

Abstract

A standard HPLC was adapted to polymer supported oligosaccharide synthesis. Solution-based reagents are delivered using a software-controlled solvent delivery system (multi-headed pump). The reaction progress and completion can be monitored in real time using a standard variable wavelength UV detector. All steps of oligosaccharide assembly including loading, glycosylation, deprotection, and cleavage can be performed using this experimental set-up.
Solid-phase synthesis1,2 has been widely utilized in the routine preparation of oligopeptides3 and oligonucleotides.4 The use of polymer supports in oligosaccharide synthesis has also been reported.5–7 The use of these techniques helps to expedite the synthesis of oligosaccharides and glycoconjugates8–16 by minimizing the necessity for purifying reaction intermediates and simplifying the removal of excess reagents that is usually achieved by filtration. To expedite solid-phase oligosaccharide synthesis, Seeberger and co-workers developed an automated approach.17–19 The automation was accomplished by using a peptide synthesizer that had been modified in order to be compatible with oligosaccharide assembly using either trichloroacetimidate or phosphate glycosyl donors at low temperature. Although being a relatively new technique, this approach has already been applied to the synthesis of a variety of oligosaccharide sequences.20–23 Other recent enhancements of the supported synthesis of oligosaccharides include, but are not limited to, approaches employing fluorous tag,24,25 ionic liquid,26–28 nanoparticle,29 and nanoporous gold30 supports.
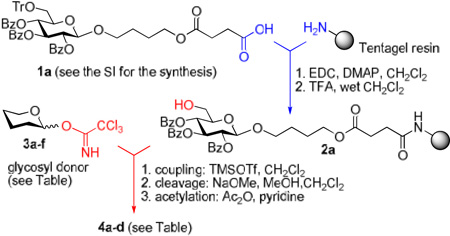
As a part of the on-going research effort in our laboratories presented herein is the development of a new experimental set-up based on a conventional HPLC for automated oligosaccharide synthesis using insoluble polymer supports. To obtain clear evidence of the advantages of the new technology in comparison to the state-of-the-art polymer-supported synthesis, we deliberately chose the most common approaches for all aspects of our synthesis.5,7 Although a vast variety of approaches have been introduced, all of the following are arguably the most commonly seen in the literature: a solution-based trichloroacemidate donor, a Tentagel resin-bound glycosyl acceptor attached via the anomeric center, TMSOTf as the activator/promoter, CH2Cl2 as the reaction solvent, and Fmoc as the temporary hydroxyl protecting group for selective deprotection.11,16,31
In accordance with the traditional manual polymer-bound synthesis, glycosyl acceptor bound to the polymer beads is shaken with an excess of the solution phase glycosyl donor and promoter (Figure 1A). The periodic monitoring of the solution phase by TLC can provide information on whether the glycosyl donor is still remaining in the solution or has been consumed/hydrolyzed. The completeness of the coupling can be determined experimentally by performing test reactions, such as the Kaiser test,32 or by cleaving the product off the polymer support followed by characterization. Routinely, the coupling step is repeated two or three times using fresh reagents to ensure that all glycosyl acceptor is consumed. Alternatively (or as necessary), the remaining hydroxyls can be capped to ensure that they will not interfere with subsequent steps during the oligosaccharide elongation.
Figure 1.

Comparison of the traditional manual polymer-supported synthesis (A) with the new experimental set-up described here (B).
The new experimental set-up is based on an unmodified HPLC, an equipment item that it readily available in practically any synthetic or analytical laboratory. In brief, a chromatography column was packed with the pre-swelled polymer resin. The column was then connected to the HPLC system containing a pump (a three-head pump was used in our experiment), a detector (a variable UV range detector was used herein), and a computer with standard HPLC-operating software installed (Figure 1B). The column was loaded with the glycosyl acceptor, purged with solvent and then two separate solutions containing glycosyl donor and promoter were delivered concomitantly. After a relatively short reaction time, typically 30–60 min, the system was purged (washed) with solvent. At this time, the resin is loaded with the disaccharide derivative that can be either cleaved off of the polymer support or the oligosaccharide elongation can be continued via alternating deprotection/glycosylation steps. All steps can be monitored using a standard HPLC detection system set to record changes in the UV absorbance of the solution eluting off the column. A solution of reagents can be recirculated to reduce the amount required for each transformation.
