

Abstract
The objective was to determine the cytochrome P450s (CYPs) responsible for the stereoselective and regiospecific hydroxylation of ketamine ((R,S)-Ket) to diastereomeric hydroxyketamines, (2S,6S;2R,6R)-HK (5a) and (2S,6R;2R,6S)-HK (5b) and norketamine ((R,S)-norKet) to hydroxynorketamines, (2S,6S;2R,6R)-HNK (4a), (2S,6R;2R,6S)-HNK (4b), (2S,5S;2R,5R)-HNK (4c), (2S,4S;2R,4R)-HNK (4d), (2S,4R;2R,4S)-HNK (4e), (2S,5R;2R,5S)-HNK (4f).
The enantiomers of Ket and norKet were incubated with HLMs and expressed CYPs. Metabolites were identified and quantified using LC/MS/MS and apparent kinetic constants estimated using single-site Michaelis-Menten, Hill or substrate inhibition equation.
5a was predominantly formed from (S)-Ket by CYP2A6 and N-demethylated to 4a by CYP2B6. 5b was formed from (R)- and (S)-Ket by CYP3A4/3A5 and N-demethylated to 4b by multiple enzymes. norKet incubation produced 4a, 4c and 4f and minor amounts of 4d and 4e. CYP2A6 and CYP2B6 were the major enzymes responsible for the formation of 4a, 4d and 4f, and CYP3A4/3A5 for the formation of 4e. The 4b metabolite was not detected in the norKet incubates.
5a and 4b were detected in plasma samples from patients receiving (R,S)-Ket, indicating that 5a and 5b are significant Ket metabolites. Large variations in HNK concentrations were observed suggesting that pharmacogenetics and/or metabolic drug interactions may play a role in therapeutic response.
Introduction
(R,S)-Ketamine ((R,S)-Ket), Fig. 1, is a chiral phencyclidine derivative that is used as a short acting anesthetic agent (Domino, 2010). Sub-anesthetic doses of the drug are currently being used in the treatment of patients with complex regional pain syndrome (CRPS) (Sabia et al., 2011), postoperative pain in opioid tolerant patients (Loftus, et al., 2010), in emergency room treatments (Lester et al., 2010), and are effective as anti-depressants in patients suffering from treatment-resistant bipolar depression (BPD) (Diazgranados et al., 2010). However, the clinical response to sub-anesthetic doses of (R,S)-Ket is highly variable with about 33% of the patients failing treatment, c.f. (Diazgranados et al., 2010; Goldberg et al., 2010; Goldberg et al., 2011). One potential explanation for the observed variations in response is inter-individual differences in (R,S)-Ket metabolism due to factors such as genetic polymorphisms and metabolic drug interactions.
Figure 1.
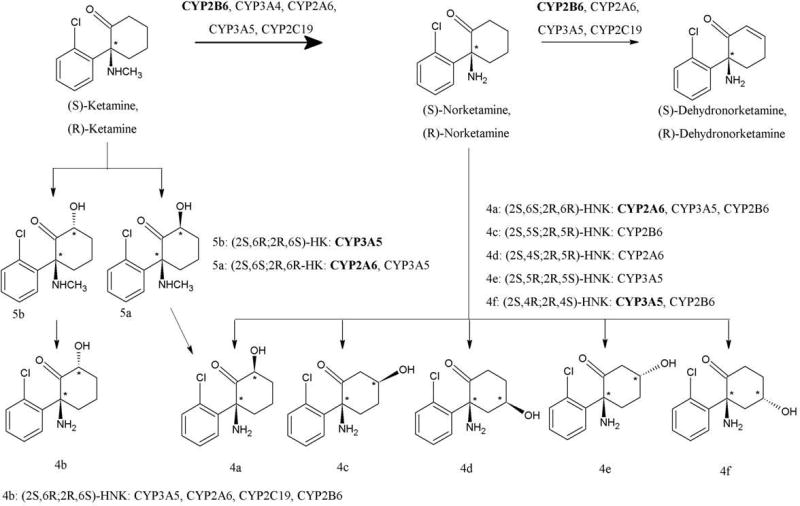
Metabolic pathways and CYP isoforms involved in the metabolism of ketamine.
(R,S)-Ket is extensively metabolized by microsomal enzymes with a major metabolic pathway involving N-demethylation to norketamine ((R,S)-norKet), Fig. 1. This transformation has been extensively studied (Trevor et al., 1983; Kharasch and Labroo, 1992; Hijazi and Boulieu, 2002; Portmann et al., 2010) and is enantioselective with the demethylation of (S)-Ket greater than that of (R)-Ket (Kharasch and Labroo, 1992; Portmann et al., 2010). (R,S)-norKet is further transformed into a series of diastereomeric hydroxynorKet (HNK) metabolites arising from hydroxylation of the cyclohexanone ring at the C6, C4 and C5 positions (Adams et al., 1981; Trevor et al., 1983; Woolf and Adams, 1987; Turfus et al., 2009; Portmann et al., 2010), Fig. 1. Initial studies using human microsomes indicate norKet biotransformation is enantioselective and regioselective with (S)-norKet preferentially converted to (2S,6S)- and (2S,6R)-HNK while (R)-norKet was converted to (2R,5S)-, (2R,5R)-, (2R,4S)- and (2R,4R)-HNK (Trevor et al., 1983). The CYP isoforms associated with the regio- and enatioselective hydroxylation of norKet have not been identified although recent studies have indicated that the oxidation of norKet is mediated by CYP2B6 and CYP2A6 (Portmann et al., 2010). (R,S)-norKet is also transformed into the enantiomeric 5,6-dehydronorketamine ((R,S)-DHNK) (Bolze and Boulieu, 1998; Turfus et al., 2009; Portmann et al., 2010) and the data from recent studies suggest that DHNK is derived from HNK derivatives produced by CYP2B6 mediated hydroxylation at the C5 position on the cyclohexanone ring of norKet (Portmann et al., 2010).
In addition to N-demethylation, (R,S)-Ket is also metabolized by hydroxylation of the cyclohexanone ring to produce diastereomeric hydroxyketamine (HKet) metabolites (Adams et al., 1981; Trevor et al., 1983; Woolf and Adams, 1987; Turfus et al., 2009; Portmann et al., 2010). It has been demonstrated that the HKet metabolites are further transformed by N-demethylation into the corresponding HNK (Woolf and Adams, 1987), however, the CYPs mediating this transformation have not been identified. In addition to the oxidation of the cyclohexanone ring, recent data indicate that the incubation of (R,S)-Ket with human microsomes also produced phenolic metabolites (Turfus,et al., 2009).
The metabolic transformations observed in the in vitro microsomal studies are consistent with the metabolism and disposition of (R,S)-Ket determined in clinical studies. (R,S)-Ket, (R,S)-norKet and (R,S)-DHNK were identified in plasma samples obtained from patients receiving (R,S)-Ket (Bolze and Boulieu, 1998), and all of the major metabolites of (R,S)-Ket were identified in urine samples obtained from volunteers who received a single 50 mg oral dose of (R,S)-Ket (Turfus et al., 2009). These metabolites were also present in the plasma and urine of CRPS patients receiving a continuous 5-day infusion of (R,S)-Ket (Moaddel et al., 2010) and in the plasma of BPD patients that had received a 50 mg/kg dose of (R,S)-Ket administered as a single 40-min infusion (Zhao et al., 2012).
