

Abstract
Summary: We developed a novel algorithm, PurityEst, to infer the tumor purity level from the allelic differential representation of heterozygous loci with somatic mutations in a human tumor sample with a matched normal tissue using next-generation sequencing data. We applied our tool to a whole cancer genome sequencing datasets and demonstrated the accuracy of PurityEst compared with DNA copy number-based estimation.
Availability: PurityEst has been implemented in PERL and is available at http://odin.mdacc.tmc.edu/~xsu1/PurityEst.html
Contact: xsu1@mdanderson.org
1 INTRODUCTION
Next-generation sequencing (NGS) provides a platform to comprehensively characterize somatic mutations, DNA copy number changes and rearrangements in tumor tissues. Because tumor tissues usually consist of a mixture of multiple tumor clones and normal cells including fibroblasts and infiltrating lymphocytes, the observed magnitude of copy number changes is diminished, which is basis of tumor clone purity estimation using SNP array data (Bengtsson et al., 2010; Carter et al., 2012; Loo et al., 2010; Sun et al., 2009; and Yu et al., 2011). Gusnanto et al. (2011) converted mapped reads to DNA copy number ratios between tumor and normal genomes for purity estimation. In principle, NGS also provides an alternative to copy number-based methods, which is to use mutant allele fractions in the heterozygous loci with somatic mutations in a tumor mixture. However, modeling the mutant allele fractions is complicated by two factors. One is that mutant allele fractions in a sample may take multiple levels. The founder mutations may have the higher levels and latent mutations lower levels. The second factor is that copy number change can also alter the observed fractions. When the mutant allele is amplified, the observed mutant allele fraction can be increased; when the wild-type allele is amplified (lost), the mutant allele fraction can be decreased (increased). When the mutant allele is lost, the mutant allele is simply not observable.
Here, we propose a simple approach to the purity estimation problem. We assume that the tumor tissue can be largely approximated by a mixture of a normal clone and a tumor clone. Our method gives a purity estimate from somatic mutations in each chromosome and takes a robust average of the chromosome-wide estimates to be the purity estimate of the tumor tissue. Since copy number changes can both enrich and deplete the fractions of mutated alleles depending on whether the copy number change occurs to the mutated allele or the wild allele, it is unlikely to affect the chromosome-wide estimate drastically.
2 METHODS
We call our method PurityEst, which estimates the fraction of tumor DNA molecules that is different from the normal matched tissue. A pure tumor sample should show a mean frequency of 0.5 for mutant alleles at heterozygous loci with somatic mutations, whereas contamination of tumor tissue with normal tissue is expected to lower the mutant allele fractions. The tumor purity 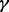 is inferred from the allelic differential representation of heterozygous loci with somatic mutations comparing a tumor sample and a matched normal tissue using the following formulation:
is inferred from the allelic differential representation of heterozygous loci with somatic mutations comparing a tumor sample and a matched normal tissue using the following formulation:
where 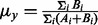 is the mutant allele fraction obtained from the tumor sample, and
is the mutant allele fraction obtained from the tumor sample, and 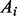 indicates the wild allele count,
indicates the wild allele count, 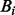 the mutant allele count in the heterozygous loci with somatic mutations, the summations include all heterozygous loci with somatic mutations;
the mutant allele count in the heterozygous loci with somatic mutations, the summations include all heterozygous loci with somatic mutations; 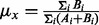 is the mutant allele fraction obtained from the normal sample and
is the mutant allele fraction obtained from the normal sample and 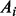 indicates the wild allele count,
indicates the wild allele count, 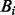 the mutant allele count in SNP heterozygous loci, the summations include all the SNP heterozygous loci attributed to germline mutations. Note that the set of the somatic mutations is assumed to be mutually exclusive with the set of germline mutations and the latter set is usually much greater than the former. Theoretically, the expected value of
the mutant allele count in SNP heterozygous loci, the summations include all the SNP heterozygous loci attributed to germline mutations. Note that the set of the somatic mutations is assumed to be mutually exclusive with the set of germline mutations and the latter set is usually much greater than the former. Theoretically, the expected value of 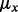 is 0.5. However, empirical data showed that the mean value is typically slightly lower than 0.5, which suggests that different alleles are not equally represented with the current sequencing technology. Hence, we choose to use the computation of the empirical value of
is 0.5. However, empirical data showed that the mean value is typically slightly lower than 0.5, which suggests that different alleles are not equally represented with the current sequencing technology. Hence, we choose to use the computation of the empirical value of 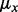 to correct for this representational bias.
to correct for this representational bias.
The above formulation does not explicitly consider effects of copy number gains and losses in tumor genomes, which can bias the tumor purity estimation. However, based on our empirical observations, the biases appear to affect only a small fraction of the tumor genome. Therefore, to minimize the effect of such biases, we choose to estimate tumor purity from each autosome 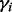 separately, and obtain a final estimate from robustly averaging the
separately, and obtain a final estimate from robustly averaging the 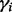 , excluding the outliers. In PurityEst, we implemented the ‘extreme studentized deviate’ (ESD) multiple-outlier procedure (Rosner, 1983) to remove the outliers. The tumor purity was estimated by:
, excluding the outliers. In PurityEst, we implemented the ‘extreme studentized deviate’ (ESD) multiple-outlier procedure (Rosner, 1983) to remove the outliers. The tumor purity was estimated by: 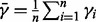 and
and 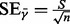 where n is the number of autosomes excluding the outliers, and
where n is the number of autosomes excluding the outliers, and 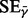 is the estimated error from the sample standard deviation.
is the estimated error from the sample standard deviation.
