

Abstract
We investigated genetic factors that govern the reduced propiconazole sensitivity of Sclerotinia homoeocarpa field isolates collected during a 2-year field efficacy study on dollar spot disease of turf in five New England sites. These isolates displayed a >50-fold range of in vitro sensitivity to a sterol demethylation inhibitor (DMI) fungicide, propiconazole, making them ideal for investigations of genetic mechanisms of reduced DMI sensitivity. The CYP51 gene homolog in S. homoeocarpa (ShCYP51B), encoding the enzyme target of DMIs, is likely a minor genetic factor for reduced propiconazole sensitivity, since there were no differences in constitutive relative expression (RE) values and only 2-fold-higher induced RE values for insensitive than for sensitive isolate groups. Next, we mined RNA-Seq transcriptome data for additional genetic factors and found evidence for the overexpression of a homolog of Botrytis cinerea atrD (BcatrD), ShatrD, a known efflux transporter of DMI fungicides. The ShatrD gene showed much higher constitutive and induced RE values for insensitive isolates. Several polymorphisms were found upstream of ShatrD but were not definitively linked to overexpression. The screening of constitutive RE values of ShCYP51B and ShatrD in isolates from two golf courses that exhibited practical field resistance to propiconazole uncovered evidence for significant population-specific overexpression of both genes. However, linear regression demonstrated that the RE of ShatrD displays a more significant relationship with propiconazole sensitivity than that of ShCYP51B. In summary, our results suggest that efflux is a major determinant of the reduced DMI sensitivity of S. homoeocarpa genotypes in New England, which may have implications for the emergence of practical field resistance in this important turfgrass pathogen.
INTRODUCTION
Dollar spot, caused by the ascomycete fungus Sclerotinia homoeocarpa (F. T. Bennett), is the most common and economically important disease of cool-season turfgrasses and, if left untreated, can lead to severe pitting of turfgrass swards (6, 41). Disease management of turf for dollar spot requires high inputs of fungicides from several chemical classes, and the sterol DMI (demethylation inhibitor) class is commonly used for preventative and curative control (14). Unfortunately, recent investigations indicate that field isolates of S. homoeocarpa have begun to exhibit decreased sensitivity to this fungicide class and others in the United States (14, 27, 32, 33).
Several recent publications reported the reduced field efficacy of the active DMI ingredient propiconazole for controlling dollar spot as well as the reduced propiconazole sensitivity of S. homoeocarpa field isolates via in vitro growth assays (15, 32, 33). Most recently, Popko et al. investigated the association of propiconazole field efficacy and the in vitro sensitivity of S. homoeocarpa isolates recovered from turf of five New England sites (four with previous DMI exposure and one baseline site) (32). That study demonstrated that practical field resistance was correlated with the presence of field isolates showing reduced in vitro sensitivity. The isolates collected by Popko et al. were sampled before and 7 days after the application of 0.44 kg propiconazole per hectare, a rate which is labeled to control dollar spot for 14 to 21 days (32). Thus, isolates of S. homoeocarpa sampled during the control period (i.e., 7 days after application) represent field isolates capable of causing practical field resistance. Fortunately, the isolates recovered by Popko et al. were kept in long-term storage and are ideal subject material for determining the molecular mechanisms behind the reduced propiconazole sensitivity and practical field resistance to DMIs of S. homoeocarpa (32).
Like other DMI fungicides, propiconazole inhibits fungal sterol synthesis by binding to the heme iron of CYP51, the 14-alpha demethylase enzyme which catalyzes the third step in ergosterol biosynthesis (the methylation of lanosterol to 4,4 dimethyl-8,14,24 trienol). Accordingly, several nonsynonymous point mutations in the gene sequence of CYP51 have been linked to decreased sensitivity to DMI fungicides in diverse groups of fungi (8, 9, 37). Moreover, the overexpression of the CYP51 gene and its paralogs has also been linked to reduced DMI sensitivity in many fungal species (11, 21, 23, 24). Another frequently reported mechanism contributing to reduced DMI sensitivity is fungicide efflux through the action of energy-dependent ABC (ATP binding cassette) and MFS (major facilitator superfamily) transporters (2, 12, 26, 29). These membrane-bound efflux transporters are present in large gene families in the genomes of a plethora of evolutionarily diverse fungi, resulting from gene duplication events that have likely allowed for their enrichment and divergence in substrate specificities (18, 19).
In this study, we utilized de novo genomics and transcriptomics as well as molecular biology tools to glean insight into potential mechanisms underlying the reduced DMI sensitivity and practical field resistance of S. homoeocarpa. Given the abundant information regarding CYP51-involved mechanisms for reduced DMI fungicide sensitivity, we first sought to test the hypothesis that coding and promoter mutations and/or the gene expression of CYP51 in S. homoeocarpa (ShCYP51B) could explain the decreased sensitivity to propiconazole of a panel of eight field isolates recovered from a pretreatment sampling of the above-mentioned five field sites described previously by Popko et al. (32). The second objective was to assay the gene expression of the ABC transporter homolog of Botrytis cinerea atrD (BcatrD) in S. homoeocarpa (ShatrD), which was mined from RNA-Seq transcriptome data. This functionally characterized gene is known to efflux DMI fungicides in B. cinerea and thus serves as a candidate determinant of the reduced DMI fungicide sensitivity of S. homoeocarpa (12). Our final objective was to test the hypothesis that the overexpression of the ShCYP51B and ShatrD genes is associated with New England field isolates exhibiting practical field resistance (32).
MATERIALS AND METHODS
Isolate selection and EC50 characterization.
Isolates were cultured from individual symptomatic leaf blades of dollar spot infection centers from five sites (four golf course sites and the University of Massachusetts Joseph Troll Turf Research Center), as described previously by Popko et al. (32). The first panel of eight isolates screened, here referred to as the “initial panel,” was chosen from an initial sampling of the five sites immediately prior to the first fungicide application described previously by Popko et al. (32). The in vitro propiconazole sensitivity was determined by the RMG (relative mycelial growth) percentage (RMG%) according to procedures described previously by Popko et al. (32). In brief, axenic cultures of isolates were grown on PDA (potato dextrose agar) (Difco Laboratories, Detroit, MI) for 2 to 3 days, and one 5-mm agar plug was transferred from the colony edge to the center of a propiconazole-amended PDA (0.1 μg ml−1) petri plate and a nonamended PDA petri plate, both in duplicate. Forty-eight hours after transfer, three radial points approximately 120° apart on the circumference of the actively growing colony edge were measured with 16EX digital calipers (Mahr, Göttingen, Germany). The average radial mycelial growth (in millimeters) on propiconazole-amended PDA was divided by the average nonamended radial mycelial growth and multiplied by 100 to give the RMG%. Isolates in the initial panel were referred to as being propiconazole “sensitive” if their RMG was <50%, and they were considered “insensitive” if it was >50% (Table 1). The rationale for these sensitivity designations stems from data reported previously by Popko et al., who reported that isolates with >50% RMG are strongly associated with practical field resistance of S. homoeocarpa (32). We stress that since the initial panel was sampled prior to treatment, isolates in the initial panel are not confirmed to exhibit practical field resistance. However, given the strong association of the >50% RMG value with practical field resistance determined previously by Popko et al., 50% RMG was used as a cutoff for the qualitative grouping of isolates as being propiconazole sensitive or insensitive (32).
