

Abstract
Viruses excreted by humans affect the commercial and recreational use of coastal water. Shellfish produced in contaminated waters have been linked to many episodes and outbreaks of viral gastroenteritis, as well as other food-borne diseases worldwide. The risk can be reduced by appropriate treatment following harvesting and by depuration. The kinetics of inactivation of murine norovirus 1 and human adenovirus 2 in natural and artificial seawater by free available chlorine was studied by quantifying genomic copies (GC) using quantitative PCR and infectious viral particles (PFU). Human JC polyomavirus Mad4 kinetics were evaluated by quantitative PCR. DNase or RNase were used to eliminate free genomes and assess potential viral infectivity when molecular detection was performed. At 30 min of assay, human adenovirus 2 showed 2.6- and 2.7-log10 GC reductions and a 2.3- and 2.4-log10 PFU reductions in natural and artificial seawater, respectively, and infectious viral particles were still observed at the end of the assay. When DNase was used prior to the nucleic acid extraction the kinetic of inactivation obtained by quantitative PCR was statistically equivalent to the one observed by infectivity assays. For murine norovirus 1, 2.5, and 3.5-log10 GC reductions were observed in natural and artificial seawater, respectively, while no viruses remained infectious after 30 min of contact with chlorine. Regarding JC polyomavirus Mad4, 1.5- and 1.1-log10 GC reductions were observed after 30 min of contact time. No infectivity assays were conducted for this virus. The results obtained provide data that might be applicable to seawater used in shellfish depuration.
INTRODUCTION
The interaction between oceans and human health is increasing partly due to the high numbers of humans living within close proximity to the oceans (15). Microbial contamination by bacteria, viruses, and protozoa is related directly and indirectly to human and animal activity and affects the safety of the seafood supply, as well as the commercial and recreational use of coastal areas (25). Human health problems associated with shellfish consumption are well described, and contaminating viruses have been linked to many episodes of gastroenteritis, as well as outbreaks of other diseases (24).
The most common viral pathogens associated with shellfish consumption are human noroviruses (NoVs), which have been implicated in numerous food-borne outbreaks (22). NoVs are responsible for up to 1 million hospitalizations and 218,000 deaths each year in children living in developing countries (34). The infectivity of NoVs detected from environmental samples cannot be assessed yet due to the absence of an in vitro cell culture system (2). This has prompted the use of viral surrogates to model the infectious nature of NoVs in environmental samples. Murine norovirus 1 (MNV-1) is considered the best surrogate for NoVs (44). Recently, MNV-1 has also been used as a surrogate for NoV in studies of heat inactivation (20), as well as chlorine and ultraviolet (UV) disinfection (9).
Human adenoviruses (HAdV) are one of the most prevalent human pathogens identified in environmental water samples, including drinking and recreational waters (29). HAdV have greater thermal stability than enteroviruses (38). They are capable of surviving for months in water, especially at low temperatures, and they show resistance to inactivation by UV light (41). JC polyomavirus (JCPyV) is a human virus that produces latent and chronic infections that persist indefinitely in individuals. Viral particles are excreted regularly in urine by healthy individuals (39). In previous studies, JCPyV was found in 98% of the 52 sewage samples collected from different geographical areas around the world (6). JCPyV has also been found in river water feeding a drinking water treatment plant (1). HAdV and JCPyV are frequently detected in the environment and have been proposed as an index of viral contamination of human origin (6).
Controlled self-purification (“depuration”) of shellfish is a method that reduces the levels of microorganisms present in mollusk meat, thus decreasing the potential for infections associated with shellfish consumption. In this process, shellfish are placed for several hours in tanks filled with clean seawater to purge contaminants from tissues by filtering (24, 27). There are two main types of shellfish depuration plants: flowthrough or recirculating (closed) systems. Depuration is effective in removing many fecal bacterial contaminants from shellfish (11, 37). However, studies focused on enteric viruses, such as HAV, NoVs, and HAdV have shown that it is difficult to remove viruses from contaminated shellfish (16, 18, 42). The flowthrough system requires a nearby, reliable, consistently clean source of seawater. In the closed depuration system, seawater is recirculated for at least 24 h. During this cycle, the water must be chemically (using chlorine and/or ozone) or physically (UV irradiation) treated to eliminate microbial contamination. For the purpose of shellfish depuration, 2 to 3 mg of free chlorine/liter is normally used for up to 1 h before the seawater is returned to the shellfish tank. Before this, the levels of free chlorine in the water need to be reduced to <0.1 mg/liter so that the shellfish and their depuration activity are not affected. This reduction is achieved by the addition of sodium thiosulfate (23).
Chlorination is one of the main disinfection methods used for water and wastewater treatment, due to its low cost, ease of application, and ability to inactivate a wide variety of pathogenic microorganisms (13). Chlorine's ability to destroy microorganisms is due to the chemical interference of HOCl. It is attributed to the ease with which HOCl can penetrate cell membranes, react with enzyme systems of bacteria, and interact with either the capsid proteins or the nucleic acids of viral pathogens (10).
In this context, the goal of the present study was to evaluate the applicability of free chlorine to inactivate selected viruses in natural and artificial seawater, using molecular and cell culture methods, to obtain information that could be useful in the disinfection of water for shellfish depuration tanks or other purposes.
MATERIALS AND METHODS
Viruses and cell lines.
The HAdV2 strain (NCPV 00213) and JCPyV strain Mad4 (kindly provided by E. O. Major, Laboratory of Molecular Medicine and Virology, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD) were grown, respectively, on A549 cells, derived from human lung adenocarcinoma (European Collection of Cell Cultures), and the human glial cell line SVGA (a gift from W. Atwood, Brown University, Providence, Rhode Island, RI). The cells were propagated in Eagle minimum essential medium (MEM; Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, and 1% l-glutamine. The titer of the viral working solution was determined by an immunofluorescence assay (8) to be ∼109 focus-forming units (FFU)/ml for HAdV2 and 106 FFU/ml for JCPyV Mad4.