While the majority of the clinical studies of (R,S)-Ket and (S)-Ket have only considered the plasma concentrations of Ket and norKet, recent data from this laboratory have suggested that the large inter-patient variation in clinical response may be associated with the exposure to Ket downstream metabolites. For example, the analysis of plasma samples obtained on Day 3 of the CRPS treatment protocol indicated that (2S,6S;2R,6R)-HNK (4a) and (2S,6R;2R,6S)-HNK (4b) were the major circulating metabolites (Moaddel et al., 2010; data in this paper) and in a study of BPD patients at 230 min post dosing (R,S)-DHNK was the major metabolite in 4 of 9 patients studied, (R,S)-norKet in 3/9 and 4a in 2/9 (Zhao et al., 2012). In addition, significant concentrations (> 8 ng/ml) of (R,S)-DHNK and 4a were present in the plasma of BPD patients 3 days after dosing.
Although neither the pharmacological activities of the hydroxylated downstream metabolites of Ket nor their relevance to observed clinical response have been definitively established, the data from a recent study of the plasma concentration of (R,S)-Ket metabolites and clinical response in 67 patients with major depressive disorder (MDD) and BPD indicated that increased plasma concentrations of (R,S)-DHNK, (2S,5S;2R,5R)-HNK (4c), and (2S,5R;2R,5R)-HNK (4e) were associated with lower psychotomimetic or dissociative side effects (Zarate, et al., 2012). Thus, the potential of significant and long term exposure to the downstream metabolites of (R,S)-Ket makes it very important to determine the metabolic pathways responsible for their formation. The primary objectives of the present study were to use human liver microsomes (HLMs) and expressed CYPs to examine the regio- and stereoselective hydroxylation of (R)- and (S)-Ket and (R)- and (S)-norKet; to identify the specific CYP enzymes involved with each of these transformations and to examine the N-demethylation of (2S,6S)-HK (5a) and (2S,6R)-HK (5b). The goal of this study is to lay the foundation for the comparison of the in vitro data and the plasma and urinary metabolic patterns obtained during clinical treatment in order to determine if pharmacogenetics and/or metabolic drug interactions play a role in therapeutic response.
Material and Methods
Chemicals
(R)-Ket, (S)-Ket, (R,S)-norKet, (R)-norKet, (S)-norKet, (R)-dehydronorketamine (DHNK), (S)-DHNK, 4a, 4b, 5a, and 5b were prepared as previously described (Moaddel et al, 2010). The internal standard, 3,4,5,6-tetradeuterophenyl-(R,S)-ketamine HCl (D4-Ket) was purchased from Cerillant (Round Rock, TX, USA). Glucose 6-phosphate, glucose-6-phosphate dehydrogenase, and NADP+ were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other chemicals were of HPLC grade.
Microsomal Preparations
Characterized human liver microsomes (HLMs) (stock, 20 mg/ml) and baculovirus-insect cell-expressed human P450s (1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4, and 3A5) (with oxidoreductase) were purchased from BD Biosciences (San Jose, CA, USA). The microsomal preparations (HLMs and CYPs) were stored at −80°C until use. The total P450 content, protein concentrations and the specific activity of each P450 isoform were as supplied by the manufacturer.
LC/MS/MS Analysis of microsomal incubates
A previously validated method was used to measure Ket and its downstream metabolites in microsomal incubates and clinical samples (Moaddel et al., 2010). Briefly, the chromatographic experiments were carried out on a Shimadzu Prominence HPLC system (Shimadzu, Columbia, MD, USA) and total analyte concentrations were determined using an Eclipse XDB-C18 guard column and a Varian Pursuit XRs 5 C18 analytical column (Varian, Inc., Palo Alto, CA, USA). The MS/MS analysis was performed using a triple quadrupole mass spectrometer model API 4000 system from Applied Biosystems/MDS Sciex equipped with Turbo Ion SprayR (TIS) (Applied Biosystems, Foster City, CA, USA). The data were acquired and analyzed using Analyst version 1.4.2 (Applied Biosystems). Positive electrospray ionization data were acquired using multiple reaction monitoring (MRM) using the following transitions: 238 → 115 (Ket); 224 → 125 (norKet); 222 → 177 (DHNK); 240 → 125 (all of the HNK metabolites); 254 → 151 (5a,5b); 254 → 141(phenolic HK metabolites). Quantification was accomplished using area ratios calculated using D4-(R,S)-Ket as the external standard, where the concentration of the external standard was set at 100 ng/ml.
A stock solution was prepared containing (R,S)-Ket (1,000 μg/ml), (R,S)-norKet (1,000 μg/ml), (R,S)-DHNK (1,000 μg/ml), 4a (100 μg/ml), 5a (10 μg/ml) and 5b (10 μg/ml). The solution was prepared in ultra-pure water and stored in polypropylene tubes at −20°C. Serial dilutions of the stock solution were used to prepare the samples for the calibration curves and quality control standards (QC).
Identification/confirmation of Ket and norKet metabolites
Pilot incubation experiments were performed to identify potentially new metabolites and/or confirm the presence of known Ket and norKet primary and secondary metabolites. Ket or norKet (in methanol) was added to a tube and solvent was evaporated. Residue was reconstituted with phosphate reaction buffer and a mixture of Ket or norKet (10 μM) and HLMs (0.5 mg/ml) in phosphate reaction buffer (pH 7.4) was allowed to equilibrate for 5 min at 37°C (final volume, 250 μl). The reaction was initiated by adding a NADPH-generating system (cofactors) (13 mM NADP, 33 mM glucose 6-phosphate, 33 mM MgCl2, and 4.0 U/mL glucose 6-phosphate dehydrogenase) and allowed to proceed for 30 min at 37°C. The reactions were terminated by placing tubes on ice and immediately adding 500 μl of acetonitrile and the mixtures were vortex mixed for 30 sec, centrifuged at 16,000 x g for 5 min in an Eppendorf model 5415D centrifuge (Brinkmann Instruments, Westbury, NY) and the supernatant was removed and extracted by shaking for 15 min with ethyl acetate under alkaline pH. After centrifugation for 15 min at 3200 × g in a Beckman Coulter-AllegraR 6R Benchtop Centrifuge, the organic layer was removed, evaporated to dryness and the residue was reconstituted with 500 μL of methanol and 10 μl of a 5 μg/ml solution of D4-Ket in methanol was added.
Control incubation (with cofactors, HLMs and substrate) and negative control incubations without cofactors, HLMs or substrate (before and after extraction) were run in parallel. The (R,S)-norKet, (S)-norKet, (R)-norKet, (R,S)-DHNK, 5a, 5b, 4a, and 4b standards were used for metabolite identification and the identification of the additional metabolites was based on the retention time of their respective standards and MRM transitions as previously described (Woolf and Adams, 1987; Turfus et al., 2009; Moaddel et al., 2010).
Linear conditions for protein and time were assessed in order to avoid the effect of sequential metabolism, and the shortest possible incubation time was selected while ensuring assay sensitivity. A final protein concentrationof 0.50 mg/ml and 30-min incubation represented linear conditions for (R,S)-norKet and its enatiomers while 0.50 mg/ml and 15-min duration of incubation represented linear conditions for (R,S)-Ket and its enantiomers. Kinetic studies for Ket and norKet metabolism were conducted in characterized HLMs (n=2) under linear conditions by incubating a concentration range of 1–2000 μM (Ket) and 1–500 μM (norKet) in duplicatefor 30 min at 37°C with HLMs (0.50 mg protein/ml) and cofactors. Reaction was terminated andprocessed as described above.
Correlation analysis
Correlation between formation rates of (R,S)-Ket and (R,S)-norKet and specific isoforms was tested by incubating the substrates (10 μM) with a panel of microsomes from 15 characterized HLMs (0.50 mg/ml) and cofactors for 30 min at 37°C. The activity of each CYP isoform in each HLM, determined by isoform-specific reaction markers, was as provided by the supplier (BD Bioscience, San Jose, CA; http://www.bdbiosciences.com/ptDatabaseList.jsp).