During our inital experimentation, the attachment of glycosyl acceptor precursor 1a (3.0 equiv. based on the theoretical loading capacity of the resin) TentaGel® MB-NH2 resin was accomplished using a conventional set-up in the presence EDC (3.0 equiv) and DMAP (1.0 equiv) in CH2Cl2. The Kaiser test32 was conducted on the resin to ensure the completion of loading (48 h). The resin was then treated with 10% trifluoroacetic acid in wet CH2Cl2 to afford polymer bound acceptor 2a (30 min). The loading of the resulting resin 2a was found to be 0.29 mmol/g approximated from all of the following: a) calculation based on the amount of triphenylcarbinol recovered from detritylation; b) recovery of the sugar material from a parallel experiment upon treatment with NaOCH3/CH3OH; c) microbalance measurement of the weight difference of resin before and after loading. Next, the preloaded acceptor bound resin 2a (190 mg, 55 µmol of the glycosyl acceptor) was packed in the Omnifit SolventPlus chromatography column equipped with an adjustable end-piece (http://www.omnifit.com). The resin was swelled in CH2Cl2 for 4–16 h, loosely packed into the column, and the column was integrated into the HPLC.
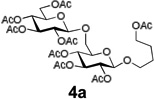
The relative inefficiency of this protocol motivated us to use the HPLC experimental set-up that has allowed us to expedite the initial loading. For instance, loading using a recirculating solution containing 1a (5 equiv), EDC (5 equiv), and DMAP (1 equiv) in CH2Cl2 (Pump B, 1.0 mL/min) was much more effective and the desired loading was achieved in 8 h. After that, the system was purged with CH2Cl2 for 10 min (Pump A, 2.0 mL/min flow rate) followed by detritylation that was accomplished using recirculating TFA/CH2Cl2/H2O (10/88/2, v/v/v, Pump C, 1.0 mL/min, 5 min) to afford the polymer bound acceptor 2a. The system was purged with CH2Cl2 for 10 min (Pump A, 2.0 mL/min flow rate). Reagent bottles, one containing a 39 mM solution of glycosyl donor 3a33 in CH2Cl2 and another one containing a 0.28 M solution of TMSOTf in CH2Cl2 were connected to inlets for pumps B and C, respectively. Pumps B/C were programmed to deliver the mixed solution of donor/promoter concomitantly in the ratio of 4/1 (v/v) at the total flow rate of 0.3 mL/min. After 60 min (18 mL total), pumps B and C were stopped and by this time about 10 mol. equiv. of donor 3a (562 µmol) has passed through the column. It should be noted that the amount of reagents used for this initial study were chosen to mimic that used in the conventional polymer-supported synthesis. Apparently, the flow rate and the ratio between the donor and promoter can be easily adjusted by simple reprogramming of the pump operation as needed. The column was purged (washed) with CH2Cl2 (pump A, flow rate 2.0 mL/min) for 10 min. The formation of disaccharide 4a was determined by cleaving off the sugar molecule from the resin using 0.1 M solution of NaOCH3 in CH3OH/CH2Cl2 (5 mL, 1/1, v/v, pump C, 1 mL/min, 60 min) carried out by continuous recirculation followed by washing with CH3OH (flow rate 2.0 mL/min) for 10 min. Standard (see the SI) neutralization and acetylation (Ac2O/pyridine) afforded product 4a in 98% yield. It should be emphasized that a glycosylation experiment using the conventional experimental set up required significantly longer reaction time (16 h). Additionally, since fresh reagents are delivered constantly, this eliminates the need for multiple reiterations of glycosylation-washing steps as found to be a frequent case for common polymer-bound glycosylations.