Table 1.
Sclerotinia homoeocarpa field isolates used in this study
| Isolatea | Sample timeb | Propiconazole EC50 | Propiconazole sensitivityc | Microsatellite genotyped |
|---|---|---|---|---|
| WBI7 | Initial | 0.448 | I | 1 |
| HRS10 | Initial | 0.018 | S | 2 |
| HRI11 | Initial | 0.839 | I | 2 |
| SMS23 | Initial | 0.130 | S | 2 |
| SMI27 | Initial | 0.258 | I | 2 |
| JTS30 | Initial | 0.016 | S | 2 |
| HFS35 | Initial | 0.032 | S | 3 |
| HFI40 | Initial | 0.500 | I | 1 |
| HRS1 | 7 DAT(C) | 0.013 | S | 2 |
| HRS2 | 7 DAT(C) | 0.008 | S | 2 |
| HRS3 | 7 DAT(C) | 0.111 | S | 2 |
| HRS4 | 7 DAT(C) | 0.020 | S | 2 |
| HRI1 | 7 DAT(T) | 0.683 | I | 2 |
| HRI2 | 7 DAT(T) | 0.516 | I | 2 |
| HRI3 | 7 DAT(T) | 0.783 | I | 2 |
| HRI4 | 7 DAT(T) | 0.830 | I | 2 |
| HFS1 | 7 DAT(C) | 0.003 | S | 3 |
| HFS2 | 7 DAT(C) | 0.014 | S | 3 |
| HFS3 | 7 DAT(C) | 0.015 | S | 3 |
| HFS4 | 7 DAT(C) | 0.009 | S | 3 |
| HFI1 | 7 DAT(T) | 0.410 | I | 1 |
| HFI2 | 7 DAT(T) | 0.390 | I | 1 |
| HFI3 | 7 DAT(T) | 0.492 | I | 1 |
| HFI4 | 7 DAT(T) | 0.771 | I | 1 |
Isolates are named as follows: location abbreviation (see Materials and Methods), followed by “I” or “S,” for propiconazole insensitive or sensitive, respectively, and the isolate collection number.
Initial sampling was done in 2009 before the initiation of the field experiment described previously by Popko et al. (32). 7 DAT(C) indicates that isolates were sampled from untreated plots, and 7 DAT(T) indicates that isolates were sampled from plots 7 days after propiconazole treatment.
Propiconazole sensitivity determined by RMG% at a propiconazole concentration of 0.1 μg/ml (I is >50% RMG, and S is <50% RMG) (32).
Multilocus genotypes were determined from seven microsatellites. Isolates of each genotype shared all alleles.
The eight isolates from the initial panel, which originated from five sites, and two additional sets of eight isolates, which originated from two of the five sites, were characterized in this study. The isolates were named as follows: location abbreviation (Hartford Golf Club [HF], Hickory Ridge Country Club [HR], Wintonbury Hills Golf Club [WB], Shuttle Meadow Country Club [SM], and University of Massachusetts Joseph Troll Turf Research Center [JT]), followed by “I” or “S” for a designation of propiconazole insensitive or sensitive, respectively, and the isolate collection number (Table 1). The latter two additional sets of isolates included four isolates observed to exhibit practical field resistance to propiconazole (termed “PFR isolates”) and four sensitive isolates sampled simultaneously from untreated control plots (total of eight isolates). These two sets were recovered from two golf course populations of S. homoeocarpa from Hartford Golf Club and Hickory Ridge Country Club (Table 1) (32). Sensitive isolates HFS1 to HFS4 from Hartford Golf Club and sensitive isolates HRS1 to HRS4 from Hickory Ridge Country Club were sampled from plots that received no fungicide treatment. Isolates HFI1 to HFI4 and HRI1 to HRI4 were sampled 7 days after the application of 0.44 kg propiconazole per hectare (in the form of Banner Maxx 1.3 ME fungicide; Syngenta Crop Protection, Greensboro, NC) and are thus considered to exhibit practical field resistance (32) (Table 1). All isolates were maintained in long-term storage according to methods described previously by Chakraborty et al. and grown out on PDA plates for each experiment (3).
The mean effective concentration for inhibiting mycelial growth by 50% (EC50) (expressed in μg propiconazole ml−1) for all isolates was calculated according to methods described previously by Jo et al. (14). The relative mycelial growth percentage was calculated by using the following concentrations: 0.001, 0.01, 0.1, and 1.0 μg propiconazole ml−1 (as described above). The EC50 for each isolate was estimated by using a linear regression of the RMG% value for the log10-transformed fungicide concentration using PROC REG (SAS v. 9.1.3; SAS Institute Inc., Cary, NC). The EC50s for all isolates are presented in Table 1.
DNA extraction, PCR, and DNA sequencing.
The extraction of DNA was performed by modifying a method described previously by Saitoh et al. (36). Isolates were grown on PDA for 5 to 6 days, and ∼200 mg of mycelium was scraped from the PDA surface by using a sterile pestle. The mycelium was treated in subsequent steps according to the method described previously by Saitoh et al. to yield nucleic acids suitable for PCR and DNA sequencing (36).
All primers for PCR, QPCR, and DNA sequencing were designed using Primer3 (35). The PCR primers for the amplification and sequencing of genomic DNA were designed with the following parameter settings for generating primers: primer length minimum of 20 bases, optimum of 25 bases, and maximum of 30 bases and annealing temperature minimum of 62°C, optimum of 67°C, and maximum of 72°C. Three primer sets were designed to amplify and sequence the ShCYP51B gene from the initial panel of eight S. homoeocarpa isolates obtained from the initial sampling of the five sites described previously by Popko et al. (32) (see Table S1 in the supplemental material). For ShatrD, primers were designed from RNA-Seq data (see below) to amplify the coding region of the gene (4,506 bp) from cDNA (see below for cDNA preparation), and additional primers were designed for the sequencing of the internal regions with Primer3. Primers for quantitative PCR (QPCR) were designed by using the Integrated DNA Technologies QPCR primer design Web tool to amplify 100- to 150-bp amplicons from the actin gene of S. homoeocarpa (Shact), ShCYP51B, and ShatrD. Two primer sets were designed to amplify and sequence the 1,000-bp region upstream of the ShCYP15B and ShatrD genes as described above. All amplicons were amplified by using the PCR protocol described below for microsatellites, with the exception of the ShatrD gene, and only primers that amplified a single band were utilized for PCR, DNA sequencing, and QPCR assays. The PCR temperature profile used to amplify ShatrD was identical for the microsatellite PCR described below, with the exception of an extension time of 1 min 15 s, and PCR volumes were 20 μl, with final concentrations of 1× Phusion HF buffer, 0.2 mM deoxynucleoside triphosphates (dNTPs), 0.5 μM each primer, ∼1 μg cDNA template, and 1.0 unit of Phusion DNA polymerase (New England BioLabs, Ipswich, MA). Amplification for all amplicons was confirmed by 1.0% (wt/vol) gel electrophoresis in 1× TBE (Tris base, boric acid, and EDTA) buffer at 100 V for 1 h with ethidium bromide poststaining (0.5 μl/ml) for 30 min, followed by 10 min of destaining in 1× TBE buffer and UV visualization. Amplicons were purified prior to sequencing by using ExoSAP-IT PCR cleanup reagents (Affymetrix, Santa Clara, CA). For sequencing, amplicons were submitted to the Genomics Resource Laboratory at the University of Massachusetts, Amherst, MA.