Inhibiton analysis
(R)- and (S)-Ket (10 μM) was incubated with pooled HLMs (0.5 mg/ml) and the NADPH-generating system at 37 °C for 15 min in the absence (control) and the presence of the following known isoform-specific inhibitors: pilocarpine (50 μM) for CYP2A6, omeprazole (5μM) for CYP2C19 and ketoconazole (1 μM) for CYP3A. (R)- and (S)-norKet (10 μM) was incubated with pooled HLMs (0.5 mg/ml) and the NADPH-generating system at 37 °C for 30 min to determine norKet hydroxylation in the absence (control) and presence of pilocarpine, omeprazole, ketoconazole and 50 μM thioTEPA (CYP2B6. Sample processing was as described above. Percentage formation rate remaining after inhibition relative to the uninhibited controls (without inhibitors or vehicle controls) was calculated.
Ket and norKet primary and secondary metabolism by expressed CYP isoforms
To obtain qualitative information as to which isoforms might catalyze Ket or norKet primary metabolism, (R)-Ket, (S)-Ket, (R,S)-norKet, (S)-norKet, or (R)-norKet (10 μM) were incubated with expressed CYP 1A2, 2A6, 2B6,2C8, 2C9, 2C19, 2D6, 2E1, 3A4, or 3A5 (13–26 pmol) and cofactors (same composition as above) at 37°C for 15min (Ket) or 30 min (norkKet). The concentration of (R)- and (S)-Ket and (R,S)-, (R)- and (S)-norKet represents saturation (Vmax) in Michaelis-Menten curve. For those isoforms that showed predominant activity towards Ket metabolism, i.e. CYP2A6 and CYP2B6, full kinetic analyses were performed by incubating (R)-Ket and (S)-Ket (1 to 2000μM) with expressed enzymes (13 pmol) and cofactors for 15 min. For those isoforms that showed predominant activity towards norKet metabolism, i.e. CYP2A6, CYP2B6 and CYP3A5, full kinetic analyses were performed by incubating (R)-norKet, or (S)-norKet (1 to 500 μM) with expressed enzymes (13 pmol) and cofactors for 30 min.
Data Analysis
Estimates of apparent kinetic constants for Ket or norKet metabolism were obtained using nonlinear regression analysis by Prism version 5.01 for Windows (GraphPad Software Inc., San Diego, CA; www.graphpad.com). The simple single-site Michaelis-Menten equation (V = Vmax * C/(Km + C), Hill equation(V = Vmax * Cn/(C50 + Cn), or substrate inhibition equation (V = Vmax/(1+Km)/C+C/Ksi) were fit to formation rates (V) of metabolites versus substrate (Ket or norKet) concentrations (C). The apparent maximum formation rate (Vmax), and apparent substrate concentration resulting in 50% of Vmax, and substrate inhibition constant (Ksi) were calculated. The models that best fit were selected based on thedispersion of residuals and standard errors of the parameter estimates. Pearson’s correlation analysis and IC50 values were calculated using the Prism Software. P<0.05 was considered statistically significant. Kinetic constants were given as mean ± SD (or ±SE of parameter estimates) when appropriate.
Analysis of clinical samples
Plasma samples from CRPS patients on Day 3 of a 5-day continuous infusion of (R,S)-Ket were assayed for Ket and metabolites as previously described (Moaddel et al, 2010). The clinical protocol was approved by the Cooper University Institutional Review Board and has been previously described (Goldberg, et al., 2010). In brief, after obtaining written informed consent the patient was admitted to a monitored telemetry unit and (R,S)-Ket, contained in a 500 ml bag of normal saline, was infused at an initial rate of 10 mg/hr and then titrated to a maximum of 40 mg/hr to achieve comfort without evidence of significant side effects or oxygen desaturation (< 92%). The infusion was maintained for 5 days with 24 h monitoring of the subject. Pre-dose and post-dose blood and urine samples were obtained as proscribed by the protocol and frozen at −80°C until analysis.
Results
The general metabolism of Ket into its primary and secondary metabolites
A series of in vitro experiments using (R,S)-Ket, (R)-Ket and (S)-Ket as substrates were performed with HLM preparations and expressed CYPs to characterize the stereo- and regio-selective oxidation of Ket to its primary and secondary metabolites. As shown in Fig. 2A, the incubation of (R,S)-Ket produced significant concentrations of (R,S)-norKet, (R,S)-DHNK, 5a, 5b, 4a, 4b, (2S,5S;2R,5R)-HNK (4c) and (2S,5R;2R,5S)-HNK (4f) while only trace amounts of (2S,4S;2R,4R)-HNK (4d) and (2S,4R;2R,4S)-HNK (4e) were detected (Fig. 2A). A phenolic Ket metabolite(s) previously described by Turfus et al., (2009) was also detected in the incubates (data not shown). The formation of these metabolites depended on NADPH, duration of incubation, microsomal protein and substrate concentrations and none of these metabolites were formed in the negative control experiments, suggesting potential involvement of CYPs.
Figure 2.

The chromatographic traces from the LC/MS/MS analysis of the incubation of (R,S)-Ket (A) and (R,S)-norKet (B) with human liver microsomal preparations in which the peaks corresponding to (R,S)-Ket, (R,S)-norKet and the internal standard D4-(R,S)-Ket have been removed. See text for details.
In this study, the CYPs isoform associated with the metabolic conversion of Ket to norKet was predominantly 2B6 (primary), with some contribution of CYP2A6, CYP2C19, CYP3A4/5(data not shown). These results are essentially consistent with previous studies that also identified CYP2B6 as the primary mediator of Ket N-demethylation to norKet with CYP3A and CYP2C9 also playing a role in this transformation (Trevor et al., 1983; Kharasch and Labroo, 1992; Hijazi and Boulieu, 2002; Portmann et al., 2010). The observed contribution of CYP2A6, 3A4 and 3A5 may reflect the oxidative hydroxylation of Ket and norKet, as CYP2B6 and CYP2A6 have been recently associated with the secondary metabolism of norKet (Portmann et al., 2010).
The metabolic formation of hydroxketamine (HK)
The kinetics for the metabolism of (R)- and (S)-Ket to the Z- or cis-(2S,6S)-HK (compound 5a) and E- or trans-(2S,6R)-HK (compound 5b) were characterized by simple Michaelis-Menten kinetics (hyperbolic saturation curves) and the derived Vmax and Km parameters are listed in Table 1. . The data indicate that the formation of 5a is enantioselective with the hydroxylation of (S)-Ket predominate relative to (R)-Ket and that the reaction is enantiospecific producing primarily a S- configuration at the C6 carbinol. Thus, the primary Z-HK metabolite from the incubation of (R)- and (S)-Ket is (2S,6S)-HK. Both (R)- and (S)-Ket were transformed into the E-HK metabolite, 5b, with the formation of (2R,6S)-HK slightly favored relative to the (2S,6R)-HK diasteromer (Table 1).
Table 1.