Encouraged by this result, we explored other series of glycosyl trichloroacetimidate donors using essentially the same experimental set-up and reaction time. Thus, glycosidation of galactosyl donor 3b34 was equally effective and disaccharide 4b was isolated in 96% yield (Table 1, entry 2). Glycosidations using mannosyl and lactosyl donors 3c35 and 3d36 were somewhat less efficient and the resulting oligosaccharides 4c and 4d were isolated in 78% and 67% yield, respectively (entries 3 and 4). Finally, glycosidations of glucosyl donors 3e and 3f equipped with an easily removable Fmoc protecting group at the C-6 and C-4 positions, respectively, were practically as efficient as 3a and disaccharide 4a was isolated in 95 and 92% yield, respectively (entries 5 and 6). Overall, these results were very indicative of the high efficiency of the new experimental set-up based on HPLC. It came to our attention that generalized reaction conditions (60 min) may not be universally applicable across all sugar series. It has now emerged as common knowledge in carbohydrate chemistry that mannose donors are less reactive than their glucose counterparts.37–39 This implies that extended reaction time would be beneficial for driving glycosidation of mannosyl donor 3c to completion, as well as for the reaction with lactose trichloroacetimidate 3d.
Table 1.
Exploratory comparative study of the coupling efficiency of glycosyl donors 3a–f with the polymer-bound acceptor 2a.
 | |||
|---|---|---|---|
| entry | glycosyl donor | product | yield |
| 1 | 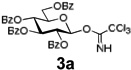 |
 |
98% |
| 2 | 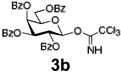 |
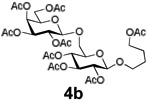 |
96% |
| 3 | 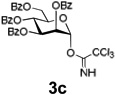 |
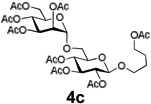 |
78% |
| 4 | 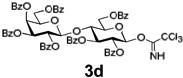 |
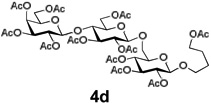 |
67% |
| 5 | 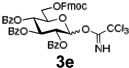 |
4a | 95% |
| 6 | 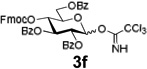 |
4a | 92% |
This called for further study, and it came to our attention that the use of an HLPC detection system would be very informative to indicate when the reaction is nearing completion. For instance, the progress of the reaction could be determined from changes in the UV absorbance of the mixture eluting off the column (measured at 254 nm) in comparison to that measured for the initial 0.39 mmol solution of the donor that is entering the column and the expected total absorbance of the donor and TMSOTf (Figure 2A). Once no change is detected (the detector trace reaches a plateau), this can serve as an indication that the reaction had ended. The real-time monitoring would help to optimize the reaction time and, consequently, reduce the amount of glycosyl donor required to drive the reaction to completion. Similar monitoring of the washing stage of the process can provide the desired information about its completion (Figure 2A). In this case, the washing can be considered as completed once the trace reaches the base-line corresponding to the standard absorbance of neat CH2Cl2.
Fjigure 2.

Idealized sketches for the UV-detector monitoring: A. glycosylation-washing steps; B. Fmoc deprotection-washing.
Using this approach, we established that the optimized reaction times for different glycosyl donors for glycosylation with the supported acceptor 2a using 0.3 mL/min combined flow rate for the donor and promoter solutions (4/1, v/v) are as follows: 3a,e,f (30 min), 3b (25 min), 3c (70 min) and 3d (85 min). Typical washing time for all reactions at 1.0 mL/min of CH2Cl2 is 5–10 min. We also found that slight lowering of the concentration of glycosyl donors or performing partial recirculation of the “used” solution by connecting the column outlet with the pump intake (recirculation) is nearly as efficient as using the standard “fresh” 39 mmol solution. However, UV-monitoring of experiments wherein reagents are continuously recirculated was found to be uninformative.
Having optimized model glycosylations we decided to undertake the synthesis of an oligosaccharide chain using the UV detection system. This synthesis began with glycosyl 2b and donor 3e equipped with the temporary Fmoc protecting group. Standard glycosylation protocol (60 min, step 1, Table 2) followed by a 10-min wash (step 2) led to the formation of disaccharide 5a. Subsequent deprotection of the Fmoc group in 5a was carried out as follows: purge with DMF for 1 min (flow rate 2.0 mL/min) and then pass a 20% solution of piperidine in DMF (flow rate 0.5 mL/min). The release of dibenzofulvene-piperidine adduct was monitored by the UV detector at 312 nm. Based on the real-time Fmoc deprotection curve presented in Figure 2B, we were able to monitor the cleavage process with good accuracy that allowed us to reduce the reaction time to 5 min (step 3).
Table 2.
Experimental data for the HPLC-assisted synthesis of pentasaccharide 8.
