structures, the mutation must have an effect on the binding of substrate and/or the conformation of the active site. Having a Phe residue on this position instead of an Ala residue appears to rescue some of the activity. The loss of activity could have been due to a structural effect and by having a Phe instead of a Ala the local conformation is less perturbed by the conservation of the ring structure. At the same time, however, the absence of the N-atoms causes a large increase in Km value and a large decrease in catalytic efficiency. This confirms the role of H42 in binding of HMBPP.
His124 was proposed to participate in the interaction with one of the diphosphate oxygens of HMBPP. Both His124 mutants show the loss of the [4Fe-4S]+ signal upon addition of HMBPP but hardly any formation of the FeSA signal. In some samples an additional paramagnetic species was detected (Fig. 10, trace A). The g values of this species (g123 = 2.041, 2.034, 2.015) are more in line with a carbon-based radical species. At the same time some of the samples show a new S = 3/2 signal upon addition of HMBPP (Fig. 10, trace B). The best way to interpret the data is to assume that an alternative cluster-HMBPP complex is formed or that HMBPP binds to the active site in close proximity to the cluster. In both cases the position of HMBPP is such that electron transfer to HMBPP still takes place but that upon transfer a free-standing radical species is formed instead of the FeSA species. As a result the enzyme becomes mostly inactive. Thus, the His124 residue is indeed important for HMBPP binding, in particular for the correct orientation of HMBPP in the active site. This result also gives a possible lead in finding inhibitors for this enzyme. Any HMBPP analogs that cause radical formation instead of the formation of the FeSA species could potentially knock out the enzyme and block the MEP/DOXP pathway.
Glu126 was proposed to participate in acid/base chemistry at the active site, and to have an important function in the reaction mechanism, either directly or through a hydrogen-bonded water molecule. The low activity in the Glu126 mutants supports this proposal. Upon addition of HMBPP to both E126A and E126Q the [4Fe-4S]+ signal disappears and a new paramagnetic species is formed that is different from FeSA detected in the WT and the other IspH mutants. Its EPR signal is identical to that detected for the E126Q species in previous work by Wang et al.32 A further discussion of this species is presented below.
In summary, the mutagenesis data supports the proposed roles of the three residues, His42, His124, and Glu126, in the binding and positioning of the substrate and in catalysis. In all cases studied here, addition of substrate to the mutant enzyme with reduced cluster caused loss of the cluster signal, demonstrating the interaction between substrate and cluster in the presence of the mutations. This is the first time that these effects are described in detail.29,32 The crystal structures of IspH show that there is an extensive hydrogen bond network between the enzyme, HMBPP and several water molecules that are present in most of the structures.29 In particular the HMBPP diphosphate group in IspH from E. coli is bonded by 7 amino acid residues: H74, H41, S269, S225, N227, S226, and H124. The H41Q and H74Q mutants were still active,29 while the H124N, S225C, and N227Q mutants were not,29 while H41A, H74A and H124A mutants afforded insoluble protein.29 With so many hydrogen bonds available, a single mutation might not be expected to have a severe effect on the binding of HMBPP. Instead, however, some of the mutants show very low levels of activity or the inability to fold properly, indicating the importance of these few residues in maintaining the local protein structure. Previous studies did not allow a conclusion as to whether the loss of activity in properly folded mutants is due to diminished binding of HMBPP or changes to the active-site structure. The present studies of the spectroscopic properties of the mutant enzyme, and in particular the [4Fe-4S] cluster provide a more in-depth understanding of the role of the (mutated) amino acid residues His42, His124, and Glu126.
Mechanistic Implications
The paramagnetic center detected in the catalytically inactive E126Q mutant incubated with dithionite and HMBPP was the first possible intermediate state of IspH to be fully characterized by EPR and ENDOR spectroscopy.32,53 13C-ENDOR studies on the E126Q species incubated with u-13C-HMBPP showed the presence of two pairs of peaks which were attributed to two different carbon atoms with couplings of 1.7 and 0.8 MHz (Table 2). Based on analogy to the bio-organometallic center characterized during reduction of alkynes by nitrogenase,54 a side-on binding of the double bond of HMBPP to the unique iron in IspH was proposed. Although the coupling values are relatively large, larger 13C couplings have been found for substrates where the 13C is not bound to the metal ion, for example in the formaldehyde-inhibited form of the molybdo-enzyme, xanthine oxidase,55 where a 13C coupling of aiso(13C)= 43 MHz was determined for a 13C derived from formaldehyde that is not directly bonded to the metal ion. Thus, a more in depth, joint analysis of the 13C and 1,2H ENDOR would be welcome confirmation. Acetylenic diphosphate compounds, however, were shown to be very strong inhibitors of IspH. But-3-ynyl diphosphate, for example, has an IC50 of 0.45 μM. In keeping with the proposed structure for the HMBPP product, these were proposed to bind side on. Hypothetical IspH mechanism II which is based on this side-on binding model is shown in Figure 11. It was proposed that the initial interaction of HMBPP with the cluster is via the oxygen of the hydroxyl group at the C4 position. Subsequent electron transfer from an outside electron donor results in the formation of a π-complex. Deoxygenation of substrate generates a water molecule and an η1-allyl complex by protonation (via Glu126) to the reduced complex. The second electron transfer yields a η3-allyl complex.