Kinetic parameters for the formation of the HK metabolites 5a and 5b for (S)-Ket and (R)-Ket in pooled HLMs. A range of S- or R-ketamine concentrations was incubated with HLMs (0.5 mg/ml) at 37°C for 15 min. Substrate concentrations versus velocity were fit using Michaelis-Menten nonlinear regression analyses where: Vmax, pmol/min/mg protein; Km, μM; and Vmax/Km (in vitro intrinsic clearance, Clint), μL/min/mg protein.
| Metabolite | (R)-Ket | (S)-Ket | ||||
|---|---|---|---|---|---|---|
| Vmax | Km | Vmax/Km | Vmax | Km | Vmax/Km | |
| 5a | 12.9 | 171 | 0.075 | 123.1 | 349.5 | 0.352 |
| 5b | 443.2 | 1095 | 0.41 | 247.9 | 471.1 | 0.53 |
| HK phenol | 6.86 | 71.5 | 0.096 | 2.69 | 54.1 | 0.05 |
The metabolism of (R)- and (S)-Ket was determined in a panel of characterized HLM preparations (n = 15) and the rates of HK metabolite formation were tested for correlations with individual CYPs (Table 2). These data confirm that CYP2B6 is the major enzyme responsible for the metabolism of (S)- and (R)-Ket, as shown by the strong correlation of norK formation rate and depletion of Ket with CYP2B6 activity. A significant relationship was observed for the formation of 5a and CYP2A6 and CYP2C19 activity (r = 0.75-to 0.86, p <0.05). While, there was a trend towards the formation of 5b and CYP2A6 and CYP2C19 (r = 0.63–0.69, p = 0.06–0.07), this did not reach a statistically significant level. The isoform specific formation of the HK metabolites was also investigated using a panel of 10 expressed CYPs. The data indicated that 5a was mainly formed by CYP2A6, with high preference towards (S)-Ket with a smaller and less enantioselective contribution from CYP3A5, Fig. 3. Subsequently, inhibition with isoform selective inhibition confirmed that CYP2A6 and 3A4 were both responsible in the formation 5a (Supplemental Table 1). Thus, a metabolic pathway of (S)-Ket is CYP2A6 mediated enantiospecific hydroxylation at the C6 position of the cyclohexanone ring that produces (2S,6S)-HK. The results also indicate that the formation of the E-HK metabolite, 5b, was mediated by CYP3A5 and CYP3A4 with the formation of (2R,6S)-HK slightly favored relative to the (2S,6R)-HK diasteromer, Fig. 3. Our inhibition data with respect to 5b was inconclusive because the metabolite formed was low, which was further decreased in the presence of the inhibitors. The rates of formation of 5a and 5b, Table 1 and Fig. 3, suggest that CYP3A5 and CYP3A4 play a greater role in the metabolic clearance of (R)-Ket than (S)-Ket.
Table 2.
Correlation of formation rates of R- and S-ketamine (10 μM) metabolism to norketamine and hydroxylated metabolites with the activities of CYP isoforms in a panel of 15 characterized HLMs.
| Metabolites | Cytochrome P450 (CYP) enzymes | FMO | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1A2 | 2A6 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 2E1 | 3A | 4A11 | ||
| R-K | 0.33 | −0.37 | −0.90** | −0.41 | 0.18 | 0.11 | −0.57 | −0.46 | −0.52 | 0.12 | 0.28 |
| S-K | 0.62 | −0.11 | −0.75* | −0.37 | 0.12 | 0.16 | −0.44 | −0.50 | −0.48 | 0.41 | 0.46 |
| NK from R-K | −0.50 | −0.09 | 0.75* | 0.42 | −0.16 | −0.47 | 0.60 | 0.45 | 0.14 | −0.41 | −0.46 |
| NK from S-K | 0.48 | 0.16 | 0.87** | 0.51 | −0.072 | −0.28 | 0.54 | 0.57 | 0.41 | −0.33 | 0.43 |
| 5a from R-K | 0.18 | 0.77* | 0.006 | −0.39 | −0.30 | 0.86** | −0.20 | −0.22 | 0.66 | 0.54 | 0.43 |
| 5a from S-K | 0.38 | 0.81* | 0.008 | −0.37 | −0.35 | 0.75* | −0.10 | −0.25 | 0.52 | 0.63 | 0.45 |
| 5b from R-K | −0.30 | 0.66 | −0.22 | −0.19 | −0.10 | 0.63 | −0.51 | 0.11 | 0.63 | −0.24 | −0.04 |
| 5b from S-K | 0.46 | 0.69 | −0.4 | −0.25 | −0.28 | 0.31 | −0.21 | −0.03 | 0.06 | 0.05 | −0.10 |
Incubation was performed in duplicate. Data were analyzed using the Pearson’s correlation test (Pearson r). The activity of each isoform was determined using standard in vitro probe substrate reactions selective for each isoform as indicated by the supplier (see Materials and Methods). Pearson r is provided.
p ≤0.05;
p<0.01
HK, hydroxyketamine; NK, norketamine, 5a, (2S,6S/2R,6R) HK1; 5b, (2S,6R/2R,6S) HK2.
Figure 3.

Kinetics for the metabolism of (R)- and (S)-Ket by a panel of expressed human CYPs to 5a and 5b. Each enantiomer (10μM) was incubated with 10 expressed CYPs (13 pmol) and cofactors at 37°C for 15 min. Each point represents average of duplicate incubations.
The previously described Ket phenolic metabolite(s) (Turfus et al., 2009) was also observed in the HLM incubates. The metabolite was mainly formed from (R)-Ket and the Clint for the formation from (R)-Ket was 1.9-fold higher than the corresponding value found with (S)-Ket, Table 1. The primary isoform associated with the formation of the phenolic metabolite from (R)-Ket was CYP2C9 with a contribution from CYP2C19, while FMO appeared to be the major factor in the formation of this metabolite from (S)-Ket (data not shown). CYP2B6 also showed activity towards the formation of the Ket phenol, although at a very low rate.
The N-demethylation of HK
Initial studies by Woolf and Adams (Woolf and Adams, 1987) demonstrated that both Z-HK (5a) and E-HK (5b) were N-demethylated by rat, rabbit and human liver microsomal preparations. In this study, 5a and 5b were incubated with the panel of expressed CYPs. When 5a was the substrate, the corresponding N-demethylated metabolite 4a, was formed by CYP2B6 at the highest rate (201.6 ng/ml) and the concentration of 5a in the incubates containing CYP2B6 was decreased by 42.5%. Other CYPs appeared to participate in the N-demethylation of 5a but to a much lesser extent with <13 ng/ml for metabolite formation and no appreciable depletion of the substrate (data not shown). All 10 expressed CYPs appear to show some activity in the N-demethylation of 5b and the formation of 4b, 2.7 to 3.5 ng/ml, but this reaction was generally very slow (data not shown). None of the CYPs tested appreciably depleted the substrate, however the data confirm that 4b is formed by N-demethylation of 5b. Of interest, is that the addition of specific inhibitors to repress each CYP’s activity demonstrated that inhibition of CYP2A6 and CYP3A4 resulted in a significant decrease in the formation NK, which appears to result in shunting the metabolic pathways in favor of ketamine hydroxylation and thus resulting in an increase in the formation of 4b (Supplemental Table 1).
The microsomal hydroxylation of (R)- and (S)-norKet
A series of in vitro experiments using (R,S)-norKet, (R)-norKet and (S)-norKet as substrates were performed with HLMs and expressed CYPs to characterize the stereo- and regio-selective oxidation of norKet to its hydroxynorKet (HNK) metabolites. The incubation of (R,S)-norKet produced significant concentrations of (R,S)-DHNK, 4a, (2S,5S;2R,5R)-HNK (4c) and (2S,5R;2R,5S)-HKet (4f), and trace amounts of (2S,4S;2R,4R)-HNK (4d) and (2S,4R;2R,4S)-HNK (4e) while 4b was not detected in the incubates, Fig 2.
The kinetics parameters for the metabolic conversion of norKet enantiomers to the HNK metabolites were determined in two HLM preparations and the substrate versus velocity data was analyzed using the Michaelis-Menten approach. The derived kinetic parameters are presented in Table 3. The data indicate that there were no significant enantioselective differences in the Km values of the various HNK diastereomers, while there were substantial differences with respect to the Vmax vlaues. Based on Clint, 4c was the most abundant followed by 4f > 4a. Kinetic parameters of 4e could only be calculated when (S)-norKet was used as a substrate. Enantioselectivities of the metabolic transformation were reflected in the relative Vmax values which were greater for the formation of 4a and 4f was higher from (R)-norKet while the value for 4d was higher from (S)-norKet and 4e was exclusively formed from (S)-norKet.