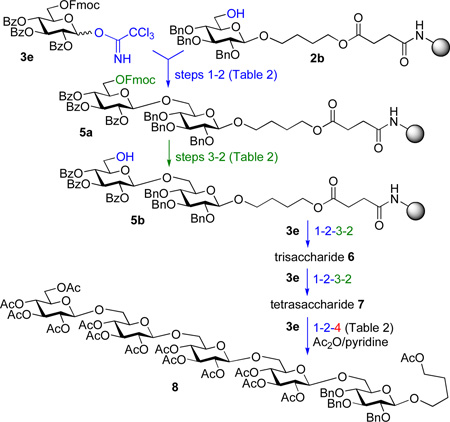 | |||||
|---|---|---|---|---|---|
| operation | action | flow rate, mL/min |
total volume |
time, min |
|
| 1. Glycosylate (acceptor 2b) | Pump B: 39 mM 3e in CH2Cl2 Pump C: 0.28 M TMSOTf in CH2Cl2 | 0.3 B/C = 4/1 | 18 mL | 60 | |
| 2. Wash | Pump A: CH2Cl2 | 2.0 | 20 mL | 10 | |
| 3. Deprotect 5a | Pump C: piperidine/DMF (1/5, v/v) | 0.5 | 2.5 mL | 5 | |
| 2. Wash | Pump A: CH2Cl2 | 2.0 | 20 mL | 10 | |
| Repeat 1 | As above, trisacccharide accceptor 6 is obtained | 0.3 | 18 mL | 60 | |
| 2 | 2.0 | 20 mL | 10 | ||
| 3 | 0.5 | 2.5 mL | 5 | ||
| 2 | 2.0 | 20 mL | 10 | ||
| Repeat 1 | As above, tetrasaccharide acceptor 7 is obtained | 0.3 | 18 mL | 60 | |
| 2 | 2.0 | 20 mL | 10 | ||
| 3 | 0.5 | 2.5 mL | 5 | ||
| 2 | 2.0 | 20 mL | 10 | ||
| Repeat 1 | Pentasaccharide 8a is obtained | 0.3 | 18 mL | 60 | |
| 2 | 2.0 | 20 mL | 10 | ||
| 4. Cleave off to obtain 8 | Pump C: 0.1 M NaOCH3 in CH3OH/CH2Cl2 | 1.0 | 5 mL (recirc.) | 60 | |
This transition was also followed by a 10-min wash with CH2Cl2 (step 2) to afford disaccharide acceptor 5b. The synthesis of trisaccharide 6 was continued using polymer-bound disaccharide acceptor 5b and glycosyl donor 3e via sequential execution of steps 1-2-3-2 (glycosylate-wash-deprotect-wash). The same sequence was repeated to obtain tetrasaccharide 7. Finally, pentasaccharide was obtained by steps 1 and 2 followed by the cleavage step 4 using recirculating 0.1 M solution of NaOCH3 in CH3OH/CH2Cl2 followed by acetylation to afford compound 8 in 62% yield. A comparative yield of pentasaccharide 8 was obtained using conventional experimental set-up, but the experimental time was significantly longer.
In conclusion, we developed a new technology for automated oligosaccharide synthesis. In principle, practically any standard HPLC or LC instrumentation can be used to create a similar set up. The new technology offers the following advantages in comparison to that of conventional oligosaccharide synthesis on polymer supports: faster reaction times, real-time reaction monitoring using an HPLC detection system, and that all steps and sequences can be automated using the standard HPLC-managing computer software. This work was greatly inspired by and is complementary to other automated approaches19,25,40,41 developed for the synthesis of oligosaccharides and related compounds including on-column,42 flow-through,24,43 microfluidic,44 and other related processes.45,46 Further optimization of the HPLC-based technology and its application to other platforms and targets are currently underway.
Supplementary Material
Acknowledgment
This work was supported by awards from the NIGMS (GM090254 and GM077170). Dr. Winter and Mr. Kramer (UM – St. Louis) are thanked for HRMS determinations.
Footnotes
Supporting Information Available: Experimental procedures, extended experimental data, 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Keith J. Stine, Email: kstine@umsl.edu.
Alexei V. Demchenko, Email: demchenkoa@umsl.edu.