MNV-1 was propagated in RAW 264.7 cells (a macrophage-like Abelson leukemia virus-transformed cell line, derived from BALB/c mice). This cell line and MNV-1 were kindly donated by Herbert W. Virgin, Department of Pathology and Immunology, Washington University School of Medicine (St. Louis, MO). RAW 264.7 cells were cultured in 1× Dulbecco MEM (DMEM; Gibco) supplemented with 10% FBS (low endotoxin serum), 1.5% HEPES, 1% penicillin-streptomycin, 1% nonessential amino acids, and 1% l-glutamine. The MNV-1 working solution titer was determined by a plaque assay (as described below) and was estimated to be approximately 4 × 108 PFU/ml.
Disinfection assays. (i) Tested waters.
All disinfection experiments were conducted in either artificial or natural seawater. Artificial seawater was prepared using commercial salts (sea salts; Sigma, St. Louis, MO) by dissolving 33.33 g of sea salts per liter of deionized chlorine demand-free water and was stored in bottles at 4°C until used. Natural seawater was collected from a coastal area south of Barcelona. The pH and conductivity were measured for both types of water tested.
(ii) Glassware treatments.
The reagents and glassware used here were prepared as described previously (41). Briefly, glassware was made chlorine demand-free by overnight soaking in a solution of at least 100 mg of free chlorine/liter. The beakers were then rinsed with chlorine demand-free water and baked for at least 2 h at 200°C. After this initial treatment, soaking in free chlorine solution and rinsing in demand-free water was the only treatment performed for all glassware.
A chlorine stock solution of approximately 150 mg/liter was prepared using bleach (commercial sodium hypochlorite solution, 42 g of free chlorine/liter) that was suitable for tap water disinfection. Dilution of this stock solution in chlorine demand-free water was carried out to achieve the free chlorine concentrations used in the disinfection experiments.
(iii) Experimental design for seawater disinfection.
Chlorine demand-free glass beakers containing 65 ml of natural or artificial seawater were placed inside a biological safety cabinet (at 23 to 25°C) and were magnetically stirred prior to each experiment. The free chlorine concentration of the stock solution was measured by the DPD (N,N-diethyl-p-phenylendiamine) method, and the volume necessary to achieve the initial free-chlorine dose in each experimental beaker was calculated. The assays were performed in duplicate for each tested water sample.
Two beakers were analyzed in every assay, and both were inoculated simultaneously with the three viruses selected (viral suspensions diluted in phosphate-buffered saline [PBS]) at a concentration that would allow detection of 4-log inactivation in either natural or artificial seawater (2 × 107 GC of HAdV2/ml; 5 × 108 of MNV-1 and 1.5 × 105 of JCPyV Mad4). One of these beakers (beaker 1) was used as a negative control of disinfection, with no chlorine added, for each of the experiments performed. This control was needed to determine the initial virus concentration and to evaluate whether virus inactivation occurred under the test conditions. Another beaker (beaker 2), containing chlorine at an initial concentration of 2.5 mg/liter, was used to evaluate the kinetics of viral disinfection. For 60 min, 5-ml seawater samples were collected at 30 s, 10, 20, 30, 45, and 60 min for viral analysis. Residual free chlorine was immediately quenched by placing the 5-ml samples into collection tubes containing 50 μl of sterile 10% sodium thiosulfate solution. These tubes were kept on ice until used for viral detection.
An additional 5 ml of seawater were collected before viral spiking (identified as 0 s) and at the same sampling times as those cited above. These samples were analyzed to determine the residual free chlorine concentration by the DPD method for each disinfection assay performed.
After chlorine disinfection, samples were ultracentrifuged at 60,000 × g for 60 min at 4°C. The resultant pellets were resuspended in sterile PBS. The nucleic acid isolation and infectivity assays were performed on the same day as the chlorine disinfection to avoid sample freezing.
Evaluation of viral inactivation by cell culture methods. (i) Cytotoxicity tests.
Cytotoxicity tests were performed to evaluate the potential toxicity caused by seawater in cell lines to be used during disinfection studies. The tests were carried out as previously described (36), with minor modifications. RAW 264.7 and A549 cell monolayers (2 × 105 cells/ml) were propagated in 24-well microplates (Nunc, Rochester, NY) for 24 h before the cytotoxicity assays were performed. Inoculation was carried out as follows: 2-fold serial dilutions of unseeded seawater ranging from 1:2 to 1:32 were prepared in serum-free culture medium (1× DMEM for RAW 264.7, and 1× MEM for A549 cells). Inoculates of 250 μl of each tested water were then adsorbed to cells that had been washed with PBS previously. All assays were carried out in duplicate. After incubation, the inoculum was removed, and the cells were supplied with 1 ml of culture medium supplemented with 2% FBS (for A549) and 10% FBS (for RAW 264.7 cells). Plates were incubated at 37°C and 5% CO2 and observed for cytopathic effect after 2 (RAW 264.7) and 4 (A549 cells) days of incubation. Each observation was compared to negative controls containing only cell monolayers and the medium but no seawater. Cell monolayers were observed under an inverted light microscope. They were fixed with 1% formalin and stained with 0.1% crystal violet to establish a first noncytotoxic dilution of seawater to be used in further viral infectivity assays. Seawater diluted 1:2 and 1:4 in serum-free culture medium were selected to be used on A549 and RAW 264.7, respectively.
(ii) Plaque assays.
In the present study, the plaque assay applied to the titration, and evaluation of MNV-1 infectivity was performed as described previously (2), with some modifications. Briefly, RAW 264.7 cells were seeded into 60-mm plates at a density of 2 × 106 cells per well. Cells were then allowed to adhere for 48 h at 37°C in the presence of 5% CO2. After chlorine disinfection, the samples were diluted in PBS at noncytotoxic dilutions to quantify infectious MNV-1. The cell culture medium was decanted, and cells were infected with 0.5 ml of each dilution. After incubation for 1 h at 37°C in the presence of 5% CO2, the inocula were aspirated and replaced with 2 ml of a solution containing 1.5% SeaPlaque Agarose (Lonza, Basel, Switzerland) and supplemented with DMEM (as described above for viruses and cell lines). The samples were allowed to solidify, and incubated at 37°C for up to 48 h until plaques were visible. To better visualize the plaques, a neutral red solution (final concentration, 0.1%) was added to 2 ml of the mixture containing 1.5% agarose and supplemented with DMEM after solidification and incubation at 37°C for 6 to 8 h. The plaques were counted, and the virus titer was expressed as PFU/ml.