Table 3.
Kinetic parameters for the metabolism of (S)- and (R)-norKet in pooled HLMs (n=2). A range of (S)- and (R)-norKet concentrations was incubated with HLMs (0.5 mg/ml) at 37°C for 30 min. Substrate concentrations versus velocity were fit using Michaelis-Menten nonlinear regression analyses, where: Vmax, pmol/min/mg protein; Km, μM; and Vmax/Km (in vitro intrinsic clearance, Clint),μL/min/mg protein.
| Metabolite | (S)-norKet | (R)-norKet | ||||
|---|---|---|---|---|---|---|
| Vmax | Km | Vmax/Km | Vmax | Km | Vmax/Km | |
| HNK | ||||||
| 4a | 196.3 | 76.2 | 2.58 | 366.7 | 96.1 | 3.82 |
| 4c | 837.3 | 75.8 | 11.0 | 825.4 | 54.3 | 15.2 |
| 4d | 437.5 | 97.0 | 4.5 | 127.2 | 199.8 | 0.64 |
| 4e | 14.3 | 62.1 | 0.23 | |||
| 4f | 213.2 | 39.6 | 5.39 | 488.8 | 111.2 | 4.39 |
| DHNK | ||||||
| DHNK | 437 | 112.6 | 3.88 | 469 | 145.2 | 2.23 |
The metabolism of (R,S)-norKet to the HNK metabolites was determined in a panel of characterized HLM preparations for correlations with the activity of individual CYPs and FMO (Table 4). The depletion of norKet from the microsomal incubates and the formation of all of the HNK metabolites was significantly correlated with CYP2B6 activity. In addition, CYP2A6, CYP2C8 and CYP3A correlated significantly with formation rate of most HNKs. This is consistent with the previous report that norKet metabolites were predominantly formed by CYP2A6 and CYP2B6 (Portman et al., 2010), although we also observed that CYP3A is active in the formation of the HNK metabolites.
Table 4.
Correlation of 10 μM NK (racemic) metabolism with cytochrome P450 (CYP) activities.
| Metabolites | Cytochrome P450 (CYP) enzymes | FMO | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1A2 | 2A6 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 2E1 | 3A | 4A11 | ||
| NK | 0.19 | −0.34 | −0.92** | −0.32 | 0.16 | −0.024 | −0.64 | −0.20 | −0.64 | −0.073 | 0.17 |
| 4a | 0.45 | 0.86**** | 0.86**** | 0.60* | 0.37 | 0.36 | −0.29 | 0.15 | 0.76*** | 0.08 | 0.30 |
| 4c | 0.50 | 0.73** | 0.92**** | 0.75*** | 0.38 | 0.12 | 0.09 | 0.28 | 0.46 | 0.004 | 0.03 |
| 4d | 0.38 | 0.69** | 0.88**** | 0.57* | 0.34 | 0.22 | −0.08 | 0.14 | 0.57* | −0.002 | 0.14 |
| 4e | 0.2 | 0.41 | 0.52* | 0.33 | 0.2 | 0.43 | −0.3 | −0.04 | 0.71** | −0.07 | 0.38 |
| 4f | 0.22 | 0.58* | 0.85**** | 0.60* | 0.16 | 0.12 | −0.15 | 0.11 | 0.64** | −0.28 | 0.1 |
| DHNK | 0.47 | 0.73** | 0.93**** | 0.73** | 0.38 | 0.13 | 0.06 | 0.28 | 0.49 | 0.004 | 0.05 |
Incubations were performed in duplicate. Data were analyzed using the Pearson’s correlation test and Pearson’s r is provided. The activity of each isoform was determined using standard in vitro probe substrate reactions selective for each isoform as indicated by the supplier (see Materials and Methods). P<0.05 is considered significant (
p = 0.05;
p<0.01;
p<0.001;
p<0.0001).
Incubation of (R)- and (S)-norKet with a panel of 10 expressed CYPs to further identify the specific CYP isoforms revealed that (R)- and (S)-norKet were efficiently depleted from the incubation mixtures by CYP2A6 (34.5% and 41.0%, respectively), CYP2B6 (47.8% and 41.6%), and CYP2C19 (35.0% and 27.3%) (Data not shown). Other isoforms showed no appreciable effect on the disappearance of either substrate using < 24% as the cutoff (Data not shown). CYP2A6 and CYP2B6 were the major enzymes responsible for the formation of 4a, 4d and 4f, and CYP3A4 and CYP3A5 contributed to the formation of 4f and were responsible for the formation of 4e, Fig 4. While there is little enantioselectivity on the CYP2B6 and CYP2A6 catalyzed formation of 4a, some of the enzymes studied showed extensive enantioselectivity and regio-specificity. The data indicate that the formation of 4c and 4f, i.e. hydroxylation of the cyclohexanone ring at the C5 position to produce the E- and Z-diastereomers, respectively, was catalyzed by CYP2B6 and preferentially from (R)-norKet, the formation of 4d, i.e., hydroxylation at the C4 position to produce the E-diastereomer, was mediated by CYP2B6 and CYP2A6 of (S)-norKet while 4e, i.e., hydroxylation at the C4 position to produce the E-diastereomer, was formed by CYP2B6 and CYP2A6 from (R)-norKet. It should be noted that 4b was not generated from (S)- or (R)-norKet by any of the CYPs used in this study.
Figure 4.

Kinetics of the metabolism of (R)- and (S)-norKet by a panel of expressed human CYPs to 4a, 4c, 4d, 4e, and 4f. Each enantiomer (10μM) was incubated with 10 expressed CYPs (13 pmol) and cofactors at 37°C for 15 min. Each point represents average of duplicate incubations.
Since CYP2B6, CYP2A6 and CYP3A5 catalyzed norKet at the highest rate (Fig 4), detailed kinetic analyses were performed with these enzymes (Table 5) and specific inhibitors were added to repress each CYP’s activity to confirm specific metabolism by the aforementioned CYPs (supplemental Table 1). Generally, the Km values for the formation of the HNK metabolites were lower for CYP2B6, suggesting that this enzyme might represent high affinity component at therapeutically relevant concentrations. The Km values obtained with CYP2A6 and CYP3A are quite high, except for the formation of 4a by CYP2A6 from (S)-norKet. Of note, Clint for the formation of 4c and 4f from (R)-norKet was ~40- and ~10-fold higher respectively, than that obtained from (S)-norKet. Inhibition of 2A6 and 3A4 resulted in a decrease in the formation of 4a, with a lesser extent from 2B6. Further, inhibition of only 2A6 resulted in a decrease in the formation of 4f.
Table 5.