References
- 1.Fruchtel JS, Jung G. Angew. Chem. Int. Ed. Engl. 1996;35:17–42. [Google Scholar]
- 2.Winter M. In: Combinatorial Peptide and Nonpeptide Libraries. Jung G, editor. VCH: Weinheim; 1996. pp. 465–510. [Google Scholar]
- 3.Merrifield B. Br. Polym. J. 1984;16:173–178. [Google Scholar]
- 4.Toy PH, Lam Y, editors. Solid-Phase Organic Synthesis. Hoboken: John Wiley & Sons, Inc; 2012. [Google Scholar]
- 5.Schmidt RR, Jonke S, Liu K. In: ACS Symp. Ser. (Frontiers in Modern Carbohydrate Chemistry) Demchenko AV, editor. Vol. 960. Oxford Univ. Press; 2007. pp. 209–237. [Google Scholar]
- 6.Seeberger PH. J. Carbohydr. Chem. 2002;21:613–643. [Google Scholar]
- 7.Seeberger PH, Haase WC. Chem. Rev. 2000;100:4349–4393. doi: 10.1021/cr9903104. [DOI] [PubMed] [Google Scholar]
- 8.Boltje TJ, Kim JH, Park J, Boons GJ. Nature Chemistry. 2010;2:552–557. doi: 10.1038/nchem.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parlato MC, Kamat MN, Wang H, Stine KJ, Demchenko AV. J. Org. Chem. 2008;73:1716–1725. doi: 10.1021/jo701902f. [DOI] [PubMed] [Google Scholar]
- 10.Kanie O, Ohtsuka I, Ako T, Daikoku S, Kanie Y, Kato R. Angew. Chem. Int. Ed. 2006;45:3851–3854. doi: 10.1002/anie.200600433. [DOI] [PubMed] [Google Scholar]
- 11.Jonke S, Liu K-g, Schmidt RR. Chem. Eur. J. 2006;12:1274–1290. doi: 10.1002/chem.200500707. [DOI] [PubMed] [Google Scholar]
- 12.Crich D, Smith M. J. Am. Chem. Soc. 2002;124:8867–8869. doi: 10.1021/ja011406u. [DOI] [PubMed] [Google Scholar]
- 13.Roberge JY, Beebe X, Danishefsky SJ. J. Am. Chem. Soc. 1998;120:3915–3927. [Google Scholar]
- 14.Randolph JT, McClure KF, Danishefsky SJ. J. Am. Chem. Soc. 1995;117:5712–5719. [Google Scholar]
- 15.Zheng C, Seeberger PH, Danishefsky SJ. Angew. Chem. Int. Ed. 1998;37:786–789. doi: 10.1002/(SICI)1521-3773(19980403)37:6<786::AID-ANIE786>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 16.Wu X, Grathwohl M, Schmidt RR. Angew. Chem. Int. Ed. 2002;41:4489–4493. doi: 10.1002/1521-3773(20021202)41:23<4489::AID-ANIE4489>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 17.Plante OJ, Palmacci ER, Seeberger PH. Science. 2001;291:1523–1527. doi: 10.1126/science.1057324. [DOI] [PubMed] [Google Scholar]
- 18.Seeberger PH. Chem. Soc. Rev. 2008;37:19–28. doi: 10.1039/b511197h. [DOI] [PubMed] [Google Scholar]
- 19.Plante OJ, Palmacci ER, Seeberger PH. Adv. Carbohydr. Chem. Biochem. 2003;58:35–54. doi: 10.1016/s0065-2318(03)58002-7. [DOI] [PubMed] [Google Scholar]
- 20.Werz DB, Castagner B, Seeberger PH. J. Am. Chem. Soc. 2007;129:2770–2771. doi: 10.1021/ja069218x. [DOI] [PubMed] [Google Scholar]
- 21.Seeberger PH, Werz DB. Nature Rev. 2005;4:751–763. doi: 10.1038/nrd1823. [DOI] [PubMed] [Google Scholar]
- 22.Krock L, Esposito D, Castagner B, Wang C-C, Bindschadler P, Seeberger PH. Chem. Sci. 2012;3:1617–1622. [Google Scholar]
- 23.Walvoort MTC, van den Elst H, Plante OJ, Krock L, Seeberger PH, Overkleeft HS, van der Marel GA, Codee JDC. Angew. Chem. Int. Ed. 2012;51:4393–4396. doi: 10.1002/anie.201108744. [DOI] [PubMed] [Google Scholar]