The assay to evaluate infectious HAdV2 followed the same principle as that described for MNV-1, with some modifications. Briefly, A549 cells in suspension (6 × 105/ml) were mixed with a seawater sample volume. This suspension was added to a 25-cm2 sterile flask and placed at 36 ± 2°C for between 4 to 6 h. A volume of overlay medium containing 3% carboxymethyl cellulose was added, and the flask was incubated at 36 ± 2°C for up to 7 days. After 7 days, the medium was removed. The cells were then fixed and stained with crystal violet. The plaques were counted, and the virus titer was expressed as PFU/ml.
Evaluation of viral inactivation by molecular methods. (i) Nucleic acid isolation.
Viral genomes from samples treated with free chlorine were isolated using the QIAmp viral RNA minikit (Qiagen, Valencia, CA) according to the manufacturer's instructions. The nucleic acids were eluted in the elution buffer provided and stored at −80°C until they were used in real-time PCR and real-time reverse transcription-PCR (RT-PCR).
(ii) Enzymatic treatment before the application of molecular assays.
In the present study, two enzymatic treatments to assess the capsid integrity of the viruses detected using real-time PCR after the disinfection of seawater with free chlorine were performed. For each sample, the enzymatic treatments were based on the use of 100 U of DNase I (molecular grade; Invitrogen, Carlsbad, CA), according to the manufacturer's instructions, and RNase A (from bovine pancreas [Invitrogen]), as previously reported (30). These treatments were performed before the nucleic acid extraction of the samples to be tested by real-time PCR, in order to quantify HAdV2, JCPyV Mad4, and MNV-1. Control assays were performed to confirm the total inactivation of DNase and RNase in the samples by using stop buffer (provided with DNase I) and 40 U of RNase inhibitor (data not shown).
Real-time PCR. (i) Preparation of DNA standard suspensions.
HAdV and JCPyV quantitation was based on the assays described previously by Hernroth et al. (19) and Pal et al. (33), respectively.
Standards for the real-time PCR assays for HAdV2 and JCPyV Mad4 were produced as described before (1). An RNA standard was used to quantify MNV-1. This standard was created using an RNA transcript from an amplicon of 1,663 bp, corresponding to the junction region of ORF1/ORF2 of the MNV-1 genome (positions 3999 to 5662) generated by conventional RT-PCR using the forward primer 5′-GAGATGGGTAAATCCATGCG-3′ and the reverse primer 5′-CAGAGACCACAAAAGACTCATC-3′ (21). This RNA was transcribed by Yorkshire Bioscience (York, United Kingdom) using a T7 RNA polymerase transcription system. Plasmid DNA was removed completely with RNase-free DNase according to the product description. RNA was purified by LiCl precipitation, followed by multiple phenol-chloroform extractions and precipitation with ethanol and dilution in Milli-Q water. The RNA concentration was determined by spectrophotometry, and the copy number of standard RNA molecules was calculated as described above. Serial 10-fold dilutions were prepared in RNA storage buffer (Ambion, Austin, TX), aliquoted, and stored at −80°C.
(ii) Quantitative PCR and quantitative reverse transcription-PCR (qPCR and RT-qPCR).
For the specific detection and quantitation of JCPyV and HAdV2 genomes, 10 μl of undiluted samples and 1:10 dilutions of every DNA extraction were tested. These dilutions were made to detect and reduce amplification inhibition due to the potential presence of inhibitory substances that may interfere with the quantitative PCR (qPCR). Amplification was performed in a 25-μl reaction mixture with TaqMan PCR master mix (Applied Biosystems, Foster City, CA). The reaction contained 10 μl of a DNA sample, 10 μl of DNA sample diluted 1:10, or 10 μl of a quantified DNA plasmid–1× TaqMan master mix, along with the corresponding primers and TaqMan probes for HAdV and JCPyV.
For the detection and quantitation of MNV-1 genomes, we assayed 5 μl of the RNA extraction and of the 10-fold dilution. Amplification was performed in a 20-μl reaction mixture with the RNA UltraSense one-step quantitative RT-PCR system (Invitrogen). The reaction contained 5 μl of an RNA sample or 5 μl of an RNA standard, 1× UltraSense mix, 1× UltraSense enzyme mix, and 1× ROX, in addition to the primers and probe.
For MNV-1, the amplification were performed after retrotranscription for 30 min at 50°C and activation of the HotStarTaq (Qiagen) for 15 min at 95°C, followed by 45 cycles of amplification (10 s at 95°C, 20 s at 55°C, and 15 s at 72°C). For HAdV2 and JCPyV, the genomes were quantified after the activation of the uracil N-glycosylase contained in the core mix (2 min at 50°C), the activation of AmpliTaq Gold for 10 min at 95°C, and 40 cycles of amplification (15 s at 95°C and 1 min at 60°C). The sets of primers and probes for each virus are described in Table 1. Quantitations were performed with an MX3000P sequence detector system (Stratagene, La Jolla, CA).
Table 1.
Primer and probe sets used to detect MNV-1, JCPyV Mad4, and HAdV2 by q(RT)-PCR
| Virus (reference) | Primer or probe | Sequence (5′–3′) |
|---|---|---|
| JCPyV (33) | JE3F | ATGTTTGCCAGTGATGATTGAAAA |
| JE3R | GGAAAGTCTTTAGGGTCTTCTACCTTT | |
| JE3P | 6-FAM-AGGATCCCAACACTCTACCCCACCTAAAAAGA-TAMRA | |
| HAdV2 (19) | AdF | CWTACATGCACATCKCSGG |
| AdR | CRCGGGCRAAYTGCACCAG | |
| AdP1 | 6-FAM-CCGGGCTCAGGTACTCCGAGGCGTCCT-TAMRA | |
| MNV-1 (3) | Fw-ORF1/ORF2 | CAC GCC ACC GAT CTG TTC TG |
| Rv-ORF1/ORF2 | GCG CTG CGC CAT CAC TC | |
| MGB-ORF1/ORF2 | 6FAM-CGC TTT GGA ACA ATG-MGBNFQ |
Kinetic modeling and statistical analysis.