Upstream sequence analysis.
Sequences of the upstream regions of ShCYP51B and ShatrD were assembled and aligned with MEGA v5 using a Clustal alignment and analyzed for polymorphisms (39). The sequences were next searched for promoters by using NNPP (Neural Network Promoter Prediction) software with default settings, which can be found at the Berkeley Drosophila Genome Project website (http://www.fruitfly.org/seq_tools/promoter.html) (34).
RNA extraction and cDNA synthesis.
Total RNA was extracted from isolates by using TRIzol reagent in a manner consistent with methods described previously by Venu et al. (40). Briefly, isolates were grown for 72 h in 25 ml of half-strength PDB (potato dextrose broth) (Difco Laboratories, Detroit, MI) in 50-ml Falcon tubes (BD Biosciences, San Jose, CA) at 23°C. After 72 h, propiconazole (in the form of Banner Maxx 1.3ME) was added to fungicide-treated tubes at 0.1 μg ml−1 PDB, and an equal volume of water was added to untreated tubes. The tubes were lightly agitated on a bench-top shaker for 1 h. The mycelium was harvested by using vacuum filtration, placed into 2.0-ml screw-cap tubes, and immediately frozen in liquid nitrogen. The frozen mycelium was stored in a −80°C freezer until further processing. For RNA extraction, mycelia were removed from the freezer, and 1 ml of TRIzol reagent was added to the frozen samples. The samples were immediately placed into a tissue disrupter for 30 s and allowed to incubate at 23°C for 5 min. Next, 200 μl of chloroform was added to the samples, which were shaken vigorously for 15 s and allowed to incubate at 23°C for 4 min. Samples were then centrifuged at 7,000 × g for 10 min at 4°C, and the aqueous layer was removed. Total nucleic acids were precipitated in a 1.2 M NaCl–0.8 M Na3C6H5O7 solution and isopropanol at 4°C for 5 min. The nucleic acids were pelleted by centrifugation at 7,000 × g for 10 min at 4°C. Pellets were air dried for 5 min and suspended in 60 μl RNase-free water at 60°C for 10 min. The total RNA was treated with DNase, and cDNA was synthesized from RNA with the QuantiTect reverse transcription kit (Qiagen Inc., Valencia, CA).
Genome sequencing and bioinformatics.
Two isolates were chosen for Illumina HiSeq2000 and 454 GS FLX Titanium transcriptome sequencing, HRS10 and HRI11, which are putative clonal genotypes, as determined by seven microsatellites (see below), but differ in propiconazole EC50s by >50-fold (Table 1; see also Fig. S1 in the supplemental material). For 454 GS FLX Titanium sequencing, total RNA was extracted from one biological replicate of untreated samples (as described above) of HRS10 and HRI11 with the Invitrogen RNA minikit with TRIzol reagent. For Illumina HiSeq2000 sequencing, total RNA was extracted from one biological replicate of fungicide-treated and untreated samples (treatments were the same as those described above) of HRS10 and HRI11, also using the Invitrogen RNA minikit with TRIzol reagent. For both Illumina and 454 sequencing, RNAs were precipitated as described above and sent to Macrogen Inc. (Seoul, South Korea) for cDNA library preparation and sequencing. Briefly, RNA samples were treated with DNase and sheared, and cDNA libraries were prepared for sequencing from the total sheared RNA. Following sequencing, low-quality data were removed, and 454 data were assembled with the GS De Novo Assembler (Newbler v. 2.5.3). Low-quality data from the Illumina HiSeq2000 data were also filtered and assembled along with the assembled 454 GS FLX Titanium contigs to give a hybrid assembly using CLC Genomics Workbench with default settings (CLC Bio, Aarhus, Denmark). An RNA-Seq analysis was next performed with CLC Genomics Workbench by the mapping of >30 million paired-end reads of Illumina data for each sample back to the hybrid transcriptome assembly and calculating the RPKM (reads per kilobase of exon model per million mapped reads) (28).
Genomic sequences were also generated by using 454 GS FLX Titanium sequencing to up to an 8× depth for S. homoeocarpa isolate JTS30 (Table 1) (Macrogen Inc.). Genomic DNA was extracted as described above and shipped to Macrogen Inc. in January 2009. Low-quality data were removed, and the remaining data were assembled by using GS De Novo Assembler (Newbler v 2.5.3).
BLASTX searches of the assembled contigs from the genomic and transcriptomic data against the NCBI nonredundant protein database were performed by using Blast2GO software (August 2011) (4).
Microsatellite genotyping.
All isolates were genotyped by using seven genomic microsatellite primer pairs, which were designed from the partial genome sequence of S. homoeocarpa isolate JTS30 (see Table S1 in the supplemental material). To locate the microsatellites, the genomic contigs were searched by using SSRIT (Simple Sequence Repeat Identification Tool) (http://www.gramene.org/db/markers/ssrtool). Search parameters specified an octamer as the maximum motif length and the minimum number of repeats to be 10. Forward and reverse PCR primers flanking the repeat sequences were designed by using the Primer3 primer design server (35). The parameter settings used to generate primer sequences were as follows: primer length minimum of 18 bases, optimum of 22 bases, and maximum of 25 bases and annealing temperature minimum of 55°C, optimum of 60°C, and maximum of 65°C. Primers were designed to amplify products ranging from 100 to 250 bp. Primers were screened by using DNAs extracted from the complete panel of 23 isolates described previously by Jo et al., which represent a collection from diverse locations across the eastern United States (16). Primers were selected for microsatellite genotyping if they amplified a single amplicon and were found to display polymorphisms among the 23 isolates of Jo et al. (16).