- 24.Carrel FR, Geyer K, Codée JDC, Seeberger PH. Org. Lett. 2007;9:2285–2288. doi: 10.1021/ol0705503. [DOI] [PubMed] [Google Scholar]
- 25.Jaipuri FA, Pohl NL. Org. Biomol. Chem. 2008;6:2686–2691. doi: 10.1039/b803451f. [DOI] [PubMed] [Google Scholar]
- 26.Yerneni CK, Pathak V, Pathak AK. J. Org. Chem. 2009;74:6307–6310. doi: 10.1021/jo901169u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tran AT, Burden R, Racys DT, Galan MC. Chem. Commun. 2011;47:4526–4528. doi: 10.1039/c0cc05580h. [DOI] [PubMed] [Google Scholar]
- 28.Galan MC, Corfield AP. Biochem. Soc. Trans. 2010;38:1368–1373. doi: 10.1042/BST0381368. [DOI] [PubMed] [Google Scholar]
- 29.Shimizu H, Sakamoto M, Nagahori N, Nishimura SI. Tetrahedron. 2007;63:2418–2425. [Google Scholar]
- 30.Pornsuriyasak P, Ranade SC, Li A, Parlato MC, Sims CR, Shulga OV, Stine KJ, Demchenko AV. Chem. Commun. 2009:1834–1836. doi: 10.1039/b817684a. [DOI] [PubMed] [Google Scholar]
- 31.Zhu T, Boons G-J. J. Am. Chem. Soc. 2000;122:10222–10223. [Google Scholar]
- 32.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Anal. Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 33.Colonna B, Harding VD, Nepogodiev SA, Raymo FM, Spencer N, Stoddart JF. Chem. Eur. J. 1998;4:1244–1254. [Google Scholar]
- 34.Rio S, Beau J-M, Jacquinet J-C. Carbohydr. Res. 1991;219:71–90. doi: 10.1016/0008-6215(91)89043-f. [DOI] [PubMed] [Google Scholar]
- 35.Bien F, Ziegler T. Tetrahedron: Asymmetry. 1998;9:781–790. [Google Scholar]
- 36.Sandbhor MS, Soya N, Albohy A, Zheng RB, Cartmell J, Bundle DR, Klassen JS, Cairo CW. Biochemistry. 2011;50:6753–6762. doi: 10.1021/bi200449j. [DOI] [PubMed] [Google Scholar]
- 37.Douglas NL, Ley SV, Lucking U, Warriner SL. J. Chem. Soc. Perkin Trans. 1998;1:51–65. [Google Scholar]
- 38.Zhang Z, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. J. Am. Chem. Soc. 1999;121:734–753. [Google Scholar]
- 39.Mydock LK, Demchenko AV. Org. Lett. 2008;10:2103–2106. doi: 10.1021/ol800345j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sears P, Wong CH. Science. 2001;291:2344–2350. doi: 10.1126/science.1058899. [DOI] [PubMed] [Google Scholar]
- 41.Machida K, Hirose Y, Fuse S, Sugawara T, Takahashi T. Chem. Pharm. Bull. 2010;58:87–93. doi: 10.1248/cpb.58.87. [DOI] [PubMed] [Google Scholar]
- 42.Mukhopadhyay B, Maurer SV, Rudolph N, van Well RM, Russell DA, Field RA. J. Org. Chem. 2005;70:9059–9062. doi: 10.1021/jo051390g. [DOI] [PubMed] [Google Scholar]
- 43.Baumann M, Baxendale IR, Ley SV. Mol. Div. 2011;15:613–630. doi: 10.1007/s11030-010-9282-1. [DOI] [PubMed] [Google Scholar]
- 44.Martin JG, Gupta M, Xu Y, Akella S, Liu J, Dordick JS, Linhardt RJ. J. Am. Chem. Soc. 2009;131:11041–11048. doi: 10.1021/ja903038d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roper KA, Lange H, Polyzos A, Berry MB, Baxendale IR, Ley SV. Beilstein J. Org. Chem. 2011;7:1648–1655. doi: 10.3762/bjoc.7.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Opalka SM, Longstreet AR, McQuade DT. Beilstein J. Org. Chem. 2011:1671–1679. doi: 10.3762/bjoc.7.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.