The chlorine disinfection was characterized by the efficiency factor Hom (EFH) model (5, 17, 41), a widely used model that takes into account the decreasing chlorine concentrations caused by the virus itself and the organics present in the solution. The chlorine concentration in each experiment is therefore modeled as a first-order kinetic equation:
| (1) |
where C(t) is the residual chlorine concentration in mg/liter at time t (in minutes), C0 is the initial chlorine concentration, and k′ is the first-order decay rate constant. C0 and k′ from equation 1 were used in the following equation to model the disinfection kinetics:
| (2) |
where k is the inactivation rate constant, n is the coefficient of dilution, m is the Hom's exponent, ln(N(t)/N0) is the natural log of the survival ratio, and N(t) is the number of organisms remaining at time t divided by N0 (the number at time zero). To determine the parameter values for both equations, Microsoft Excel Solver (Microsoft Excel 2010; Microsoft Corp.) was used to minimize the error sum of squares between the observed and predicted C(t) and ln(N(t)/N0) for each viral disinfection replicate of each experiment.
For every experiment, an F-approximate test was conducted to assess the homogeneity of the disinfection kinetic on the two replicates. If homogeneous replicates were found, 12 pairs of previously selected experiments were also compared to an F-test to assess the homogeneity of the overall pair. The P values of these multiple comparison tests were adjusted with the Benjamini and Hochberg correction (4).
Once the parameters of the EFH model equation had been estimated for all of the experiments, they could be used to predict the time values for 2- and 3-log inactivation of the viruses. These times, expressed in minutes, are noted, respectively, as t99 and t99.9.
RESULTS
Free chlorine decay in seawater.
The rate and extent of free chlorine demand in tested waters before and after spiking with viral stocks was determined. As shown in Fig. 1, both types of seawater exhibited a demand for free chlorine. The residual concentration of the disinfectant decreased over time. At an initial dosage of ∼2.5 mg/liter, free chlorine was consumed rapidly by both test waters within 30 s. After 60 min, at the end of the assay, an 80% reduction in the initial dose of chlorine was observed.
Fig 1.
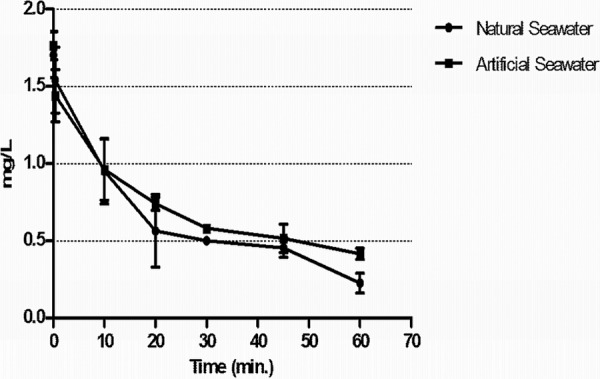
Free chlorine decay curve in disinfection assays using natural and artificial seawater. The dots represent an average of two replicates for each type of water tested.
Viral inactivation by chlorination of seawater.
The inactivation kinetics of the three viruses by 2.5 mg of free chlorine (initial dose)/liter in natural and artificial seawater was based on the results obtained in two independent experiments for each type of water. Virus disinfection data, in PFU/ml and GC/ml for HAdV2 and MNV-1 and in GC/ml for JCPyV Mad4, are presented in the figure as the log reduction values obtained in each of the assays performed (Fig. 2).
Fig 2.

Log reduction curves for seeded HAdV2, MNV-1, and JCPyV in natural (A) and artificial (B) seawater treated with free chlorine at 2.5 mg/liter (initial concentration). Each data point is an average of two independent experiments, except in the case of HAdV2 infectivity, where the values correspond to one experiment. ↓, Detection limit.
The initial concentration of HAdV2 in the tested waters was of the order of 107 GC/ml being free DNA present since after DNase I treatment applied before nucleic acid extraction a reduction of nearly 2 logs was observed by qPCR (enzymatic treatment [ET]-qPCR) being the concentration of the order of 105 GC/ml. Infectious HAdV2 at the beginning of the experiment were of the order of 104 PFU/ml and thus 1 log less than when quantifying by ET-qPCR. Based on ET-qPCR results, HAdV2 showed an ∼2.5-log10 reduction for both natural and artificial seawater after 30 min of contact with chlorine. At the end of the assay, the reduction reached 2.7- and 3.3-log10 reductions for natural and artificial seawater, respectively, which indicates a reduction of 99.9% of the initial viral load. Regarding plaque assay, HAdV2 remained infectious throughout the assay and 1.7 × 102 PFU/ml in natural seawater and 4.5 × 101 PFU/ml in artificial seawater were observed (reductions of 2.3 log10 and 3.30 log10, respectively) after 60 min of contact with chlorine. The results obtained for MNV-1 disinfection (Fig. 2) indicate that this virus was more sensitive to chlorine than HAdV2 in both types of seawater tested. The initial 108 and 107 GC/ml observed by ET-qPCR for natural and artificial seawater, respectively, decrease to 104 and 102 after 60 min of contact with chlorine (4-log10 and 5-log10 reductions, respectively). However, when a plaque assay was performed, infectious MNV-1 was only observed after 20 min of contact (1.25 × 101 PFU/ml, in natural seawater, 4.2-log10 reduction; 1.65 × 101 PFU/ml, in artificial seawater, 4.4-log10 reduction). Treatment with RNase before nucleic acid extraction allowed us to eliminate at least 0.5 to 1 log10 of RNA accessible for RNases present in the samples.