Table 2.
Comparison of the coupling constants of selected atoms for the different proposed reaction intermediates.
| IspH E126Q (‘species X’) | Atom | aiso (MHz) | ref |
|---|---|---|---|
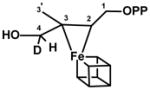
|
D4 | 0.4 | 32 |
| 13C2,3 | 1.7 and 0.8 | 32 | |
| 31P | <0.1 | 32 | |
| IspH WT-FeSA | |||
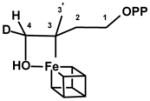
|
H4 (D4) | 5.2 (0.8) | This study |
| 31P | 0.17 | This study | |
| IspH WT-FeSA | |||
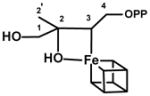
|
H2′(D2′) | 12 (1.8) | 33, 58, 61 |
| 13C2 | 17.7 | 33, 61 | |
| 13C3 | 3.0 | 33, 61 | |
| 17O | n.d. | 33, 61 |
n.d., not determined
Figure 11. IspH Mechanism II.

Hypothetical reaction mechanism II for the conversion of HMBPP by IspH. Adapted from [32]. See text for details.
Here we describe the formation of the FeSA species in one-electron-reduced catalytically active enzyme upon incubation with HMBPP. The ENDOR data presented for FeSA show a weak coupling from 31P of the diphosphate group, with a maximum of 0.17 MHz (Table 2). An approximate point dipole calculation gave an effective distance between the FeS cluster and the 31P of ~7 Å. The lack of any significant isotropic contribution, and the large distance indicate that the phosphate group is in the vicinity of the cluster, but not directly bound to it.
The ENDOR studies using HMBPP labeled with deuterium at the C4 position show the presence of 2H-peaks corresponding to the two deuterium positions of the racemate mixture. Simulations give an excellent estimate for the hyperfine tensor of the more strongly coupled deuteron, assuming that it is H1. The isotropic 2H1 coupling is equivalent to a 1H1 coupling of, aiso = 5.2 MHz and an effective H1-to-cluster distance of ~3.4 Å, indicative that C4 is adjacent to the site of linkage between the FeS cluster and HMBPP, or its reaction product (Table 2). It is not possible to determine the precise hyperfine and quadrupole interaction tensors for the second deuterium. However, all these findings are consistent with an HMBPP bound to the cluster not through phosphate, but via attachment through the hydroxyl group or as a π-complex.
How can we explain the differences between the FeSA species and the species detected in the E126Q mutant, considering both the different g-values and the observed coupling constants (Table 2)? It is possible that both represent a reaction intermediate at different points in the reaction cycle. Given that the Glu126 mutation is catalytically inactive, it is also possible, however, that the Glu126 mutation resulted in the formation of a dead-end product not directly related to the reaction mechanism. More studies are needed to discern between these two options.
Where would we place the FeSA species in IspH mechanism II (Fig. 11)? It might be just the complex formed by binding the hydroxyl of HMBPP to the reduced 4Fe cluster. However, with all the ingredients present for the conversion of HMBPP there is no obvious reason why the reaction would not proceed from there all the way to the η1-complex. This complex, however, is not paramagnetic and therefore cannot be the FeSA species. This puzzle, along with the finding that the E126Q mutant is not active and that the putative side-on bound state might not represent a catalytic intermediate, leads us to next explore other mechanistic models.
Alternative Models
As mentioned earlier there are two other enzymes that show EPR signals from catalytic intermediates that are very similar to the FeSA signal observed in WT IspH. The first enzyme is FTR. This enzyme functions as a 1e−/2e− switch, using single electrons donated by ferredoxin for a two electron reduction of a disulfide bond present on the substrate enzyme thioredoxin. The active site of FTR contains the unique combination of a [4Fe-4S] cluster in close proximity to an active-site disulfide.56 Transfer of the first electron from the [4Fe-4S]+ cluster to the disulfide would theoretically cause the formation of a thiolate and a thiyl radical. The formation of the radical species, however, is prevented by forming a bond between this sulfur atom and an iron atom of the 4Fe cluster.48,49,57 The unique iron site ends up being coordinated by two cysteinates. This species is best described as a [4Fe-4S]3+ cluster. The other enzyme is IspG. This enzyme is the penultimate enzyme in the MEP/DOXP pathway and converts 2-C-methyl-D-erythritol-2,4-cyclodiphosphate (MEcPP) into HMBPP (Fig. 12). MEcPP is a cyclic compound and the reaction involves the opening of the ring and removal of the C3 hydroxyl group consuming the total of two electrons. The enzyme contains a single [4Fe-4S] cluster in its active site.