Microsatellites were amplified in a 10-μl reaction mixture volume with final concentrations of 1× PCR buffer, 0.2 mM dNTP with 2.5 nM MgCl2, 0.2 μM each primer, 1 unit Taq DNA polymerase (New England BioLabs, Ipswich, MA), and ∼200 ng of genomic DNA template. The PCR regimen was as follows: an initial denaturation step at 95°C for 60 s, followed by 35 cycles of denaturation (at 95°C for 30 s) and annealing (at 72°C for 1 min, decreasing at increments of 1.0°C per cycle for the first eight cycles, and at 65°C for the remaining 27 cycles), with a final extension step at 72°C for 2 min. The PCRs were performed in 96-well plates on an MJ PTC-200 thermocycler (MJ Research, Watertown, MA), with one reaction per primer pair without a template as a negative control. Eight microliters of the PCR products and 2 μl of 6× loading dye per reaction were mixed, and amplicons were separated by gel electrophoresis on 3% (wt/vol) MetaPhor agarose (Cambrex Bio Science Rockland Inc., Charles City, IA) gels in 1× TBE buffer at 160 V for 2.5 h. Gels were poststained with ethidium bromide (0.5 μl/ml) for 30 min, followed by 10 min of destaining in 1× TBE buffer. Gel bands were visualized under UV light and photographed, and gel pictures were captured as TIFF files. Allelic data from the seven microsatellites were coded into a binary data matrix and used to construct a pairwise distance matrix in GeneAlEx (31). This distance matrix was exported and used to build an unweighted-pair group method using average linkages (UPGMA) dendrogram in MEGA v5 (39).
Phylogenetic analyses.
A phylogenetic analysis was conducted with the CYP51 amino acid sequence from S. homoeocarpa (ShCYP51B) and a subset of CYP51 amino acid sequences from five fungal species that have genomes encoding one to three CYP51 gene paralogs (CYP51A, CYP51B, and CYP51C) described previously by Becher et al. (1). Additionally, CYP51B sequences from three representatives of the Sclerotiniaceae (Sclerotinia sclerotiorum, B. cinerea, and Monilinia fructicola) were included, which are not known to encode multiple CYP51 gene paralogs. Amino acid sequences were aligned by the MAFFT server using automated method selection and a Blosum 62-amino-acid scoring matrix (17). The amino acid alignment was imported into MEGA v5 for neighbor joining phylogenetic analysis with the Poisson model, gamma-distributed rates among sites, a complete deletion of missing data, and 1,000 bootstrap replicates (39).
An additional phylogenetic analysis using the above-mentioned strategy was conducted for the ShatrD amino acid sequence and included amino acid sequences from the phylogeny of the G subfamily of ABC (ABC-G) transporters reported previously by Hayashi et al. (12), with the addition of the amino acid sequence of the ABC-G gene ABC1 from M. fructicola (23).
Quantitative PCR analyses.
Two quantitative PCR assays were designed and carried out by using real-time PCR technology with SYBR green reporter dye to examine the relative gene expression levels of ShCYP51B and ShatrD in S. homoeocarpa isolates. The Shact gene was chosen as a housekeeping gene, and primers were designed to amplify a portion of the actin gene overlapping that of M. fructicola (Mfactin) (GenBank accession number AF420305) used previously by Luo and Schnabel for quantitative reverse transcription (RT)-PCR (see Table S1 in the supplemental material) (23). Real-time PCR was performed with 25-μl reaction mixtures that included 12.5 μl of Absolute Blue SYBR QPCR MasterMix (Thermo Fisher Scientific, Waltham, MA), 1 μl of cDNA at 250 ng/μl, 1.75 μl of each primer at 1 μM to give a final primer concentration of 70 nM, and 8 μl microbiological-grade water (Thermo Fisher Scientific, Waltham, MA). Reactions were run with a Mastercycler Ep Realplex real-time thermocycler (Eppendorf, Hauppauge, NY) with the following PCR temperature regimen: 1 cycle of 15 min at 95°C and 40 cycles of 15 s at 95°C, 30 s at 60°C, and 30 s at 72°C. A final melt curve analysis was also included to ensure proper amplification, and gel electrophoresis and staining of RT-PCR amplicons confirmed the amplification of a single amplicon. Relative gene expression (RE) was calculated by the comparative threshold cycle (CT) method described previously by Livak and Schmittgen (22). For calculating the RE of each data set, the isolate with the highest mean ΔCT values between target and housekeeping genes was chosen as a calibrator for calculating RE. Two biological replicates were performed for each set of isolates/treatments, with three technical replicates per each biological replicate.
Statistical analysis of quantitative RT-PCR data.
Independent RE data sets for ShCYP51B and ShatrD were generated for the three sets of isolates: the initial panel of 8 isolates from five sites and the two additional sets of 8 isolates, one from Hickory Ridge Country Club and the other from Hartford Golf Club (Table 1) (32). Analysis of variance was conducted by using the JMP, version 9.0, software package on RE data for ShCYP51B and ShatrD from the initial panel and from the two sets to examine differences in RE levels between the sensitive and the insensitive groups of isolates and between the two biological replications of RE data (SAS Institute Inc., Cary, NC).
Linear regression analysis was performed with EC50s (x axis) and log10-transformed mean RE values (y axis) for ShatrD and ShCYP51B constitutive expression for all 24 isolates by using the JMP software package. Relative expression values for both biological replicates were combined for the linear regression, since no significant differences were observed between the biological replicates.
Nucleotide sequence accession numbers.
The assembled transcriptome contig sequences were submitted to the Transcriptome Shotgun Assembly Sequence Database (GenBank accession numbers JW813327 to JW839590), and assembled genome contig sequences were submitted to the Whole Genome Shotgun Sequence Database, deposited at DDBJ/EMBL/GenBank under the accession number AKKO00000000. The version described in this paper is the first version, AKKO01000000. The following gene and genomic sequences generated for this study were also submitted to GenBank: ShatrD, JQ612525; ShCYP51B, JN712909; Shact, JQ627642; ShCYPB upstream sequences, JX067995 and JX067994; and ShatrD upstream sequences, JX067993 and JX067992.
RESULTS
Characterization of isolates: microsatellite genotyping, EC50, and DNA sequencing.
Three hundred nine simple sequence repeats (SSRs) were identified, and 96 of these were selected for further screening. Following screening, seven primer pairs were selected, and microsatellite genotyping indicated the presence of 17 alleles with the seven microsatellites for 24 isolates. Three putative clonal genotypes of S. homoeocarpa were found and are indicated in Table 1. The UPGMA dendrogram of microsatellite data is presented in Fig. S1 in the supplemental material. Microsatellites SSR5848, SSR8045, SSR8400, and SSR30326 gave two alleles each and differentiated genotype 1 from genotypes 2 and 3. Microsatellites SSR20624, SSR8569, and SSR21791 gave three alleles each and differentiated the three genotypes from one another (Table 1). The propiconazole EC50s for these isolates ranged from 0.003 to 0.839 μg/ml and did not correlate with microsatellite genotypes (Table 1).
The ShCYP51B sequences of all isolates were identical, and upstream sequencing from the initial panel yielded one SNP (single-nucleotide polymorphism) at base pair position +590 for two insensitive isolates, WBI7 and HFI40, and for the four PFR isolates from Hartford Golf Club (HFI1 to HFI4). Upstream ShCYP51B sequences for all other isolates were identical. The above-mentioned six isolates with an SNP upstream of ShCYP51B also showed eight SNPs upstream of ShatrD at the following base pair positions: +17, +29, +490, +531, +561, +587, +826, and + 923. An insertion at position +385 and a deletion at position +367 were detected in these isolates as well. All other isolates had identical upstream ShatrD sequences. Promoter prediction with NNPP software found two putative promoters in ShCYP51B (the first from positions +165 to +116 and the second from positions +872 to +823); however, the SNP was not colocalized with either of the putative promoter regions (34). No putative promoters were detected in the 1,000-bp region upstream of ShatrD.