JCPyV showed higher genome stability than the other two viruses analyzed (Fig. 2). Initial concentrations quantified by ET-qPCR showed 2.1 × 104 and 5.2 × 103 GC/ml for natural and artificial seawater, respectively (1 log less than when no DNase treatment was applied). At the end of the assay (60 min), 6 × 102 and 9.5 × 102 GC/ml were observed for natural and artificial seawater, respectively (1.45- and 1.33-log10 reductions). The infectivity of JCPyV was not evaluated, due to the technical limitations of JCPyV immunofluorescence assay (8) when applied to seawater samples.
F-approximated tests conducted to assess the homogeneity of the disinfection kinetic on the two replicates of every experiment provided the series of P values represented in Table 2. According to these P values, only assays in which the decay of JCPyV and HAdV2 was quantified by qPCR in natural seawater showed no homogeneity between replicates.
Table 2.
P values calculated by F-approximate tests to assess the homogeneity of the disinfection kinetics on the replicates of every experiment for each virus
| Expta |
Pb |
||
|---|---|---|---|
| MNV-1 | HAdV2 | JCPyV | |
| NS infectivity | 0.55 | 1R | NT |
| AS infectivity | 0.09 | 1R | NT |
| NS qPCR | 0.13 | 0.001 | <0.001 |
| AS qPCR | 0.16 | 0.05 | 0.69 |
| NS ET-qPCR | 0.92 | 0.44 | 0.15 |
| AS ET-qPCR | 0.35 | 0.09 | 0.84 |
NS, natural seawater; AS, artificial seawater.
NT, not tested; 1R, one replicate.
We conducted a statistical comparison of 12 selected pairs of homogeneous replicates with an F-test (P = 0.05) to assess the homogeneity of the overall pair and thus to know whether the two inactivation kinetics were equivalent or not. From these comparisons we may draw several conclusions. (i) The decay in the infectivity of HAdV2 follows kinetics similar to the decay of the HAdV2 genome copies (GC), as measured by ET-qPCR titers (Fig. 3) for both types of waters tested (P = 0.67152 and 0.1334, respectively). (ii) For MNV-1, the kinetics observed when disinfection was measured by qPCR were different when enzymatic treatment was or was not applied to both types of water (P = 0.00030 and 0.00861, respectively). However, the kinetics observed by ET-qPCR do not correlate to those observed by infectivity assays (P < 0.00001 and 0.00034, respectively) (Fig. 3). (iii) The disinfection kinetics of HAdV2 and JCPyV measured by ET-qPCR in natural seawater follow equivalent kinetics (P = 0.1842), whereas different kinetics were observed between these two viruses when artificial seawater was studied by qPCR, with or without enzymatic treatment (P = 0.00004 and 0.00951, respectively).
Fig 3.

Log reduction of HAdV2 and MNV-1 infectivity and ET-qPCR in natural (A) and artificial (B) seawater treated with free chlorine at 2.5 mg/liter (initial concentration). For HAdV2, the common EFH model is plotted as a dotted line; for MNV-1, the EFH model of infectivity is plotted as a dotted line, and the EFH model of ET-qPCR is plotted as a dashed line. The observed replicates of each series, of both viruses, are plotted as squares (ET-qPCR) or circles (infectivity).
The C(t), the product of the disinfectant concentration (C; in mg/liter) × the contact time (t; in min), and the t99.9 and t99.99 values were calculated to evaluate the disinfection effectiveness for all viruses (Table 3). The C(t) values of chlorine disinfection for a 3-log reduction C(t)[t99.9%] of HAdV2 by plaque assay in natural seawater were not reached. For artificial seawater, the value was 42.24 mg/liter × min. The C(t) t99.9% of JCPyV Mad4 could not be calculated in any replicate assayed, since the reduction rate did not reach 3 logs. The C(t)t99.9% values for MNV-1, as determined by plaque assay in natural and artificial seawater, were 11.49 and 20.82 mg/liter × min, respectively. This indicates that MNV-1 is more susceptible than HAdV2 and JCPyV Mad4 to chlorine treatment.
Table 3.
t99.9 and t99.99 values predicted to inactivate 3 and 4 logs, respectively, of each virus tested, along with the calculated C(t)a
| Expt | MNV-1 |
HAdV2 |
JCPyV Mad4 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3-Log inactivation |
4-Log inactivation |
3-Log inactivation |
4-Log inactivation |
3-Log inactivation |
4-Log inactivation |
|||||||
| t99.9 | C(t) | t99.99 | C(t) | t99.9 | C(t) | t99.99 | C(t) | t99.9 | C(t) | t99.99 | C(t) | |
| NS infectivity | 6.72 | 11.49 | 22.77 | 59.90 | NR | NR | NT | |||||
| AS infectivity | 11.83 | 20.82 | 16.70 | 41.70 | 24.69 | 42.24 | NR | NT | ||||
| NS qPCR | NR | NR | NR | NR | NR | NR | ||||||
| AS qPCR | 28.63 | 50.38 | 45.17 | 113 | 21.64 | 37.00 | 70.23 | 175 | NR | NR | ||
| NS ET-qPCR | 1.58 | 2.70 | NR | NR | NR | NR | NR | |||||
| AS ET-qPCR | 0.94 | 3.20 | 2.13 | 5.30 | NR | NR | NR | NR | ||||
NS, natural seawater; AS, artificial seawater; C(t), disinfectant concentration (in mg/liter) × the contact time (in minutes); NR, not reached; NT, not tested.
DISCUSSION
Chlorine was one of the earliest means used to disinfect seawater for depuration. Addition of chlorine is usually undertaken by the use of sodium hypochlorite solution at 2 to 3 mg of free chlorine/liter for a contact time of up to an hour (23).
The aim of the present study was to investigate the ability of chlorine in natural and artificial seawater to inactivate RNA and DNA viruses. MNV-1 was evaluated since it has been used as surrogate of human NoV, an RNA pathogen commonly involved in shellfish originated outbreaks, and HAdV and JCPyV were evaluated since they have been proposed as human fecal viral indicators (6). Differences were observed in the disinfection kinetics between the viruses used for genome persistence and for viral infectivity.