Figure 12. IspG Mechanism I.

Hypothetical reaction mechanism for the conversion of MEcPP by IspG. See text for details.
In a recent study on IspG 58 we showed that several paramagnetic species could be observed in steady-state and pre-steady-state kinetic experiments. One of these signals was from a transient species that was dubbed FeSA, which displayed a rhombic EPR signal with g123 = 2.087, 2.019, and 2.000. It is because the g-values for FeSA of IspH reported here are so similar to those for IspG, g123 = 2.173, 2.013, and 1.997, that the signal induced in one-electron-reduced IspH incubated with HMBPP was also dubbed FeSA. 31P-ENDOR measurements on the IspG-FeSA species, like IspH-FeSA, showed a weak 31P coupling which is in line with binding of the substrate to the enzyme in close proximity of the active-site cluster.58 Based on EPR/ENDOR measurements it was proposed that the substrate binds directly to the [4Fe-4S] cluster during the reaction. Parallel to this, labeling studies by Wang et al.33 with the alternative substrate [2,3-17O]-HMBPP epoxide showed that there is a Fe-O bond, in line with binding models for MEcPP via one of its hydroxyl groups or the formation of a more tightly bound ferraoxetane complex (Table 2). Thus we inferred that the iron-sulfur cluster is directly involved in a reductive elimination of a hydroxyl group in IspG, which thus suggests the same for IspH.
IspH and IspG catalyze a very similar reaction, the reductive elimination of a hydroxyl group. In both cases a very similar paramagnetic species, thus denoted FeSA, is formed during the reaction mechanism. We propose that in both enzymes the FeSA species represent a reaction intermediate that is bound to the 4Fe cluster via a hydroxyl group. Based on the similarities with the EPR signal detected in FTR we also propose that the FeSA signals are due to an [4Fe-4S]3+ species. This is supported by the fact that the FeSA signals have an average g value > ge, which is strongly indicative of a [4Fe-4S]3+ species.59,60
Key to proposing any reaction mechanism for IspH is that a completely radical based mechanism should be avoided, because the carbon-oxygen bond connecting the diphosphate group to the rest of the molecule is more labile than the one connecting the hydroxyl group and would therefore have a higher chance of being broken in such a mechanism. IspH mechanism II (Fig. 11) does avoid such a situation, but binding of the hydroxyl group to the cluster would make it a much better leaving group, favoring removal of the cluster-bound hydroxyl group. The IspG mechanism I which we recently proposed is shown in Figure 12. In this case the substrate MEcPP starts by binding to the cluster via the hydroxyl group that is ultimately eliminated. Protonation of MEcPP results in ring opening and the formation of a carbocation. Internal electron transfer from the cluster to the substrate results in the formation of a carbon radical. There are several possibilities of how the cluster can stabilize this radical species. One option would be the transfer of an additional electron, making the cluster formally 3+ (reaction I). However, this would create a carbanion species that could be very reactive. The ferraoxetane structure as proposed by Wang et al.61 would provide a way to stabilize the carbanion (and even the radical species in the previous step) by forming an additional bond to the unique iron (via reaction III). At this point it is not clear, however, if there is a direct bond between the C3 carbon and the unique iron. As an alternative (reaction II) a π/η-type complex can be proposed, perhaps accompanied by interaction with the hydroxyl oxygen. In either case, transfer of the second electron from the outside electron donor to the active-site cluster results in the release of the hydroxyl group and double bond formation. The hydroxyl group can stay bound to the cluster and comes off later since it is only weakly bound as shown for example for aconitase.62
A similar approach is followed in the IspH mechanism III (Fig. 13). HMBPP can bind to either the [4Fe-4S]2+ or [4Fe-4S]+ forms of the enzyme. Transfer of two electrons from the cluster to the bound substrate results in the formation of a carbanion (reaction I). This could be stabilized by an additional interaction with the unique Fe of the cluster. In the most extreme case there could be a direct bond between the iron and the C3-carbon (reaction II), similar to the ferraoxetane species proposed for IspG. Rereduction of the cluster to the 2+ state by an outside electron donor causes the cleavage of the C-O(H) bond and the formation of either IPP or DMAPP.
Figure 13. IspH Mechanism III.

Hypothetical reaction mechanism III for the conversion of HMBPP by IspH. See text for details.