Genome and transcriptome sequencing and bioinformatics.
Illumina HiSeq 2000 sequence reads for each of four samples yielded a total of ∼160 million paired-end reads of Illumina sequence data (>30 million paired-end reads per sample library). 454 GS FLX Titanium sequencing of two libraries gave just over 100,000 reads for each of the two libraries. The hybrid transcriptome assembly was composed of 26,263 contigs over 500 bp in length. Assembled contigs were searched for the overexpression of candidate DMI resistance factors, and the BcatrD homolog in S. homoeocarpa was identified in the BLAST results by an expect (E) value of 0 and a query coverage of 99% (12). The hybrid transcriptome assembly provided coding sequences of the ShatrD gene, which was used for primer design, PCR, and DNA sequencing (see below) to confirm the full-length coding sequence of 4,506 bp (ShatrD). The ShatrD protein sequence shares 83% amino acid identity (1,246 out of 1,501 amino acids) with the BcatrD protein sequence (GenBank accession number CAC41639). Additionally, there are 92% positive amino acid matches (1,376 out of 1,501). Further support for the involvement of ShatrD in the reduced DMI sensitivity of S. homoeocarpa was found with the digital gene expression values, where we found >20-fold-higher RPKM values for HRI11 than for HRS10 for both the treated and untreated samples. The assembled transcriptome contigs provided the full-length coding sequence of the ShCYP51B gene, which was 1,569 bp in length, encoding 522 amino acids. The top BLASTX hit for the ShCYP51B coding sequence was CYP51 from M. fructicola (GenBank accession number ACF06196), with an E value of 0 and 100% query coverage. The ShCYP51B sequence shares 87% amino acid identity (454 out of 524 amino acids) and 93% positive amino acid matches (488 out of 524) with CYP51 from M. fructicola.
The assembled 454 GS FLX Titanium genome sequence data represented were composed of 31,623 contigs, each over 200 bp in size. The assembled genomic contigs provided the partial 5′ and 3′ genomic sequences of the ShCYP51B gene, which were used for primer design, PCR, and DNA sequencing to give the full-length genomic sequence. The genomic sequence totaled 1,680 bp in length. Following a Clustal alignment of ShCYP51B genomic and transcriptomic sequences with MEGA v5, two introns in the genomic sequences were identified, the first from bp 247 to 299 and the second beginning at bp 498 and ending at bp 555 (39).
A phylogenetic analysis showed clustering and high bootstrap values to confirm the similarity of the CYP51 gene of S. homoeocarpa with the sequences of CYP51B from Sclerotiniaceae taxa included in the analysis (see Fig. S2 in the supplemental material). Phylogenetic analysis of the ShatrD homolog displays its similarity with BcatrD from B. cinerea, as opposed to other ABC-G transporters of the Sclerotiniaceae (see Fig. S3 in the supplemental material). A sequence comparison of the N-terminal and C-terminal Walker A, ABC signature, and Walker B loops indicated a nearly 99% match with BcatrD, with 1 residue out of 103 total amino acids that was not an identical match. Two nucleotide binding domains (NBDs) were detected by a Pfam domain search, in addition to two ABC2 domains and a pleiotropic drug resistance (PDR) domain. The overall domain configuration is congruent with that of BcatrD and matches those of other full-size ABC-G transporters, where the first N-terminal domain is an NBD (12, 18, 19).
Quantitative PCR of ShCYP51 and ShatrD on the initial panel of eight isolates.
No significant differences (P = 0.0608) were found for constitutive RE values of ShCYP51B between the sensitive (mean = 1.5 ± 0.1 standard errors [SE]) and insensitive (mean = 1.7 ± 0.1 SE) groups in the initial panel of eight isolates (Fig. 1A). However, significant differences (P < 0.0001) were found between the sensitive (mean = 4.4 ± 0.3 SE) and insensitive (mean = 9.3 ± 0.3 SE) groups for induced RE (Fig. 1A). The differences in the constitutive RE values of ShatrD were highly significant (P < 0.0001) between the sensitive (mean = 2.0 ± 1.3 SE) and insensitive (mean = 16.3 ± 1.2 SE) groups of isolates for untreated samples (Fig. 1B). The differences in the induced REs of ShatrD were also highly significant (P < 0.0001) between the sensitive (mean = 4.8 ± 5.0 SE) and insensitive (mean = 58.2 ± 5.1 SE) groups of isolates (Fig. 1B).
Fig 1.

Relative expression (RE) values for ShCYP51B and ShatrD in eight S. homoeocarpa isolates from five sites in New England. All bars (gray, constitutive RE; black, induced RE) indicate mean RE values, and error bars represent 1 standard error from the mean. Isolates indicated with an “I” or “S” are propiconazole insensitive or sensitive, respectively. (A) Constitutive and induced RE of ShCYP51B. (B) Constitutive and induced RE of ShatrD.
Quantitative PCR of ShCYP51 and ShatrD in isolates with practical field resistance.
No significant differences (P = 0.0682) in constitutive RE values of ShCYP51B were found for the PFR isolates from Hickory Ridge Country Club (HRI1 to HRI4) (mean = 2.4 ± 0.2 SE) compared to the sensitive isolates (HRS1 to HRS4) (mean = 2.0 ± 0.2 SE) (Fig. 2A). In contrast, significant differences (P < 0.0001) in the RE values of ShCYP51B were found for PFR isolates from Hartford Golf Club (HFI1 to HFI4) (mean = 3.4 ± 0.2 SE) compared to the sensitive isolate group (HFS1 to HFS4) (mean = 1.3 ± 0.2 SE) (Fig. 2A). Significant differences (P < 0.0001) in RE values of ShatrD were found for PFR isolates (HFI1 to HFI4) (mean = 17.2 ± 2.0 SE) compared to the sensitive isolate group (HFS1 to HFS4) (mean = 1.8 ± 2.0 SE) (Fig. 2B). Significant differences (P < 0.0001) in RE values of ShatrD were also found for PFR isolates (HRI1 to HRI4) (mean = 85.0 ± 11.7 SE) compared to the sensitive isolate group (HRS1 to HRS4) (mean = 3.1 ± 11.7 SE) (Fig. 2B). Hartford Golf Club PFR isolates significantly overexpressed ShCYP51B (P < 0.0001) compared to Hickory Ridge Country Club PFR isolates (Fig. 2A). In contrast, PFR isolates from Hickory Ridge Country Club significantly overexpressed ShatrD (P < 0.0006) compared to Hartford Golf Club PFR isolates (Fig. 2B).
Fig 2.