Kinetic parameters for the metabolism of (S)- and (R)-norKet) by expressed CYP2B6, CYP2A6 and CYP3A5. A range of (S)- and (R)-norKet concentrations was incubated induplicate with expressed CYPs (13 pmol) at 37°C for 15 min. Substrate concentrations versus velocity were fit using Michaelis-Menten nonlinear regression analyses, where: Vmax, pmol/min/pmol P450; Km, μM; and Vmax/Km (in vitro intrinsic clearance, Clint), μL/min/pmol P450.
| Metab. | (S)-norKet | (R)-norKet | ||||
|---|---|---|---|---|---|---|
| Vmax | Km | Vmax/Km | Vmax | Km | Vmax/Km | |
| CYP2B6 | ||||||
| 4a | 0.56 | 53.0 | 0.011 | 0.76 | 50.0 | 0.45 |
| 4c | 16.3 | 36.1 | 0.45 | 16.72 | 21.6 | 0.77 |
| 4d | 3.34 | 45.6 | 0.07 | 0.78 | 42.5 | 0.018 |
| 4f | 0.30 | 54.3 | 0.0055 | 3.98 | 69.4 | 0.057 |
| DHNK | 7.61 | 146.8 | 0.052 | 13.0 | 286.5 | 0.045 |
| CYP2A6 | ||||||
| 4a | 3.9 | 67.0 | 0.059 | 8.18 | 101.5 | 0.081 |
| 4c | 1.48 | 106.5 | 0.014 | 1.25 | 263.6 | 0.005 |
| 4d | 4.67 | 88.6 | 0.053 | 1.28 | 237.2 | 0.005 |
| 4f | 0.33 | 120.8 | 0.003 | 0.44 | 338.4 | 0.001 |
| DHK | 0.92 | 128.3 | 0.007 | 0.98 | 328.6 | 0.003 |
| CYP3A5 | ||||||
| 4a | 3.87 | 606.6 | 0.0064 | 0.29 | 226.2 | 0.0013 |
| 4c | 0.30 | 318.9 | 0.001 | 0.038 | 189.5 | 0.0002 |
| 4d | 0.20 | 261.6 | 0.0008 | 0.029 | 312.0 | 0.0001 |
| 4e | 3.14 | 224.9 | 0.014 | 0.087 | 86.4 | 0.001 |
| 4f | 5.71 | 278.2 | 0.021 | 1.69 | 270.0 | 0.006 |
| DHK | 1.96 | 2497 | 0.0008 | 0.079 | 216.4 | 0.0004 |
The metabolic formation of dehydronorketamine (DHNK)
The incubation of (R,S)-Ket, (R)-Ket and (S)-Ket with HLM preparations produced significant concentrations of (R)- and (S)-DHNK (Fig. 2A) as did the incubation of (R)- and (S)-norKet (Fig. 2B), Table 3. Incubation of (R)- and (S)-norKet with 10 expressed enzymes indicated that CYP2B6 is the major isoform in the formation of DHNK, with some contribution of CYP2A6 and CYP3A4 (Data not shown). The correlation analysis depicted in Table 4 indicates significant correlations between DHNK formation and CYP2A6, CYP2B6 and CYP2C8 activity. Further experiments of incubation of (R)- and (S)-norKet with the panel of characterized CYPs demonstrated that CYP2B6 was the key isoform involved in the formation of the DHNK with contributions from CYP2A6 and CYP3A5, Table 5. . The involvement of CYP2B6 is consistent with previous data (Portmann et al., 2010). However, the data from the incubations with HLM preparations, Table 3, and characterized CYPs, Table 4, indicate that the transformation of (R)- and (S)-norKet to the corresponding DHNK metabolites is not significantly enantioselective, which differs from the earlier observations that the DHNK is formed in a stereoselective manner (Portmann et al., 2010).
CRPS patient data
Plasma samples were obtained from two CRPS patients, P17 and P18, on Day 3 of a 5-day continuous infusion of (R,S)-Ket at a rate of 40 mg/hr. Based upon the data from earlier studies (Goldberg et al., 2010a, Goldberg, et al. 2010b), the plasma concentration of (R,S)-Ket was assumed to have achieved steady-concentrations and the samples were assayed for the concentrations of Ket and its major metabolites. The plasma samples contained significant concentrations (> 50 ng/ml) of the N-demethylated metabolites and measurable concentrations (< 10 ng/ml) of 5a (Fig 5). However, there were significant differences in the relative concentrations of (R,S)-norKet, (R,S)-DHNK, 4a and 4b. As had been previously reported, the relative concentrations of these metabolites in patient P17 were 4b > 4a ≫ (R,S)-DHNK ≫ (R,S)-norKet (Moaddel et al., 2010) while in patient P18 the relative relationship was 4a ≫ (R,S)-norKet > 4b > DHNK. Based upon the microsomal incubations of Ket, norKet, 5a and 5b, the significant plasma concentrations of 4b demonstrate that the hydroxylation of Ket to the HK metabolites 5a and 5b followed by N-demethylation to 4a and 4b is an important in vivo metabolic pathway. This observation is consistent with the data from the analysis of plasma samples from 9 patients suffering from bipolar depression who received a single 40-min infusion of 50 mg/kg of (R,S)-Ket (Zhao et al., 2012). In the bipolar depression study, the plasma samples obtained 40 – 230 min after the completion of the administration of (R,S)-Ket contained significant concentrations of 4b (≥ 10.0 ng/ml) and measurable concentrations of 5a (≤ 3.0 ng/ml). It is also interesting to note that at 230 min, (R,S)-DHNK was the major metabolite in 4/9 patients, (R,S)-norKet (3/9) and 4a (2/9) indicating that the primary norKet and HK metabolites are rapidly converted into the secondary DHNK and HNK metabolites.
Figure 5.

Plasma concentrations of the metabolites of (R,S)-Ket in CRPS patients P17 and P18 on Day 3 of a 5-day continuous infusion of 40 mg/kg of (R,S)-Ket.
Discussion
In the initial pharmacological studies of Ket, norKet and 4a in Sprague-Dawley rats, Leung and Baillie (1986) demonstrated that the i.v. administration of Ket produced significant plasma and brain tissue concentrations of norKet and 4a, that the administration of norKet produced 4a and that when 4a was administered the compound readily passed the blood brain barrier (Leung and Baillie, 1986). The study also demonstrated that Ket and norKet had CNS activity associated with general anesthesia and increased spontaneous locomotor activity during the postanesthetic recovery phase, while 4a was inactive in both of these tests. Thus, Ket was identified as the active anesthetic agent and norKet as the “active” metabolite, and almost all of the subsequent metabolic, pharmacokinetic, pharmacological and clinical studies have concentrated on these two compounds. However, while this approach may be appropriate for the study of Ket induced anesthesia, recent studies of the therapeutic effects of sub-anesthetic doses of (R,S)-Ket and (S)-Ket in CRPS patients (Goldberg et al., 2011, Sigtermans et al., 2009) and of (R,S)-Ket in BPD (Diazgranados et al., 2010) were unable to establish a relationship between Ket and norKet pharmacokinetics and clinical response. These studies were confined to the determination of the PK/PD of Ket and norKet and the results suggest that it may be necessary to expand the scope of these studies. We have recently reported the results of an expanded PK/response study following a single administration of (R,S)-Ket to patients suffering from BPD and major depressive disorder and demonstrated thatsingificant concentrations of the downstream metabolites of (R,S)-Ket are present in the plasma of these patients and that the concentrations of (R,S)-DHNK, 4c and 4e were associated with a positive response (Zarate et al., 2012).
Based on the initial data obtained in the treatment of depression, it is reasonable to assume that the Ket downstream metabolites play a role in the clinical activity of Ket. Thus, the objective of this study was to identify the CYP isoforms that contribute to the stereoselective and regiospecific formation of the HNK metabolites. The results demonstrate that the key enzymes involved in this process are CYP2A6, 2B6, 2C19, 3A4 and 3A5. The data also indicate that CYP2A6 and CYP3A5 play a previously unrecognized role in the formation the HNK metabolites 4a and 4b, respectively, via the formation of the HK metabolites 5a and 5b. While it has been that the direct hydroxylation of Ket occurs to a “marginal extent” (Portmann, et al., 2010) the presence of 4b in the HLM preparation incubates of (R,S)-Ket but not in the corresponding (R,S)-norKet incubates indicate that the hydroxylation of Ket followed by the N-demethylation of the resulting HK metabolite makes a significant contribution to the overall metabolism of Ket. This is supported by the presence of significant concentrations of 4b in the plasma of CRPS patients (Fig 5), inpatients with BPD (Zhao et al., 2012) and in patients receiving ket for the treatment of BPD and major depressive disorder (Diazgranados et al., 2010).