IspH mechanism III (Fig. 13) is a working model that includes the following assumptions: a direct Fe-O bond; a 3+ oxidation state for the cluster. These will be tested through a series of labeling studies with 13C, 17O, and 57Fe, using a combination of ENDOR and Mössbauer spectroscopy. IspH mechanism III also does not explain why (E)-3-(fluoromethyl)-2-butenyl diphosphate (C4-F instead of C4-OH) is a substrate for this enzyme, as this compound has no hydroxyl group.39 It is well known that enzymes can employ different reaction mechanisms for substrates with different properties and the possibility that this compound reacts via a different reaction mechanism will be tested by examining the EPR signals induced by incubation of one-electron reduced WT or E126A/Q IspH with (E)-3-(fluoromethyl)-2-butenyl diphosphate.
Summary
This paper presents evidence for the important role of a [4Fe-4S] cluster in the reaction mechanism of the IspH enzyme. The cluster is involved in direct binding of HMBPP which stays bound during the subsequent catalysis. A paramagnetic reaction intermediate, denoted FeSA, is generated upon the incubation of one-electron-reduced enzyme with HMBPP, with EPR and ENDOR studies revealing that it contains an HMBPP-derived intermediate bound to the cluster. Similarities of this species with other paramagnetic intermediates associated with [4Fe-4S]-containing enzymes, in particular a very similar species in the previous enzyme in the MEP/DOXP pathway, IspG, indicates an association via the C4-(hydroxyl) oxygen. Studies with mutant enzyme confirmed the roles of His42 and His124 in binding of HMBPP and the role of Glu126 in the reaction mechanism. The role of His124 is particularly interesting since it appears to play a role in keeping the substrate HMBPP in the correct orientation so that the stable HMBPP-cluster complex can be formed. Mutating this residue resulted in the formation of a HMBPP-based radical instead.
Supplementary Material
Abbreviations
- HMBPP
(E)-4-hydroxy-3-methyl-but-2-enyl diphosphate
- IPP
isopentenyl diphosphate
- DMAPP
dimethylallyl diphosphate
- FTR
ferredoxin:thioredoxin reductase
- MEcPP
2-C-methyl-D-erythritol-2,4-cyclodiphosphate
Footnotes
This work was supported by the National Science Foundation, 0848196 to ECD and by the National Institutes of Health, HL 13531, to BMH.
Additional EPR, electron absorption and CD spectra for the three IspH proteins and the A. aeolicus mutants can be found in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Altincicek B, Duin EC, Reichenberg A, Hedderich R, Kollas AK, Hintz M, Wagner S, Wiesner J, Beck E, Jomaa H. LytB protein catalyzes the terminal step of the 2-C-metyhl-D-erythritol-4-phosphate pathway of isoprenoid biosynthesis. FEBS Lett. 2002;532:437–440. doi: 10.1016/s0014-5793(02)03726-2. [DOI] [PubMed] [Google Scholar]
- 2.Rohdich F, Zepeck F, Adam P, Hecht S, Kaiser J, Laupitz R, Gräwert T, Amslinger S, Eisenreich W, Bacher A, Arigoni D. The deoxyxylulose phosphate pathway of isoprenoid biosynthesis: Studies on the mechanisms of the reactions catalyzed by IspG and IspH protein. Proc Natl Acad Sci USA. 2003;100:1586–1591. doi: 10.1073/pnas.0337742100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolff M, Seemann M, Bui BTS, Frapart Y, Tritsch D, Estrabot AG, Rodríguez-Concepción M, Boronat A, Marquet A, Rohmer M. Isoprenoid biosynthesis via the methylerythritol phosphate pathway: the (E)-4-hydroxy-3-methylbut-2-enyl diphosphate reductase (LytB/IspH) from Escherichia coli is a [4Fe-4S] protein. FEBS Lett. 2003;541:115–120. doi: 10.1016/s0014-5793(03)00317-x. [DOI] [PubMed] [Google Scholar]
- 4.Rohmer M. The discovery of a mevalonate-independent pathway for isoprenoid biosynthesis in bacteria, algae and higher plants. Nat Prod Rep. 1999;16:565–574. doi: 10.1039/a709175c. [DOI] [PubMed] [Google Scholar]