When chlorine is added to seawater, the concentration of residual chlorine decreases over time in two phases (14). In the first phase, the concentration decrease rapidly. This is followed by a much slower but steady decrease in concentration over time. A significant fraction of the chlorine demand observed in our study was caused by the reaction between the added chlorine and naturally occurring organic compounds in seawater, which reduced the availability of free chlorine for disinfection. Inorganic substances, such as ammonia (NH3) and iron (Fe2+), react rapidly with chlorine and create an instant chlorine demand (43). If seawater is considered to be at around pH 8, the addition of chlorine will result in the presence of HOCl−, OCl−, HOBr, and OBr−, which will create a hostile environment for living organisms (14). This may explain why chlorine inactivated the selected viruses assayed, even at low concentrations. It is widely accepted that free chlorine loses its virucidal and bactericidal activity in the presence of organic substances (40), such as host cell components and proteins in the suspension media (for instance, MEM contains reducing reagents, such as cysteine, which interfere with the action of sodium hypochlorite). Therefore, the use of a viral suspension in PBS prevented higher chlorine consumption in all of the experiments. Although a higher amount of naturally organic compounds in natural seawater might cause differences in inactivation between viruses in natural or artificial seawater in our experiments, the behaviors of viruses in both water types show no statistically significant differences.
Our findings suggest that MNV-1 is more sensitive to chlorine disinfection than is HAdV2 and is initially inactivated rapidly, as measured by a plaque assay in both types of tested waters. These results are in agreement with previous published studies (9, 12, 40). The reduction probably reflects the greater sensitivity of MNV-1 to free chlorine than to combined chlorine residuals. Although MNV-1 is inactivated rapidly, the time required for 2-log reduction was greater than in the studies cited above. This suggests that environmental factors, such as ionic strength and compounds naturally found in seawater, may slow the disinfection process but not affect its efficiency. More experimental data should be accumulated to define suitable disinfection treatments for NoVs.
HAdV2 was inactivated by free chlorine within the first min of contact time and might have been inactivated by combined chlorine, although not completely, up to 60 min (t99.9 not reached and of 24.69 min for natural and artificial seawater, respectively, as measured by plaque assays and not reached t99.99 for both types of water). HAdV2 behavior may follow bi- and/or triphasic inactivation curves during chlorine disinfection experiments, with an initial rapid inactivation phase, followed by more gradual inactivation, depending on water quality (32). This type of inactivation curve may occur if microorganisms are more resistant to disinfection or if the presence of aggregated individuals provides higher resistance to inactivation than dispersed organisms. Another possibility is that more than one mechanism of inactivation may affect HAdV2, which presents a more complex capsid than MNV-1 (12). A more recent study (31) suggests that free chlorine-treated HAdV2 retained their ability to bind to A549 cell receptors, despite being unable to form plaques. This indicates that the free chlorine inactivation mechanism involves the inhibition of postbinding events in the HAdV2 life cycle, such as early viral protein expression. In that same study, DNA isolated from HAdV2 that had been inactivated by free chlorine was amplified by PCR, which indicates that genome damage was not the only cause of inactivation.
The C(t) values observed here in analyzing seawater were higher than other published results using different water matrices (12, 26). The homogeneous composition and high salt concentration in this matrix may affect viral dispersion, aggregation, and stability.
Although JCPyV has been described as a potential human fecal indicator (6), data on its inactivation by chlorine have not been reported. The presence of human polyomaviruses has been reported (28) in 2/9 tertiary chlorine wastewater samples. Previous studies showed great prevalence of this virus in the environment (7, 28) and in source water (1). We present here the first data on the stability and chemical disinfection of JCPyV in seawater. According to our results, JCPyV genomes might be highly resistant to chlorine disinfection when measured by qPCR. We could only calculate the t99 on the basis of qPCR data in one of the eight qPCR replicates assayed (qPCR and ET-qPCR in both artificial and natural seawater), since no t99 values were reached in the other replicates in the evaluated period of time. Further studies on survival of JCPyV after disinfection should be conducted in the future.
Although qPCR and RT-qPCR are sensitive and specific assay systems, they appear to underestimate virus inactivation by free chlorine, since the nucleic acids of inactivated viruses may be amplified (35). Enzymatic treatments may differentiate intact from damaged viruses on the basis of differences in the ability of the viral proteic capsids to protect the genomes from proteases and nucleases. When the capsid is degraded, the naked DNA/RNA is more susceptible to nuclease degradation than capsid-enclosed DNA/RNA (35). This would allow disinfection studies of viruses that do not grow in cell culture, such as human NoVs. In the present study, enzymatic treatment solved part of this problem and generated results that were more similar to those obtained in infectivity assays (30). Our data, taking into account the limited number of replicates assayed, indicate that the kinetics of disinfection of HAdV2, as measured by infectivity assays or by ET-qPCR, are similar. Hence, ET-qPCR could be used to infer the infectiousness of HAdVs in the conditions assayed. However, this correlation was not observed for MNV-1, although ET-qPCR yielded results more similar to the data on infectivity than to the qPCR data. Further work on these treatments should be carried out in the future to develop a molecular method that can provide results that are as similar as possible to those obtained from infectivity assays, which are usually time-consuming and expensive. The data obtained here suggest that the times applied to the disinfection of water to be used in shellfish depuration should be revised since, after 60 min of contact with chlorine, infectious adenovirus was still detected. Further studies should focus on evaluating the time needed for the efficient removal of all viruses present in water used for depuration. Since different viruses present different inactivation kinetics, those more stable to chlorine action should be used in these studies. Alternative disinfectants such us UV irradiation or ozone may also be compared to chlorine treatment, although infectious HAdV has also been detected in shellfish samples depurated with waters treated with these agents (J. Rodriguez-Manzano, unpublished data).