In our studies of the use of subanesthetic doses of (R,S)-Ket for the treatment of CRPS and depression (Goldberg et al, 2010; Goldberg et al., 2011; Zhao et al., 2012), we have found a wide variation in the plasma concentrations of the (R,S)-Ket downstream metabolites, in which is highlighted by the data from the CRPS patients P17 and P18 (Fig. 5). One potential explanation of these observations is genetic differences in the highly polymorphic CYP2A6, CYP2B6, CYP2C19 and CYP3A5, all of which exist as many alleles and allelic subvariants that affect enzymatic activity (Human Cytochrome P450 (CYP) Allele Nomenclature Committee: www.cypalleles.ki.se/). This suggests that pre-treatment determination of metabolic genotype may help optimize clinical treatment with (R,S)-Ket. Retrospective and prospective studies addressing this issue are in progress and the data will be reported elsewhere. However, the CYP genotype does not always predict the corresponding expressed phenotype as the functional expression of the CYP expression is affected by disease states, e.g. type 2 diabetes (Matzke et al., 2000; Wang et al., 2003), inflammation (Renton, 2005), nutritional status (O’Neill et al., 1997; Williams and Wainer, 2002) and aging (Tanaka, 1998; Serpia et al., 2010). Indeed, we have recently demonstrated that protein malnourishment in rats decreased the metabolic clearance of (R)- and (S)-norKet and (R)- and (S)-DHNK but did not significantly affect the clearance of (R)- or (S)-Ket (Williams et al., 2004).
An addition potential source of the metabolite pattern observed in patients P17 and P18 is metabolic drug-drug interactions. A retrospective examination of the clinical records of these patients revealed that 12 medications were co-administered to P17 and 13 to P18 during the course of their treatment with (R,S)-Ket (Supplemental Material, Table S2) A review of the CYPs involved in the metabolism of these agents using P450 Drug Interaction Table (http://medicine.iupui.edu/clinpharm/ddis/table.aspx) and Drugbank (http://www.drugbank.ca/; Knox et al., 2011) indicated multiple potential sources of the induction or inhibition of (R,S)-Ket metabolism. This situation is common in the treatment of CRPS patients as a retrospective chart review of the 16 CRPS patients studied in our initial pharmacokinetic/pharmacodynamic study of (R,S)-Ket (Goldberg et al, 2011a) indicated that these patients received 73 different concomitant medications at different dosages along with their (R,S)-Ket infusion; i.e., an average of 9.4 medications per individual (Supplemental Data, Table S3). Metabolic drug interactions involving (R,S)-Ket and (S)-Ket have previously been reported involving medetomidine (Kharasch et al., 1992), dexmedeomidine (Nama et al., 2010) and rifampicin (Noppers et al., 2011) and Ket itself has been shown to induce CYPs in rats (Chan et al., 2005; Chen and Chen, 2010). As the use of (R,S)-Ket increases in the management of postoperative and perioperative pain and in opioid tolerant patients (Loftus et al., 2010; Angst and Clark, 2010) and in emergency room treatments (Lester et al., 2010) it is likely that the reported interactions will also increase.
The data from the rapidly expanding clinical use of (R,S)-Ket and (S)-Ket and the associated laboratory studies examining the pharmacological effects and mechanisms of the drug suggest that it may be necessary to expand the scope of these studies to include other primary and secondary metabolites and the metabolic transformation associated with their formation. This knowledge will help optimize and individualize the use of Ket, even though the pharmacological activities and clinical relevance of the downstream metabolites have not been definitively established.
Supplementary Material
Abbreviations
- Ket
Ketamine
- norKet
norketamine
- DHNK
dehydronorketamine
- HK
hydroxyketamine
- 5a
(2S,6S;2R,6R)-HK
- 5b
(2S,6R;2R,6S)-HK
- HNK
hydroxynorketamine
- 4a
(2S,6S;2R,6R)-HNK
- 4b
(2S,6R;2R,6S)-HNK
- 4c
(2S,5S;2R,5R)-HNK
- 4d
(2S,4S;2R,4R)-HNK
- 4e
(2S,4R;2R,4S)-HNK
- 4f
(2S,5R;2R,5S)-HNK
- CRPS
complex regional pain syndrome
- BPD
treatment-resistant bipolar depression
- HLM
characterized human liver microsomes
References
- Adams JD, Jr, Baillie TA, Trevor AJ, Castagnoli N. Studies on the biotransformation of ketamine 1-Identification of metabolites produced in vitro from rat liver microsomal preparations. Biomed Mass Spec. 1981;8:527–538. doi: 10.1002/bms.1200081103. [DOI] [PubMed] [Google Scholar]
- Angst MS, Clark JD. Ketamine for managing perioperative pain in opioid-dependent patients with chronic pain: a unique indication? Anesthesiology. 2010;113:514–551. doi: 10.1097/ALN.0b013e3181e9092d. [DOI] [PubMed] [Google Scholar]
- Bolze S, Boulieu R. HPLC determination of ketamine, norketamine and dehydronorketamine in plasma with a high-purity reversed-phase sorbent. Clin Chem. 1998;44:560–564. [PubMed] [Google Scholar]
- Chan WH, Sun WZ, Ueng TH. Induction of rat hepatic cytochrome P-450 by ketamine and its toxicological implications. J Toxicol Environ Health A. 2005;68:1581–1597. doi: 10.1080/15287390590967522. [DOI] [PubMed] [Google Scholar]
- Chen JT, Chen RM. Mechanisms of ketamine-involved regulation of cytochrome P450 gene expression. Expert Opin Drug Metab Toxicol. 2010;6:273–281. doi: 10.1517/17425250903505108. [DOI] [PubMed] [Google Scholar]
- Diazgranados N, Ibraham L, Brutsche NE, Newberg A, Kronstein P, Khalife S, Kammerer WA, Quezado Z, Luckenbaugh DA, Salvadore G, Machado-Vieira R, Manji KM, Zarate CA. A randomized add-on trial of an N-methyl-D-asparate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry. 2010;67:793–801. doi: 10.1001/archgenpsychiatry.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domino EF. Taming the ketamine tiger. Anesthesiology. 2010;113:678–686. doi: 10.1097/ALN.0b013e3181ed09a2. [DOI] [PubMed] [Google Scholar]
- Goldberg ME, Torjman MC, Schwartzman RJ, Mager DE, Wainer IW. Pharmacodynamic profiles of ketamine (R)-(−)- and (S)-(+) with 5 day inpatient infusion for the treatment of complex regional pain syndrome. Pain Phys. 2010;13:379–387. [PMC free article] [PubMed] [Google Scholar]
- Goldberg ME, Torjman MC, Schwartzman RJ, Mager DE, Wainer IW. Enantioselective pharmacokinetics of (R)- and (S)-ketamine after a 5-day infusion in patients with complex regional pain syndrome. Chirality. 2011;23:138–143. doi: 10.1002/chir.20890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijazi Y, Boulieu R. Contribution of CYP3A4, CYP2B6, and CYP2C9 isoforms to N-demethylation of ketamine in human liver microsomes. Drug Metab Dispos. 2002;30:853–858. doi: 10.1124/dmd.30.7.853. [DOI] [PubMed] [Google Scholar]
- Kharasch ED, Labroo R. Metabolism of ketamine stereoisomers by human liver microsomes. Anesthesiology. 1992;77:1201–1207. doi: 10.1097/00000542-199212000-00022. [DOI] [PubMed] [Google Scholar]