- 5.Eisenreich W, Bacher A, Arigoni D, Rohdich F. Biosynthesis of isoprenoids via the non-mevalonate pathway. Cell Mol Life Sci. 2004;61:1401–1426. doi: 10.1007/s00018-004-3381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rohdich F, Bacher A, Eisenreich W. Isoprenoid biosynthetic pathways as anti-infective drug targets. Biochem Soc Trans. 2005;33:785–791. doi: 10.1042/BST0330785. [DOI] [PubMed] [Google Scholar]
- 7.Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Turbachova I, Eberl M, Zeidler J, Lichtenthaler HK, Soldati D, Beck E. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science. 1999;285:1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- 8.Kuzuyama T, Shimizu T, Takahashi S, Seto H. Direct formation of 2-C-methyl-D-erythritol 4-phosphate from 1-deoxy-D-xylulose 5-phosphate by 1-deoxy-D-xylulose 5-phosphate reductoisomerase, a new enzyme in the non-mevalonate pathway to isopentenyl diphosphate. Tetrahedron Lett. 1998;39:7913–7916. [Google Scholar]
- 9.Zeidler J, Schwender J, Muller C, Wiesner J, Weidemeyer C, Beck E, Jomaa H, Lichtenthaler HK. Inhibition of the non-mevalonate 1-deoxy-D-xylulose-5-phosphate pathway of plant isoprenoid biosynthesis by fosmidomycin. Z Naturforsch. 1998;53c:980–986. [Google Scholar]
- 10.Koppisch AT, Fox DT, Blagg BSJ, Poulter DC. E-coli MEP synthase: Steady-state kinetic analysis and substrate binding. Biochemistry. 2002;41:236–243. doi: 10.1021/bi0118207. [DOI] [PubMed] [Google Scholar]
- 11.Missinou MA, Borrmann S, Schindler A, Issifou S, Adegnika AA, Matsiegui PB, Binder R, Lell B, Wiesner J, Baranek T, Jomaa H, Kremsner PG. Fosmidomycin for malaria. The Lancet. 2002;360:1941–1942. doi: 10.1016/S0140-6736(02)11860-5. [DOI] [PubMed] [Google Scholar]
- 12.Lell B, Ruangweerayut R, Wiesner J, Missinou MA, Schindler A, Baranek T, Hintz M, Hutchinson D, Jomaa H, Kremsner PG. Fosmidomycin, a novel chemotherapeutic agent for malaria. Antimicrob Agents Chemother. 2003;47:735–738. doi: 10.1128/AAC.47.2.735-738.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borrmann S, Issifou S, Esser G, Adegnika AA, Ramharter M, Matsiegui PB, Oyakhirome S, Mawili-Mboumba DP, Missinou MA, Kun JFJ, Jomaa H, Kremsner PG. Fosmidomycin-clindamycin for the treatment of Plasmodium falciparum malaria. Journal of Infectious Diseases. 2004;190:1534–1540. doi: 10.1086/424603. [DOI] [PubMed] [Google Scholar]
- 14.Giessmann D, Heidler P, Haemers T, Van Calenbergh S, Reichenberg A, Jomaa H, Weidemeyerd C, Sanderbrand S, Wiesner J, Link A. Towards new antimalarial drugs: Synthesis of non-hydrolyzable phosphate mimics as feed for a predictive QSAR study on 1-deoxy-D-xylulose-5-phosphate reductoisomerase inhibitors. Chem Biodivers. 2008;5:643–656. doi: 10.1002/cbdv.200890060. [DOI] [PubMed] [Google Scholar]
- 15.Haemers T, Wiesner J, Giessmann D, Verbrugghen T, Hillaert U, Ortmann R, Jomaa H, Link A, Schlitzer M, Van Calenbergh S. Synthesis of beta- and gamma-oxa isosteres of fosmidomycin and FR900098 as antimalarial candidates. Bioorg Med Chem. 2008;16:3361–3371. doi: 10.1016/j.bmc.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 16.Wiesner J, Ortmann R, Jomaa H, Schlitzer M. Double ester prodrugs of FR900098 display enhanced In-Vitro antimalarial activity. Archiv der Pharmazie. 2007;340:667–669. doi: 10.1002/ardp.200700069. [DOI] [PubMed] [Google Scholar]
- 17.Ortmann R, Wiesner J, Silber K, Klebe G, Jomaa H, Schlitzer M. Novel deoxyxylulosephosphate-reductoisomerase inhibitors: Fosmidomycin derivatives with spacious acyl residues. Archiv der Pharmazie. 2007;340:483–490. doi: 10.1002/ardp.200700149. [DOI] [PubMed] [Google Scholar]
- 18.Devreux V, Wiesner J, Jomaa H, Van der Eycken J, Van Calenbergh S. Synthesis and evaluation of alpha, beta-unsaturated alpha-aryl-substituted fosmidomycin analogues as DXR inhibitors. Bioorg Med Chem Lett. 2007;17:4920–4923. doi: 10.1016/j.bmcl.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 19.Devreux V, Wiesner J, Jomaa H, Rozenski J, Van der Eycken J, Van Calenbergh S. Divergent strategy for the synthesis of alpha-aryl-substituted fosmidomycin analogues. J Org Chem. 2007;72:3783–3789. doi: 10.1021/jo0700981. [DOI] [PubMed] [Google Scholar]