In summary, our study has shown that the use of 2.5 mg of free chlorine/liter in seawater requires a long contact time in order to significantly decrease the viral load. We found that the genome of JCPyV Mad4 is more resistant to chlorine disinfection than those of HAdV2 and MNV-1, when molecular methods were applied, providing new information about JCPyV stability in seawater. This could be because JCPyV presents a complex genomic structure.
HAdV2 show high resistance to chlorine treatment, and infectious viral particles were still observed at the end of the experiment. MNV-1 capsid integrity is more affected by chlorine treatment than its genome compared to the other viruses tested. According to the statistical analysis, the enzymatic treatment by DNase applied before qPCR was useful to infer the HAdV2 infectivity decay caused by chlorine. In contrast, further studies are needed to improve the use of RNase treatment.
Chlorine application to the disinfection of water for shellfish depuration purposes should be further evaluated in order to increase the contact times currently applied. All of the results obtained were statistically equivalent when artificial and natural seawater were studied, suggesting that artificial seawater could also be used in shellfish depuration tanks.
ACKNOWLEDGMENTS
This study was supported by a European Commission Framework Program 7 project, Integrated Monitoring and Control of Food-Borne Viruses in European Food Supply Chains (VITAL; grant KBBE 213178), led by the coordination team of Nigel Cook (FERA, United Kingdom), Martin D'Agostino (FERA, United Kingdom), and Franco M. Ruggeri (ISS, Italy); by project AGL2008-05275-C03-01/ALI, Integrated Risk Analysis Related to the Consumption of Pathogenic Viruses Present in Water and Food, which was funded by the Spanish Ministry of Science; and by project A/017172/08, Development of Methods to Evaluate the Risk Associated with Shellfish Polluted with Human Viruses, funded by the Spanish Ministry of Cooperation. During this study, A.D.A.C. was a fellow of the Conselho de Aperfeiçoamento de Pessoal de Ensino Superior, CAPES-PDEE, Brazil (with a 1-year period of collaboration in the University of Barcelona, project number 4038-08-8). A.C. was a fellow of the Spanish Ministry of Science.
We are grateful to Carlos Maluquer de Motes for critically reviewing the manuscript.
Footnotes
Published ahead of print 6 July 2012
REFERENCES
- 1. Albinana-Gimenez N, et al. 2009. Analysis of adenoviruses and polyomaviruses quantified by qPCR as indicators of water quality in source and drinking water-treatment plants. Water Res. 43:2011–2019 [DOI] [PubMed] [Google Scholar]
- 2. Bae J, Schwab KJ. 2008. Evaluation of murine norovirus, feline calicivirus, poliovirus, and MS2 as surrogates for human norovirus in a model of viral persistence in surface water and groundwater. Appl. Environ. Microbiol. 74:477–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baert L, et al. 2008. Detection of murine norovirus 1 by using plaque assay, transfection assay, and real-time reverse transcription-PCR before and after heat exposure. Appl. Environ. Microbiol. 74:543–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat Soc. Ser. B Stat. Methodol. 57:289–300 [Google Scholar]
- 5. Black S, Thurston-Enriquez JA, Gerba CP. 2009. Determination of CT values for chlorine of resistant enteroviruses. J. Environ. Sci. Health A Tox. Hazard Subst. Environ. Eng. 44:336–339. [DOI] [PubMed] [Google Scholar]
- 6. Bofill-Mas S, Pina S, Girones R. 2000. Documenting the epidemiologic patterns of polyomaviruses in human populations by studying their presence in urban sewage. Appl. Environ. Microbiol. 66:238–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bofill-Mas S, et al. 2006. Quantification and stability of human adenoviruses and polyomavirus JCPyV in wastewater matrices. Appl. Environ. Microbiol. 72:7894–7896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Calgua B, Barardi CR, Bofill-Mas S, Rodriguez-Manzano J, Girones R. 2011. Detection and quantitation of infectious human adenoviruses and JC polyomaviruses in water by immunofluorescence assay. J. Virol. Methods 171:1–7 [DOI] [PubMed] [Google Scholar]
- 9. Cannon JLE, et al. 2006. Surrogates for the study of norovirus stability and inactivation in the environment: a comparison of murine norovirus and feline calicivirus. J. Food Prot. 69:2761–2765 [DOI] [PubMed] [Google Scholar]
- 10. Cheremisinoff NP. 2002. Handbook of water and wasterwater treatment technologies. Butterworth Heinemann, Woburn, MA [Google Scholar]
- 11. Correa AA, et al. 2007. Depuration dynamics of oysters (Crassostrea gigas) artificially contaminated by Salmonella enterica serovar Typhimurium. Mar. Environ. Res. 63:479–489 [DOI] [PubMed] [Google Scholar]
- 12. Cromeans TL, Kahler AM, Hill VR. 2010. Inactivation of adenoviruses, enteroviruses, and murine norovirus in water by free chlorine and monochloramine. Appl. Environ. Microbiol. 76:1028–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deborde M, Von Gunten U. 2008. Reactions of chlorine with inorganic and organic compounds during water treatment–kinetics and mechanisms: a critical review. Water Res. 42:13–51 [DOI] [PubMed] [Google Scholar]
- 14. Din AMS, Rasheed AA, Hammoud AA. 2000. On the chlorination of seawater. Desalination 129:53–62 [Google Scholar]
- 15. Fleming LE, et al. 2006. Oceans and human health: emerging public health risks in the marine environment. Mar. Pollut. Bull. 53:45–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fitzgerald A, Syvret M, Hamilton A, Pyke M. 2011. Review for industry of reduced depuration times for the mussel Mytilus edulis, p. 97 Scottish Aquaculture Research Forum, Pitlochry, Scotland: http://www.sarf.org.uk/cms-assets/documents/29383-342050.sarf066.pdf [Google Scholar]