3 RESULTS AND DISCUSSION
To test the accuracy of PurityEst method, we re-analyzed a publically available dataset (Berger et al., 2011). The dataset was generated with Illumina GAII, containing 7 matched prostate cancer samples with the paired-end 76 nt reads. We used MOSAIK (Hiller et al., 2008) to align the reads to the reference genome (GRCh37/hg19) and used GigaBayes (Marth et al., 1999) to detect the single-nucleotide variations (SNVs). We filtered out all known SNVs based on two public databases: UCSC dbSNP 135 and the 1000 Genomes Project SNP database. We then determined the somatic status of each SNV by comparing the genotypes between matched normal tissue and tumor samples. Both wild and mutant allele counts at each heterozygous loci of both SNPs and somatic mutations were generated by GigaBayes. The tumor samples have a mean genomic coverage ranging from 29.5 to 35.8, and the matched normal tissue samples with a mean coverage ranging from 18.8 to 34.9.
To estimate the tumor purity for the samples, we first estimated the tumor purity level for each autosomal chromosome in a sample. We removed the outliers in the overall tumor purity estimation when the outliers were detected. Figure 1A showed the autosomal purity levels of one of the samples. The purity level estimated from chromosome 10 was found to be substantially lower than other chromosomes. It is not clear what caused this outlier, as no major copy number change in the chromosome. One possible cause is that the tumor was made of multiple clones, and chromosome 10 was protected from mutation. Alternatively, it was caused by representational bias. We note that had the chromosome corresponded to an outlier with high fraction of mutated alleles, we would have attributed the results to founder mutations.
Fig. 1.

(A) Purity estimations by PurityEst for each autosomal chromosome in sample PR-1701. The error bars were estimated by bootstrap sampling. Chromosome 10 was found to be an outlier. The mean purity excluding the outlier is 0.535. (B) Scatter plot of tumor purity estimates from ABSOLUTE and PurityEst. The correlation coefficient between two kinds of estimates is 0.91 from seven patient samples
Figure 1B showed the PurityEst estimates along with the estimates reported by Berger et al. (2011), who used copy number changes derived from SNP array data of the same seven prostate cancer samples. The correlation coefficient between the two kinds of estimates is 0.91, demonstrating that PurityEst estimates are consistent with that from DNA copy number data.
In summary, we showed that PurityEst can be used to estimate tumor purity based on mutant allele fractions in a mixture of a tumor clone and a normal clone. Multiple factors, such as coverage, copy number changes and representational bias can all potentially affect the purity estimation. Our method can handle some, but not all of the effects. When adequate coverage is available, our method may be extended to model multiple mutant allele fractions that reflect intra-tumor heterogeneity. We expect our method to be a simple and effective solution for tumor purity estimation in cancer studies and invite users to test our software.
Funding: National Cancer Institute (U24CA143883, in part); and National Center for Research Resources (3UL1RR024148 and ULTR000371, in part and a grant in part from the H.A. and Mary K. Chapman Foundation and the Michael & Susan Dell Foundation).
Conflict of Interest: none declared.
REFERENCES
- Bengtsson H, et al. TumorBoost: Normalization of allele-specific tumor copy numbers from a single pair of tumor-normal genotyping microarrays. BMC Bioinformatics. 2010;11:1471–2105. doi: 10.1186/1471-2105-11-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotech. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusnanto A, et al. Correcting for cancer genome size and tumor cell content enables better estimation of copy number alterations from next-generation sequence data. Bioinformatics. 2011;28:40–47. doi: 10.1093/bioinformatics/btr593. [DOI] [PubMed] [Google Scholar]
- Hiller LW, et al. Whole-genome sequencing and variant discovery in C. elegans. Nat. Methods. 2008;5:183–188. doi: 10.1038/nmeth.1179. [DOI] [PubMed] [Google Scholar]
- Loo PV, et al. Allele-specific copy number analysis of tumors. PNAS. 2010;107:16910–16915. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth GT, et al. A general approach to single-nucleotide polymorphism discovery. Nat. Genet. 1999;23:452–456. doi: 10.1038/70570. [DOI] [PubMed] [Google Scholar]
- Rosner B. Percentage points for a generalized ESD many outlier procedure. Technometrics. 1983;25:165–172. [Google Scholar]
- Sun W, et al. Integrated study of copy number states and genotype calls using high-density SNP arrays. Nucleic Acids Res. 2009;37:5365–5377. doi: 10.1093/nar/gkp493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, et al. BACOM: in silico detection of genomic deletion types and correction of normal cell contamination in copy number data. Bioinformatics. 2011;27:1473–1480. doi: 10.1093/bioinformatics/btr183. [DOI] [PMC free article] [PubMed] [Google Scholar]