- 20.Haemers T, Wiesner J, Busson R, Jomaa H, Van Calenbergh S. Synthesis of alpha-aryl-substituted and conformationally restricted fosmidomycin analogues as promising antimalarials. Eur J Org Chem. 2006:3856–3863. [Google Scholar]
- 21.Devreux V, Wiesner J, Goeman JL, Van der Eycken J, Jomaa H, Van Calenbergh S. Synthesis and biological evaluation of cyclopropyl analogues of fosmidomycin as potent Plasmodium falciparum growth inhibitors. Journal of Medicinal Chemistry. 2006;49:2656–2660. doi: 10.1021/jm051177c. [DOI] [PubMed] [Google Scholar]
- 22.Haemers T, Wiesner J, Van Poecke S, Goeman J, Henschker D, Beck E, Jomaa H, Van Calenbergh S. Synthesis of alpha-substituted fosmidomycin analogues as highly potent Plasmodium falciparum growth inhibitors. Bioorg Med Chem Lett. 2006;16:1888–1891. doi: 10.1016/j.bmcl.2005.12.082. [DOI] [PubMed] [Google Scholar]
- 23.Matthews PD, Wurtzel ET. Metabolic engineering of carotenoid accumulation in E. coli by modulation of the isoprenoid precursor pool with expression of deoxyxylulose phosphate synthase. Appl Microbiol Biotechnol. 2000;53:396–400. doi: 10.1007/s002530051632. [DOI] [PubMed] [Google Scholar]
- 24.Rodríguez-Concepción M, Querol J, Lois LM, Imperial S, Boronat A. Bioinformatic and molecular analysis of hydroxymethylbutenyl diphosphate synthase (GCPE) gene expression during carotenoid accumulation in ripening tomato fruit. Planta. 2003;217:476–482. doi: 10.1007/s00425-003-1008-5. [DOI] [PubMed] [Google Scholar]
- 25.Ajikumar PK, Xiao WH, Tyo KEJ, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G. Isoprenoid Pathway Optimization for Taxol Precursor Overproduction in Escherichia coli. Science. 2010;330:70–74. doi: 10.1126/science.1191652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lichtenthaler HK, Zeidler J, Schwender J, Muller C. The non-mevalonate isoprenoid biosynthesis of plants as a test system for new herbicides and drugs against pathogenic bacteria and the malaria parasite. Z Naturforsch, C: Biosci. 2000;55:305–313. doi: 10.1515/znc-2000-5-601. [DOI] [PubMed] [Google Scholar]
- 27.Fellermeier M, Kis K, Sagner S, Maier U, Bacher A, Zenk MH. Cell-free conversion of 1-deoxy-D-xylulose 5-phosphate and 2-C-methyl-D-erythritol 4-phosphate into beta-carotene in higher plants and its inhibition by fosmidomycin. Tetrahedron Lett. 1999;40:2743–2746. [Google Scholar]
- 28.Rekittke I, Wiesner J, Rohrich R, Demmer U, Warkentin E, Xu WY, Troschke K, Hintz M, No JH, Duin EC, Oldfield E, Jomaa H, Ermler U. Structure of (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate reductase, the terminal enzyme of the non-mevalonate pathway. J Am Chem Soc. 2008;130:17206–17207. doi: 10.1021/ja806668q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gräwert T, Rohdich F, Span I, Bacher A, Eisenreich W, Eppinger J, Groll M. Structure of active IspH enzyme from Escherichia coli provides mechanistic insight into substrate reduction. Angew Chem Int Ed. 2009;48:5756–5759. doi: 10.1002/anie.200900548. [DOI] [PubMed] [Google Scholar]
- 30.Gräwert T, Span I, Eisenreich W, Rohdich F, Eppinger J, Bacher A, Groll M. Probing the reaction mechanism of IspH protein by X-ray structure analysis. Proc Natl Acad Sci USA. 2010;107:1077–1081. doi: 10.1073/pnas.0913045107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gräwert T, Span I, Bacher A, Groll M. Reductive Dehydroxylation of Allyl Alcohols by IspH Protein. Angew Chem Int Ed. 2010;49:8802–8809. doi: 10.1002/anie.201000833. [DOI] [PubMed] [Google Scholar]
- 32.Wang WX, Wang K, Liu YL, No JH, Li JK, Nilges MJ, Oldfield E. Bioorganometallic mechanism of action, and inhibition, of IspH. Proc Natl Acad Sci USA. 2010;107:4522–4527. doi: 10.1073/pnas.0911087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang WX, Li JK, Wang K, Huang CC, Zhang Y, Oldfield E. Organometallic mechanism of action and inhibition of the 4Fe-4S isoprenoid biosynthesis protein GcpE (IspG) Proc Natl Acad Sci USA. 2010;107:11189–11193. doi: 10.1073/pnas.1000264107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gräwert T, Kaiser J, Zepeck F, Laupitz R, Hecht S, Amslinger S, Schramek N, Schleicher E, Weber S, Haslbeck M, Buchner J, Rieder C, Arigoni D, Bacher A, Eisenreich W, Rohdich F. IspH protein of Escherichia coli: Studies on iron-sulfur cluster implementation and catalysis. J Am Chem Soc. 2004;126:12847–12855. doi: 10.1021/ja0471727. [DOI] [PubMed] [Google Scholar]