Relative expression (RE) of ShCYP51B and ShatrD in S. homoeocarpa isolates exhibiting practical field resistance (PFR) to propiconazole on two New England golf courses. All bars (gray, RE of sensitive isolates from control plots; black, RE of PFR isolates) indicate mean RE values, and error bars represent 1 standard error from the mean. (A) Constitutive RE of ShCYP51B. (B) Constitutive RE of ShatrD.
Linear regression of ShatrD expression and EC50 for propiconazole.
The linear regression of the EC50 and log10 RE values of ShCYP51B revealed a significant relationship (P = 0.0478) (Fig. 3A). A highly significant linear regression of the EC50 and log10 RE values of ShatrD was observed (P < 0.0001) (Fig. 3B).
Fig 3.
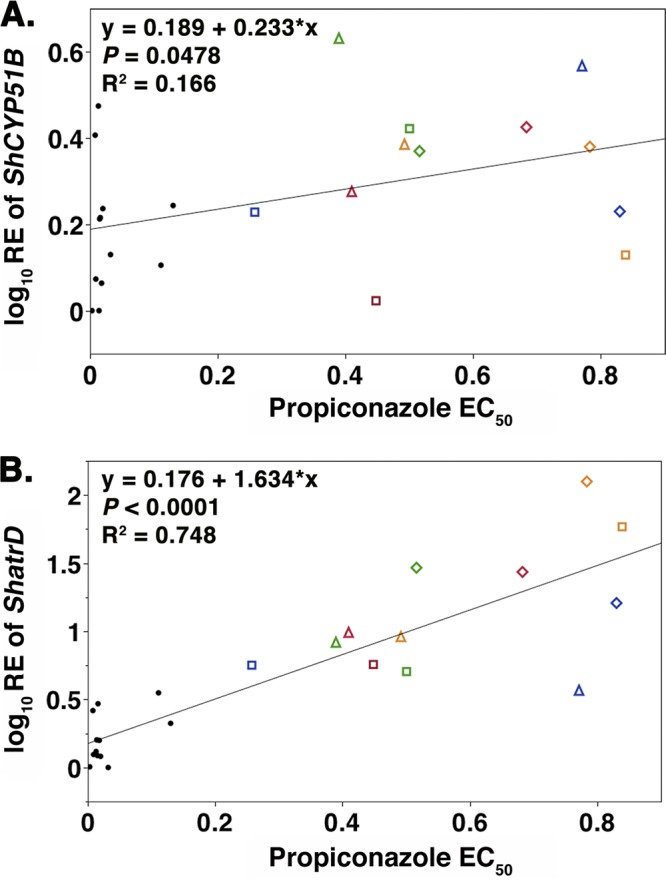
Regression lines showing the linear relationship of log10 RE values of ShCYP51B (A) or log10 RE values of ShatrD (B) and sensitivity to propiconazole (as measured by the EC50) for 24 isolates from five New England sites. For both panels, triangles denote PFR isolates from Hartford Golf Club, diamonds denote PFR isolates from Hickory Ridge Country Club, and squares denote insensitive isolates from four sites. Each colored shape represents the same isolate for both panels A and B. Sensitive isolates from five sites and from control plots are indicated by black dots.
DISCUSSION
The emergence of decreased sensitivity to DMI fungicides can occur by several common mechanisms as demonstrated in diverse fungal lineages (7, 26). A reduced sensitivity to DMIs is often attributed to the constitutive overexpression of CYP51 and/or multiple gene paralogs of CYP51, which have also been shown to play a significant role (1, 21). However, the genomes of members of the Sclerotiniaceae are known to encode only one CYP51 ortholog (CYP51B), and ShCYP51B clusters with other CYP51Bs of the Sclerotiniaceae with high bootstrap support (see Fig. S2 in the supplemental material) (1). Thus, we hypothesize that S. homoeocarpa likely belongs to an evolutionary lineage that contains a single CYP51 gene ortholog.
In other studies of CYP51-involved DMI resistance mechanisms, CYP51 overexpression was associated with SNPs in the gene coding region, with increases in gene copy numbers through aneuploidy events or with upstream promoter rearrangements (23–25, 38). No SNPs were found in ShCYP51B, but an SNP was present upstream in isolates WBI7 and HFI40 from the initial panel of isolates which we screened and in PFR isolates from Hartford Golf Club. Although this SNP was not located in the predicted promoter regions, it was linked to the overexpression of ShCYP51B in the PFR isolates from Hartford Golf Club. However, the overexpression was less than 3-fold higher than that of the isolates from the nontreated plots, and this difference in ShCYP51B expression cannot fully account for the disparity of EC50s exhibited by the two isolate groups (Table 1). The level of induced expression of ShCYP51B was higher for insensitive isolates screened from the initial panel of isolates, but the difference was also minor, at just over 2-fold higher. It remains to be determined if gene copy number variations could explain the observed levels of overexpression of ShCYP51B.
Even though significant constitutive and induced overexpressions of ShCYP51B were found, the numerical differences in mean values were minimal and probably have limited biological significance. The ShatrD gene is likely the predominant genetic determinant of reduced DMI sensitivity in S. homoeocarpa populations, exhibiting overexpression in all of the insensitive and PFR isolate groups and to a much higher degree than ShCYP51B. This hypothesis is further validated by the highly significant linear relationship between the EC50 and log10 RE values of ShatrD (Fig. 3B). Furthermore, BcatrD, with which ShatrD shares homology, encodes a membrane-bound ABC transporter known to efflux DMI fungicides (12). The ABC transporters encoded by BcatrD and ShatrD belong to the G subfamily of ABC transporters, which are known as PDR (pleiotropic drug resistance) transporters and are implicated in multidrug resistance in many fungal pathogens (see Fig. S3 in the supplemental material) (2, 12, 18). In fact, ABC-G transporters are the least evolutionarily conserved of all ABC transporters and have undergone differential duplication and expansion in fungal lineages (18, 19). The number of PDRs in Sclerotiniaceae genomes was found by Lamping et al. to be variable, with Botrytis fuckeliana (B. cinerea) having the most at 6, S. sclerotiorum having the next most at 5, and M. fructicola being known to encode only 1 (19). Within our RNA-Seq transcriptome data from S. homoeocarpa, we detected at least four additional ABC-G transporters with putative homologs in B. cinerea, which is not completely surprising in light of the high amino acid sequence similarity and phylogenetic clustering of ShatrD with BcatrD (see Fig. S3 in the supplemental material). Further work is needed to examine the putative role of other ABC-G transporters, as well as additional genetic factors, in the decreased DMI fungicide sensitivity of S. homoeocarpa. In the plant pathogens B. cinerea and Mycosphaerella graminicola, the overexpression of multiple ABC and MFS transporters with known redundant functions gives reduced sensitivity to DMIs and other fungicide chemistries, which may be the case for S. homoeocarpa (26, 42).