- Kharasch ED, Herrmann S, Labroo R. Ketamine as a probe of medetomidine stereoisomer inhibition of human liver microsomal drug metabolism. Anesthesiology. 1992;77:1208–1214. doi: 10.1097/00000542-199212000-00023. [DOI] [PubMed] [Google Scholar]
- Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C, Neveu V, Djoumbou Y, Eisner R, Guo AC, Wishart DS. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2011;39(Database issue):D1035–41. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester L, Braude DA, Niles C, Crandall CS. Low-dose ketamine for analgesia in the ED: a retrospective case series. Am J Emerg Med. 2010;28:820–827. doi: 10.1016/j.ajem.2009.07.023. [DOI] [PubMed] [Google Scholar]
- Leung LY, Baillie TA. Comparative pharmacology in the rat of ketamine and its two principal metabolites, norketamine and (Z)-6-hydroxynorketamine. J Med Chem. 1986;29:2396–2399. doi: 10.1021/jm00161a043. [DOI] [PubMed] [Google Scholar]
- Lester L, Braude DA, Niles C, Crandall CS. Low-dose ketamine for analgesia in the ED: a retrospective case series. Am J Emerg Med. 2010;28:820–827. doi: 10.1016/j.ajem.2009.07.023. [DOI] [PubMed] [Google Scholar]
- Loftus RW, Yeager MP, Clark JA, Brown JR, Abdu WA, Sengupta DK, Beach ML. Intraoperative ketamine reduces perioperative opiate consumption in opiate-dependent patients with chronic back pain undergoing back surgery. Anesthesiology. 2010;113:639–646. doi: 10.1097/ALN.0b013e3181e90914. [DOI] [PubMed] [Google Scholar]
- Matzke G, Frye RF, Early JJ, Straka RJ, Carson SW. Evaluation of the influence of diabetes mellitus on antipyrine metabolism and CYP1A2 and CYP2D6 activity. Pharmacotherapy. 2000;20:182–190. doi: 10.1592/phco.20.3.182.34775. [DOI] [PubMed] [Google Scholar]
- Moaddel R, Venkata SLV, Tanga MJ, Bupp JE, Green CE, LaIyer L, Furimsky A, Goldberg ME, Torjman MC, Wainer IW. A parallel chiral-achiral liquid chromatographic method for the determination of the stereoisomers of ketamine and ketamine metabolites in the plasma and urine of patients with complex regional pain syndrome. Talanta. 2010;82:1892–1904. doi: 10.1016/j.talanta.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nama S, Meenan DR, Fritz WT. The use of sub-anesthetic Intravenous ketamine and adjuvant dexmedetomidine when treating acute pain from CRPS. Pain Phys. 2010;13:365–368. [PubMed] [Google Scholar]
- Noppers I, Olofsen E, Niesters M, Aarts L, Mooren R, Dahan A, Kharasch E, Sarton E. Effect of rifampicin on S-ketamine and S-norketamine plasma concentrations in healthy volunteers after intravenous S-ketamine administration. Anesthesiology. 2011;114:1435–1445. doi: 10.1097/ALN.0b013e318218a881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill WM, Gilfix BM, DiGirolamo A, Tsoukas CM, Wainer IW. N-Acetylation among HIV positive and patients with AIDS: When is fast, fast and slow, slow? Clin Pharmacol Ther. 1997;62:261–271. doi: 10.1016/S0009-9236(97)90028-X. [DOI] [PubMed] [Google Scholar]
- Portmann S, Kwan HT, Theurillat R, Schmitz A, Mevissen M, Thormann W. Enantioselective capillary electrophoresis for the identification and characterization of human cytochrome P450 enzymes which metabolize ketamine and norketamine in vitro. J Chromatog A. 2010;1217:7942–7948. doi: 10.1016/j.chroma.2010.06.028. [DOI] [PubMed] [Google Scholar]
- Renton KW. Regulation of drug metabolism and disposition during inflammation and infection. Expert Opin Drug Metab Toxicol. 2005;1:629–640. doi: 10.1517/17425255.1.4.629. [DOI] [PubMed] [Google Scholar]
- Sabia M, Hirsh RA, Torjman MC, Wainer IW, Cooper N, Domsky R, Goldberg ME. Advances in translational neuropathic research: Example of enantioselective pharmacokinetic-pharmacodynamic modeling of ketamine-induced pain relief in complex regional pain syndrome. Curr Pain Headache Rep. 2011;15:207–14. doi: 10.1007/s11916-011-0185-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seripa D, Pilotto A, Penzaz F, Matera MG, Pilotto A. Pharmacogenetics of cytochrome P450 (CYP) in the elderly. Ageing Res Rev. 2010;9:457–474. doi: 10.1016/j.arr.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Sigtermans M, Dahan A, Mooren R, Bauer M, Kest B, Sarton E, Olofson E. S(+)-Ketamine effect on experimental pain and cardiac output. Anesthesiology. 2009;111:892–903. doi: 10.1097/ALN.0b013e3181b437b1. [DOI] [PubMed] [Google Scholar]
- Tanaka E. In vivo age-related changes in hepatic drug-oxidizing capacity in humans. J Clin Pharm Ther. 1998;23:247–255. doi: 10.1046/j.1365-2710.1998.00164.x. [DOI] [PubMed] [Google Scholar]
- Trevor AJ, Woolf TF, Baillie TA, Adams JD, Castagnoli N. Stereoselective metabolism of ketamine enantiomers. In: Kamenka JM, Domino EF, Geneste P, editors. Phencyclidine and Related Arylcyclohexylamines, Present and Future Applications. Ann Arbor, MI: NPP Books; 1983. pp. 279–289. [Google Scholar]
- Turfus SC, Parkin MC, Cowan DA, Halket JM, Smith NW, Braithwaite RA, Elliot SP, Steventon GB, Kicman AT. Use of human microsomes and deuterated substrates; an alternative approach for the identification of novel metabolites of ketamine by mass spectrometry. Drug Metab Dispos. 2009;37:1769–1778. doi: 10.1124/dmd.108.026328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Hall SD, Maya JF, Li L, Asghar A, Gorski JC. Diabetes mellitus increases the in vivo activity of cytochrome P450 2E1 in humans. J Clin Pharmacol. 2003;55:77–85. doi: 10.1046/j.1365-2125.2003.01731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ML, Wainer IW. Genotype/phenotype comparisons: A probe for the effect of disease progression on drug metabolism. Current Opin Drug Discov Devel. 2002;5:144–149. [PubMed] [Google Scholar]
- Williams ML, Mager DE, Parenteau H, Gudi G, Tracy T, Mulheran M, Wainer IW. Effects of protein calorie malnutrition on the pharmacokinetics of ketamine in rats. Drug Metab Dispos. 2004;32:786–793. doi: 10.1124/dmd.32.8.786. [DOI] [PubMed] [Google Scholar]
- Woolf TF, Adams JD. Biotransformation of ketamine, (Z)-6-hydroxyketamine, and (E)-6-hydroxyketamine by rat, rabbit, and human liver microsomal preparations. Xenobiotica. 1987;17:839–847. doi: 10.3109/00498258709043993. [DOI] [PubMed] [Google Scholar]
- Zhao X, Venkata SLV, Moaddel R, Luckenbaugh DA, Brutsche NE, Ibrahim L, Zarate CA, Jr, Mager DE, Wainer IW. Simultaneous Population Pharmacokinetic Modeling of Ketamine and Three Major Metabolites in Patients with Treatment-Resistant Bipolar Depression. Br J Clin Pharmacol. 2012 doi: 10.1111/j.1365-2125.2012.04198.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Jr, Brutsche N, Laje G, Luckenbaugh DA, Venkata SLV, Ramamoorthy A, Moaddel R, Wainer IW. Relationship of Ketamine’s Plasma Metabolites with Response, Diagnosis, and Side Effects in Major Depression. Biological Psychiatry. 2012 doi: 10.1016/j.biopsych.2012.03.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.