- 17. Haas CN, Joffe J. 1994. Disinfection under dynamic conditions: modification of Hom's model for decay. Environ. Sci. Technol. 28:1367–1369 [DOI] [PubMed] [Google Scholar]
- 18. Hernroth BE, Allard A. 2007. The persistence of infectious adenovirus (type 35) in mussels (Mytilus edulis) and oysters (Ostrea edulis). Int. J. Food Microbiol. 13:296–302 [DOI] [PubMed] [Google Scholar]
- 19. Hernroth BE, Conden-Hansson AC, Rehnstam-Holm AS, Girones R, Allard AK. 2002. Environmental factors influencing human viral pathogens and their potential indicator organisms in the blue mussel, Mytilus edulis: the first Scandinavian report. Appl. Environ. Microbiol. 68:4523–4533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hewitt J, Rivera-Aban M, Greening GE. 2009. Evaluation of murine norovirus as a surrogate for human norovirus and hepatitis A virus in heat inactivation studies. J. Appl. Microbiol. 107:65–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hsu CC, Riley LK, Livingston RS. 2007. Molecular characterization of three novel murine norovirus. Virus Genes 34:47–155 [DOI] [PubMed] [Google Scholar]
- 22. Koopmans M, Duizer E. 2004. Foodborne viruses: an emerging problem. Int. J. Food Microbiol. 90:23–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee R, Lovatelli A, Ababouch L. 2008. Bivalve depuration: fundamental and practical aspects. Fisheries technical paper 511. Food and Agricultural Organization of the United Nations, Rome, Italy [Google Scholar]
- 24. Lee CY, Panicker G, Bej AK. 2003. Detection of pathogenic bacteria in shellfish using multiplex PCR followed by CovaLinkTM NH microwell plate sandwich hybridization. J. Microbiol. Methods 53:199–209 [DOI] [PubMed] [Google Scholar]
- 25. Letson D. 2008. Oceans and human health: human dimensions, p 91–98 In Walsh PJ, et al. (ed), Oceans and human health: risks and remedies from the sea. Elsevier, Burlington, MA [Google Scholar]
- 26. Lim MY, Kim JM, Ko G. 2010. Disinfection kinetics of murine norovirus using chlorine and chlorine dioxide. Water Res. 44:3243–3251 [DOI] [PubMed] [Google Scholar]
- 27. Marino A, Lombardo L, Fiorentino C. 2005. Uptake of Escherichia coli, Vibrio cholerae non-o1, and Enterococcus durans by and depuration of mussels (Mytilus galloprovincialis). J. Food Microbiol. 99:281–286 [DOI] [PubMed] [Google Scholar]
- 28. McQuaig SM, Scott TM, Lukasik JO, Paul JH, Harwood VJ. 2009. Quantification of human polyomaviruses JC virus and BK virus by TaqMan quantitative PCR and comparison to other water quality indicators in water and fecal samples. Appl. Environ. Microbiol. 75:3379–3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mena KD, Gerba CP. 2009. Waterborne adenovirus. Rev. Environ. Contam. Toxicol. 198:133–167 [DOI] [PubMed] [Google Scholar]
- 30. Nuanualsuwan S, Cliver DO. 2002. Pretreatment to avoid positive RT-PCR results with inactivated viruses. J. Virol. Methods 104:217–225 [DOI] [PubMed] [Google Scholar]
- 31. Page MA, Shisler JL, Mariñas BJ. 2010. Mechanistic aspects of adenovirus serotype 2 inactivation with free chlorine. Appl. Environ. Microbiol. 76:2946–2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Page MA, Shisler JL, Mariñas BJ. 2009. Kinetics of adenovirus type 2 inactivation with free chlorine. Water Res. 43:2916–2926 [DOI] [PubMed] [Google Scholar]
- 33. Pal A, Sirota L, Maudru T, Peden K, Lewis AM. 2006. Real-time, quantitative PCR assays for the detection of virus-specific DNA in samples with mixed populations of polyomaviruses. J. Virol. Methods 135:32–42 [DOI] [PubMed] [Google Scholar]
- 34. Patel MM, et al. 2008. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg. Infect. Dis. 14:1224–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pecson BM, Martin LV, Kohn T. 2009. Quantitative PCR for determining the infectivity of bacteriophage MS2 upon inactivation by heat, UV-B radiation, and singlet oxygen: advantages and limitations of an enzymatic treatment to reduce false-positive results. Appl. Environ. Microbiol. 75:5544–5554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rigotto C, Sincero TC, Simões CMO, Barardi CRM. 2005. Detection of adenoviruses in shellfish by means of conventional PCR, nested-PCR and integrated cell culture PCR (ICC/PCR). Water Res. 39:297–304 [DOI] [PubMed] [Google Scholar]
- 37. Roderick GE, Schneider KR. 1994. Depuration and relaying of molluscan shellfish, p 331–363 In Hackney CK, Pierson MD. (ed), Environmental indicators and shellfish safety. Chapman & Hall, New York, NY [Google Scholar]
- 38. Sauerbrei A, Wutzler P. 2009. Testing thermal resistance of viruses. Arch. Virol. 154:115–119 [DOI] [PubMed] [Google Scholar]
- 39. Shah KV. 1995. Polyomaviruses, p 2027–2043 In Fields BN, Knipe DM, Howley PM. (ed), Fields virology, 3rd ed Raven Publishers, Philadelphia, PA [Google Scholar]
- 40. Shin GA, Sobsey MD. 2008. Inactivation of norovirus by chlorine disinfection of water. Water Res. 42:4562–4568 [DOI] [PubMed] [Google Scholar]
- 41. Thurston-Enriquez JA, Haas CN, Jacangelo J, Gerba CP. 2003. Chlorine inactivation of adenovirus type 40 and feline calicivirus. Appl. Environ. Microbiol. 69:3979–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ueki Y, et al. 2007. Persistence of calicivirus in artificially contaminated oysters during depuration. Appl. Environ. Microbiol. 73:5698–5701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Warton B, Heitz A, Joll C, Kagi R. 2006. A new method for calculation of the chlorine demand of natural and treated waters. Water Res. 40:2877–2884 [DOI] [PubMed] [Google Scholar]
- 44. Wobus CE, Thackray LB, Virgin HW. 2006. Murine norovirus: a model system to study norovirus biology and pathogenesis. J. Virol. 80:5104–5112 [DOI] [PMC free article] [PubMed] [Google Scholar]