From the data presented, the degree to which ShatrD and ShCYP51B are associated with reduced propiconazole sensitivity and practical field resistance may be population and/or genotype specific (Table 1 and Fig. 1 and 2). The RE data for Hartford Golf Club PFR isolates suggest that ShCYP51B and ShatrD, when constitutively overexpressed, each confer some proportion to the reduction in propiconazole sensitivity or perhaps have synergy together to this effect (Fig. 2). The cooccurrence of CYP51 and ABC-G overexpressions in fungi with reduced DMI sensitivity is widespread and known for field isolates of fungal plant pathogens and clinical isolates of a human fungal pathogen (5, 20). In Candida albicans, chromosomal aneuploidy and a subsequent loss of heterozygosity (LOH) give not only higher CYP51 gene expression levels but also simultaneous increases in expression levels of a transcriptional regulator, TAC1, of an ABC-G efflux gene (5, 38). Since S. homoeocarpa is thought to be an asexual haploid fungus capable of forming heterokaryons and without known diploid or sexual stages, LOH is not a viable explanation (16). However, there may be the potential for mitotic recombination in the heterokaryotic mycelium of S. homoeocarpa through a parasexual cycle, which was demonstrated previously for the fungal barley pathogen Rhynchosporium secalis with antibiotic transgenes between nuclei in heterokaryons (10). Such a mechanism might allow for the recombination of multiple independently arising point mutations, culminating in the accumulation of upstream mutations leading to the overexpression of multiple genes, such as ShCYP51B and ShatrD. Multiple SNPs, an insertion, and a deletion were found upstream of ShatrD in conjunction with an SNP upstream of ShCYP51B. However, it remains to be determined if these polymorphisms confer overexpression, since they were not located in predicted promoter regions and since an equal number of isolates overexpressing ShatrD showed no upstream SNPs compared to sensitive isolates.
The increased constitutive overexpression of ShatrD in PFR isolates from Hickory Ridge Country Club and the simultaneous lack of significant ShCYP51B overexpression compared to Hartford Golf Club PFR isolates suggest that the gene expression patterns of ShCYP51B and ShatrD can exhibit variability in a complementary fashion. Perhaps, the enhanced ShatrD overexpression in Hickory Ridge Country Club PFR isolates is necessary to compensate for the absence of ShCYP51B overexpression in order to give the level of reduced sensitivity required for practical field resistance (and vice versa for Hartford Golf Club PFR isolates). Also of interest, Hickory Ridge Country Club and Hartford Golf Club PFR isolates are each of different clonal genotypes, which leaves open the possibility that the gene expression patterns are population and/or genotype specific and may reflect the unique chemical exposure histories of the populations of S. homoeocarpa sampled (Fig. 2 and Table 1). The apparent differential overexpression of ShatrD between Hartford Golf Club and Hickory Ridge Country Club PFR isolates could not be attributed to upstream SNPs but could be due to nonsynonymous SNPs in a transcription factor controlling ShatrD expression. This is the case for nonsynonymous TAC1 transcription factor polymorphisms in C. albicans, one of which results in the hyperexpression of the ABC-G efflux transporters CDR1 and CDR2, while other nonsynonymous SNPs in TAC1 result in overexpression to a much lesser extent (5). Additionally, field isolates of B. cinerea have been shown to harbor polymorphisms in a transcriptional regulator of ABC efflux transporters, Mrr1, which can confer the overexpression of ABC efflux genes and decreased sensitivity to DMI fungicides (26). It is currently not clear whether increases in copy numbers or mutations in transcription factors and upstream elements are behind the observed gene expression patterns of S. homoeocarpa, but additional genome-level investigations of isolates from this study could shed light on how these genotypes have evolved the observed gene expression patterns.
Bearing in mind the parallels in genetic mechanisms for reduced DMI sensitivity shared between diverse groups of fungal pathogens, turf practitioners should prepare for resistance to new fungicide chemistries and for the emergence of novel multiple- and cross-resistance phenotypes in S. homoeocarpa. Cross-resistance to multiple triazole DMIs as well as to triazole PGRs (plant growth regulators) was recently reported for this pathogen (30). From our findings, the overexpression of ShatrD is likely involved, since its homolog in B. cinerea is capable of mediating sensitivity to all classes of DMI fungicides (12). To confirm this, the substrate specificity of ShatrD should be assayed with a yeast complementation system to demonstrate its capacity to efflux propiconazole as well as other triazole DMIs and PGRs (12, 42).
The involvement of a fungicide efflux transporter in the practical field resistance of geographically disparate populations of S. homoeocarpa stresses the need to develop and test available chemistries that have a blocking effect on ABC transporter-mediated drug efflux. Previous reports have confirmed the synergistic inhibitory activity of efflux blockers and DMI fungicides on the mycelial growth of fungal plant pathogens with ABC transporter-mediated fungicide resistance (13, 20). Thus, our findings provide grounds to investigate the potential for ABC transporter efflux blockers in the treatment of dollar spot epidemics that cannot be adequately controlled by DMI fungicides alone. In summary, a more thorough understanding of efflux mechanisms and transporter substrate specificities, as well as the potential use of efflux blockers in combination with DMIs, will afford insights that translate to better resistance management practices, giving optimal turf quality while limiting the emergence of practical field resistance.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Li-Jun Ma of the Department of Plant, Soil, and Insect Sciences for providing guidance and computing tools for this study. We also acknowledge Katie Campbell-Nelson, former research associate, for her careful review in the preparation of the manuscript. Finally, we thank additional laboratory associates who contributed to this study, Leah Zivalic, who participated in the sequencing of the ShCYP51B gene and upstream regions, and Frank Dixon, who assisted in the development and screening of microsatellites for S. homoeocarpa.
Funding for this work comes from the National Institute of Food and Agriculture, U.S. Department of Agriculture, the Massachusetts Agricultural Experiment Station, and the Department of Plant, Soil, and Insect Sciences under project number MAS00925.