- 35.Zepeck F, Gräwert T, Kaiser J, Schramek N, Eisenreich W, Bacher A, Rohdich F. Biosynthesis of isoprenoids. Purification and properties of IspG protein from Escherichia coli. J Org Chem. 2005;70:9168–9174. doi: 10.1021/jo0510787. [DOI] [PubMed] [Google Scholar]
- 36.Puan KJ, Wang H, Dairi T, Kuzuyama T, Morita CT. fldA is an essential gene required in the 2-C-methyl-D-erythritol 4-phosphate pathway for isoprenoid biosynthesis. FEBS Lett. 2005;579:3802–3806. doi: 10.1016/j.febslet.2005.05.047. [DOI] [PubMed] [Google Scholar]
- 37.Wolff M, Seemann M, Grosdemange-Billiard C, Tritsch D, Campos N, Rodríguez-Concepción M, Boronat A, Rohmer M. Isoprenoid biosynthesis via the methylerythritol phosphate pathway. (E)-4-Hydroxy-3-methylbut-2-enyl diphosphate: chemical synthesis and formation from methylerythritol cyclodiphosphate by a cell-free system from Escherichia coli. Tetrahedron. 2002;43:2555–2559. [Google Scholar]
- 38.Röhrich RC, Englert N, Troschke K, Reichenberg A, Hintz M, Seeber F, Balconi E, Aliverti A, Zanetti G, Köhler U, Pfeiffer M, Beck E, Jomaa H, Wiesner J. Reconstitution of an apicoplast-localised electron transfer pathway involved in the isoprenoid biosynthesis of Plasmodium falciparum. FEBS Lett. 2005;579:6433–6438. doi: 10.1016/j.febslet.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 39.Xiao YL, Zhao ZK, Liu PH. Mechanistic studies of IspH in the deoxyxylulose phosphate pathway: Heterolytic C-O bond cleavage at C-4 position. J Am Chem Soc. 2008;130:2164–2165. doi: 10.1021/ja710245d. [DOI] [PubMed] [Google Scholar]
- 40.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 41.Fish WW. Rapid colorimetric micromethod for the quantitation of complexed iron in biological sample. Meth Enzymol. 1988;158:357–364. doi: 10.1016/0076-6879(88)58067-9. [DOI] [PubMed] [Google Scholar]
- 42.Davoust CE, Doan PE, Hoffman BM. J Mag Res A. 1996;119:38–44. [Google Scholar]
- 43.Zipse H, Artin E, Wnuk S, Lohman GJS, Martino D, Griffin RG, Kacprzak S, Kaupp M, Hoffman B, Bennati M, Stubbe J, Lees N. Structure of the nucleotide radical formed during reaction of CDP/TTP with the E441Q-α2β2 of E. coli ribonucleotide reductase. J Am Chem Soc. 2009;131:200–211. doi: 10.1021/ja806693s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hagen WR. EPR spectroscopy of iron-sulfur proteins. Adv Inorg Chem. 1992;38:165–222. [Google Scholar]
- 45.Adam P, Hecht S, Eisenreich W, Kaiser J, Gräwert T, Arigoni D, Bacher A, Rohdich F. Biosynthesis of terpenes: Studies on 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase. Proc Natl Acad Sci USA. 2005;99:12108–12113. doi: 10.1073/pnas.182412599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seemann M, Janthawornpong K, Schweizer J, Bottger LH, Janoschka A, Ahrens-Botzong A, Tambou MN, Rotthaus O, Trautwein AX, Rohmer M, Schunemann V. Isoprenoid biosynthesis via the MEP pathway: In vivo Mössbauer spectroscopy identifies a [4Fe-4S]2+ center with unusual coordination sphere in the LytB protein. J Am Chem Soc. 2009;131:13184–13185. doi: 10.1021/ja9012408. [DOI] [PubMed] [Google Scholar]
- 47.Cammack R, Patil DS, Fernandez VM. Electron-spin-resonance/electron-paramagnetic-resonance spectroscopy of iron-sulphur enzymes. Biochem Soc Trans. 1985;13:572–578. doi: 10.1042/bst0130572. [DOI] [PubMed] [Google Scholar]
- 48.Staples CR, Ameyibor E, Fu W, Gardet-Salvi L, Stritt-Etter AL, Schürmann P, Knaff DB, Johnson MK. The function and properties of the iron-sulfur center in spinach ferredoxin:thioredoxin reductase: A new biological role for iron-sulfur clusters. Biochemistry. 1996;35:11425–11434. doi: 10.1021/bi961007p. [DOI] [PubMed] [Google Scholar]