Footnotes
Published ahead of print 13 July 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Becher R, Weihmann F, Deising HB, Wirsel SGR. 2011. Development of a novel multiplex DNA microarray for Fusarium graminearum and analysis of azole fungicide responses. BMC Genomics 12:52 doi:10.1186/1471-2164-12-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cannon RD, et al. 2009. Efflux-mediated antifungal drug resistance. Clin. Microbiol. Rev. 22:291–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chakraborty N, Chang T, Casler MD, Jung G. 2006. Response of bentgrass cultivars to Sclerotinia homoeocarpa isolates representing 10 vegetative compatibility groups. Crop Sci. 46:1237–1244 [Google Scholar]
- 4. Conesa A, et al. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676 [DOI] [PubMed] [Google Scholar]
- 5. Coste A, et al. 2006. A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in Candida albicans. Genetics 172:2139–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Couch HB. 1995. Diseases of turfgrasses, 3rd ed. Krieger Publishing Company, Malabar, FL [Google Scholar]
- 7. Cowen LE, et al. 2002. Population genomics of drug resistance in Candida albicans. Proc. Natl. Acad. Sci. U. S. A. 99:9284–9289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Delye C, Bousset L, Corio-Costet MF. 1998. PCR cloning and detection of point mutations in the eburicol 14-demethylase (CYP51) gene from Erysiphe graminis f. sp. hordei, a “recalcitrant” fungus. Curr. Genet. 34:399–403 [DOI] [PubMed] [Google Scholar]
- 9. Delye C, Laigret F, Corio-Costet MF. 1997. A mutation in the 14-demethylase gene of Uncinula necator that correlates with resistance to a sterol biosynthesis inhibitor. Appl. Environ. Microbiol. 63:2966–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Forgan AH, Knogge W, Anderson PA. 2007. Asexual genetic exchange in the barley pathogen Rhynchosporium secalis. Phytopathology 97:650–654 [DOI] [PubMed] [Google Scholar]
- 11. Hamamoto H, et al. 2000. Tandem repeat of a transcriptional enhancer upstream of the sterol 14 alpha-demethylase gene (CYP51A1) in Penicillium digitatum. Appl. Environ. Microbiol. 66:3421–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hayashi K, Schoonbeek H, De Waard MA. 2002. Expression of the ABC transporter BcatrD from Botrytis cinerea reduces sensitivity to sterol demethylation inhibitor fungicides. Pestic. Biochem. Physiol. 73:110–121 [Google Scholar]
- 13. Hayashi K, Schoonbeek H, De Waard MA. 2003. Modulators of membrane drug transporters potentiate the activity of the DMI fungicide oxpoconazole against Botrytis cinerea. Pest Manag. Sci. 59:294–302 [DOI] [PubMed] [Google Scholar]
- 14. Jo YK, Niver AL, Rimelspach JW, Boehm MJ. 2006. Fungicide sensitivity of Sclerotinia homoeocarpa from golf courses in Ohio. Plant Dis. 90:807–813 [DOI] [PubMed] [Google Scholar]
- 15. Jo YK, Chang SW, Boehm M, Jung G. 2008. Rapid development of fungicide resistance by Sclerotinia homoeocarpa on turfgrass. Phytopathology 98:1297–1304 [DOI] [PubMed] [Google Scholar]
- 16. Jo YK, Chang SW, Rees J, Jung G. 2008. Reassessment of vegetative compatibility of Sclerotinia homoeocarpa using nitrate-nonutilizing mutants. Phytopathology 98:108–114 [DOI] [PubMed] [Google Scholar]
- 17. Katoh T. 2008. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinformatics 9:286–298 [DOI] [PubMed] [Google Scholar]
- 18. Kovalchuk A, Driessen AJM. 2010. Phylogenetic analysis of fungal ABC transporters. BMC Genomics 11:177 doi:10.1186/1471-2164-11-177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lamping E, et al. 2010. Fungal PDR transporters: phylogeny, topology, motifs and function. Fungal Genet. Biol. 47:127–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leroux P, Walker AS. 2011. Multiple mechanisms account for resistance to sterol 14α-demethylation inhibitors in field isolates of Mycosphaerella graminicola. Pest Manag. Sci. 67:44–59 [DOI] [PubMed] [Google Scholar]
- 21. Liu X, et al. 2011. Paralogous CYP51 genes in Fusarium graminearum mediate differential sensitivity to sterol demethylation inhibitors. Fungal Genet. Biol. 48:113–123 [DOI] [PubMed] [Google Scholar]
- 22. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 23. Luo CX, Schnabel G. 2008. The cytochrome P450 lanosterol 14α-demethylase gene is a demethylation inhibitor fungicide resistance determinant in Monilinia fructicola field isolates from Georgia. Appl. Environ. Microbiol. 74:359–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ma Z, Proffer TJ, Jacobs JL, Sundin GW. 2006. Overexpression of the 14α-demethylase target gene (CYP51) mediates fungicide resistance in Blumeriella jaapii. Appl. Environ. Microbiol. 72:2581–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mellado E, et al. 2007. A new Aspergillus fumigatus resistance mechanism conferring in vitro cross-resistance to azole antifungals involves a combination of cyp51A alterations. Antimicrob. Agents Chemother. 51:1897–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mernke D, et al. 2011. Two promoter rearrangements in a drug efflux transporter gene are responsible for the appearance and spread of multidrug resistance phenotype MDR2 in B. cinerea isolates in French and German vineyards. Phytopathology 101:1176–1183 [DOI] [PubMed] [Google Scholar]
- 27. Miller L, Stevenson KL, Burpee LL. 2002. Sensitivity of Sclerotinia homoeocarpa isolates to propiconazole and impact on control of dollar spot. Plant Dis. 86:1240–1246 [DOI] [PubMed] [Google Scholar]
- 28. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5:621–628 [DOI] [PubMed] [Google Scholar]
- 29. Nakaune R, et al. 1998. A novel ATP-binding cassette transporter involved in multidrug resistance in the phytopathogenic fungus Penicillium digitatum. Appl. Environ. Microbiol. 64:3983–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ok C-H, Popko J, Campbell-Nelson K, Jung G. 2011. In vitro assessment of Sclerotinia homoeocarpa resistance to fungicides and plant growth regulators. Plant Dis. 95:51–56 [DOI] [PubMed] [Google Scholar]
- 31. Peakall R, Smouse PE. 2006. GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6:288–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Popko JT, Ok C-H, Campbell-Nelson K, Jung G. 2012. The association between in vitro propiconazole sensitivity and field efficacy of five New England Sclerotinia homoeocarpa populations. Plant Dis. 96:552–561 [DOI] [PubMed] [Google Scholar]
- 33. Putman AI, Jung G, Kaminski JE. 2010. Geographic distribution of fungicide-insensitive Sclerotinia homoeocarpa isolates from golf courses in the northeastern United States. Plant Dis. 94:186–195 [DOI] [PubMed] [Google Scholar]
- 34. Reese MG. 2001. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem. 26:51–56 [DOI] [PubMed] [Google Scholar]
- 35. Rozen S, Skaletsky H. 2000. Primer 3 on the WWW for general users and biologist programmers. Methods Mol. Biol. 132:365–386 [DOI] [PubMed] [Google Scholar]
- 36. Saitoh K, Togashi K, Arie T, Teraoka T. 2006. A simple method for a mini-preparation of fungal DNA. J. Gen. Plant Pathol. 72:348–350 [Google Scholar]
- 37. Sanglard D, Ischer F, Koymans L, Bille J. 1998. Amino acid substitutions in the cytochrome P-450 lanosterol 14α-demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob. Agents Chemother. 42:241–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Selmecki A, Forche A, Berman J. 2006. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313:367–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Venu RC, et al. 2011. Large scale identification of genes involved in plant-fungal interactions using Illumina's sequencing-by-synthesis technology. Methods Mol. Biol. 722:167–178 [DOI] [PubMed] [Google Scholar]
- 41. Walsh B, Ikeda SS, Boland GJ. 1999. Biology and management of dollar spot (Sclerotinia homoeocarpa); an important disease of turfgrass. HortScience 34:13–21 [Google Scholar]
- 42. Zwiers LH, Stergiopoulos I, Gielkens MMC, Goodall SD, De Waard MA. 2003. ABC transporters of the wheat pathogen Mycosphaerella graminicola function as protectants against biotic and xenobiotic compounds. Mol. Gen. Genomics 269:499–507 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.