- 49.Staples CR, Gaymard E, Stritt-Etter AL, Telser J, Hoffman BM, Schürmann P, Knaff DB, Johnson MK. Role of the [Fe4S4] cluster in mediating disulfide reduction in spinach ferredoxin:thioredoxin reductase. Biochemistry. 1998;37:4612–4620. doi: 10.1021/bi9729763. [DOI] [PubMed] [Google Scholar]
- 50.Dai S, Schwendtmayer C, Schürmann P, Ramaswamy S, Eklund H. Redox signaling in chloroplasts: Cleavage of disulfides by an iron-sulfur cluster. Science. 2000;287:655–658. doi: 10.1126/science.287.5453.655. [DOI] [PubMed] [Google Scholar]
- 51.Jameson GNL, Walters EM, Manieri W, Schürmann P, Johnson MK, Huynh BH. Spectroscopic evidence for site specific chemistry at a unique iron site of the [4Fe-4S] cluster in ferredoxin:thioredoxin reductase. J Am Chem Soc. 2003;125:1146–1147. doi: 10.1021/ja029338e. [DOI] [PubMed] [Google Scholar]
- 52.Walters EM, Garcia-Serres R, Jameson GNL, Glauser DA, Bourquin F, Manieri W, Schürmann P, Johnson MK, Huynh BH. Spectroscopic characterization of site-specific [Fe4S4] cluster chemistry in ferredoxin:thioredoxin reductase: Implications for the catalytic mechanism. J Am Chem Soc. 2005;127:9612–9624. doi: 10.1021/ja051909q. [DOI] [PubMed] [Google Scholar]
- 53.Wang K, Wang WX, No JH, Zhang YH, Zhang Y, Oldfield E. Inhibition of the Fe4S4-Cluster-Containing Protein IspH (LytB): Electron Paramagnetic Resonance, Metallacycles, and Mechanisms. J Am Chem Soc. 2010;132:6719–6727. doi: 10.1021/ja909664j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee HI, Igarashi RY, Laryukhin M, Doan PE, Dos Santos PC, Dean DR, Seefeldt LC, Hoffman BM. An organometallic intermediate during alkyne reduction by nitrogenase. J Am Chem Soc. 2004;126:9563–9569. doi: 10.1021/ja048714n. [DOI] [PubMed] [Google Scholar]
- 55.Shanmugam M, Zhang B, McNaughton RL, Kinney RA, Hille R, Hoffman BM. The Structure of Formaldehyde-Inhibited Xanthine Oxidase Determined by 35 GHz 2H ENDOR Spectroscopy. J Am Chem Soc. 2010;132:14015–14017. doi: 10.1021/ja106432h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dai S, Saarinen M, Ramaswamy S, Meyer Y, Jacquot JP, Eklund H. Crystal structure of Arabidopsis thaliana NADPH dependent thioredoxin reductase at 2.5 Å resolution. J Mol Biol. 1996;264:1044–1057. doi: 10.1006/jmbi.1996.0695. [DOI] [PubMed] [Google Scholar]
- 57.Walters EM, Garcia-Serres R, Jameson GNL, Glauser DA, Bourquin F, Manieri W, Schürmann P, Johnson MK, Huynh BH. Spectroscopic characterization of site-specific [Fe4S4] cluster chemistry in ferredoxin:thioredoxin reductase: Implications for the catalytic mechanism. J Am Chem Soc. 2005;127:9612–9624. doi: 10.1021/ja051909q. [DOI] [PubMed] [Google Scholar]
- 58.Xu W, Lees NS, Adedeji D, Wiesner J, Jomaa H, Hoffman BM, Duin EC. Paramagnetic Intermediates of (E)-4-Hydroxy-3-methylbut-2-enyl Diphosphate Synthase (GcpE/IspG) under Steady-State and Pre-Steady-State Conditions. J Am Chem Soc. 2010;132:14509–14520. doi: 10.1021/ja101764w. [DOI] [PubMed] [Google Scholar]
- 59.Mouesca JM, Lamotte B. Iron-sulfur clusters and their electronic and magnetic properties. Coordination Chemistry Reviews. 1998;178:1573–1614. [Google Scholar]
- 60.Lepape L, Lamotte B, Mouesca JM, Rius G. Paramagnetic states of four iron four sulfur clusters. 1 EPR single-crystal study of 3+ and 1+ clusters of an asymmetrical model compound and general model for the interpretation of the g-tensors of these two redox states. J Am Chem Soc. 1997;119:9757–9770. [Google Scholar]
- 61.Wang W, Wang K, Li J, Nellutla S, Smirnova TI, Oldfield E. An ENDOR and HYSCORE Investigation of a Reaction Intermediate in IspG (GcpE) Catalysis. J Am Chem Soc. 2011;133:8400–8403. doi: 10.1021/ja200763a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beinert H, Kennedy MC, Stout CD. Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chem Rev. 1996;97:2315–2334. doi: 10.1021/cr950040z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.