

Abstract
Hemoglobin is released from lysed red blood cells in numerous clinical settings. HMGB1 is a nuclear and cytosolic DNA-binding protein released from injured cells that has been shown to play an important role in inducing inflammation. Because both of these endogenous molecules are frequently present in sites of necrosis and inflammation, we studied their interaction on the activation of macrophages. We report here that hemoglobin and HMGB1 synergize to activate mouse macrophages to release significantly increased pro-inflammatory cytokines. Addition of microbial ligands that activate through TLR2 or TLR4 resulted in further significant increases, in a “3-way” synergy between endogenous and microbial ligands. The synergy was strongly suppressed by hemopexin, an endogenous heme-binding plasma protein. The findings suggest that hemoglobin may play an important role in sterile as well as infectious inflammation, and that endogenous hemopexin can modulate this response. Administration of hemopexin may be beneficial in clinical settings characterized by elevated extracellular hemoglobin and HMGB1.
INTRODUCTION
Hemoglobin, the predominant oxygen carrying protein in red blood cells, is released into the extracellular space when red blood cells undergo lysis. High concentrations of extracellular hemoglobin occur in tissues after injury or necrosis from any etiology, and in the vascular space in clinical settings that include hemolytic syndromes, ischemia-reperfusion injury, cardiopulmonary bypass, renal dialysis, and after transfusion of blood products. A series of early studies revealed that hemoglobin synergistically increases the production of TNF and other cytokines by macrophages exposed to LPS and other activators(1-3). It has previously been proposed that hemoglobin enhances the presentation of LPS to immune cells, resulting in a synergistic increase in cytokine release (3-5). Other studies, however, implicated a critical role of the heme moiety of hemoglobin in the mechanism that enhances macrophage TNF release during endotoxemia (6). This synergistic activation of immune cells by heme and LPS has recently been independently confirmed (7). During the course of studying the influence of hemoglobin and free heme on macrophage responses to LPS, we recently observed that these molecules also synergistically enhance the macrophage response to TLR4-independent ligands of microbial origin, and to killed E. coli and S. aureus (8). The synergy was significantly suppressed by a plasma glycoprotein, hemopexin (Hx) that binds to free heme with extraordinary affinity (Kd < 1 pM) (9;10). Accordingly, we reasoned that the mechanism of synergy might extend beyond a direct interaction between hemoglobin and the LPS molecule to involve endogenous inflammatory mediators, and that if such a synergy was present that hemopexin might suppress these synergistic interactions as well.
HMGB1, a nuclear and cytosolic DNA-binding protein, is actively secreted from stimulated immune cells, and passively released from injured cells (11). Extracellular HMGB1 occupies a critical role in the pathogenesis of inflammation in the setting of tissue necrosis, with or without infection (11). Recent evidence indicates that TLR4 signaling is required for HMGB1 dependent stimulation of macrophage cytokine release (12). Increased concentrations of extracellular hemoglobin are ubiquitous when capillaries are breached, and secondary breakdown of the red cell membrane occurs. Therefore, high concentrations of both HMGB1 and hemoglobin accumulate at sites of tissue injury, inflammation, ischemia, cell injury, hemorrhage and disruption of the microenvironment. As these scenarios frequently complicate surgery, trauma, or tissue necrosis, or illnesses that involve primary or secondary inflammation or fragility of blood vessels, we hypothesized that hemoglobin may synergistically enhance HMGB1-induced release of TNF and other cytokines.
We studied the interactions of hemoglobin and HMGB1 on the activation of macrophages. The results indicate that these two endogenous molecules act synergistically to enhance cytokine production from macrophages. Moreover, adding microbial ligands results in significant “3-way” synergy. Importantly, we found that all of these responses were reversed by hemopexin.
MATERIALS AND METHODS
Materials
The following TLR agonists were purchased: smooth LPS from E.coli O55:B5 (List Biologicals), Pam3Cys (EMC Microcollections). All these TLR agonists were dissolved in pyrogen-free H2O and saved as aliquots at −80°C. Diphenyleneiodonium (DPI), N-acetyl-L-systeine (NAC), allopurinol, L-NAME, and piceatannol were obtained from Sigma. C57BL/6, TLR2 knockout, C3H/HeN, and C3H/HeJ mice were obtained from Charles River Labs. MyD88 knockout mice were a kind gift from Dr. Joseph El-Khoury, (Department of Medicine, Massachusetts General Hospital, Boston, MA). TRIF knockout mice were obtained from Jackson Labs. RAGE knockout mice were obtained from Helena Erlandsson-Harris (Department of Medicine, Karolinska Institute, Stockholm, Sweden). The Institutional Animal Care and Use Committee at Massachusetts General Hospital approved the animal protocols used in this study. Recombinant rat HMGB1 was expressed in E. coli and purified to homogeneity as previously described (13;14).
Purification of mouse and human hemoglobin
Hemoglobin was purified as previously described with modifications (15), utilizing pyrogen-free conditions. Briefly, mouse blood was collected from C57BL/6 mice by cardiac puncture. Human blood was collected aseptically from healthy human volunteers. The blood was washed with an equal weight of isotonic saline solution (0.9% NaCl, w/v) three times by centrifugation at 1,000×g to remove serum proteins. Equal volumes of saline were added to the pellet containing red blood cells and this solution was sonicated 5 × 10 sec at amplitude 40% with 1 min laps between pulses in Branson 450 sonicator from Branson Ultrasonics Corporation (Danbury, CT). The hemoglobin solution was diluted with an equal volume of saline and subjected to a second centrifugation at 2,000×g for 1 h. The resulting hemoglobin solution, removed from the center layer, was filtered through 0.22 μm Millipore membranes and saved at −20°C in the dark. The concentration of purified hemoglobin was measured by Micro-BCA. The purity of the hemoglobin was confirmed to be > 99% by non-denaturing PAGE and high pressure liquid chromatography.
Purification of hemopexin from mouse or human serum
Mouse serum Hx (mHx) or human serum Hx (hHx) was purified by using heme affinity chromatography essentially as we have described (16). Briefly, serum was filtered through 0.22 μm Millipore membranes, and then albumin was precipitated and removed by adding cold 1.68% rivanol solution (pH 8.0). The resultant post-rivanol precipitation sample was dialyzed against pyrogen-free PBS. Protease inhibitors (0.5 mM AEBSF, 10 μM E-64, 2 μg/ml aprotinin and 1 μM pepstatin A) were added to the dialyzed post-rivanol precipitation for 15 minutes with gentle agitation at 4°C. The mixture was then applied to a 6 ml hemin-agarose column (Sigma) 3 times, followed by extensive washing with 1,200 ml PBS containing 0.5 M NaCl overnight at 4°C to remove unbound proteins. Hx bound to the column was eluted using 0.2 M citric acid (pH 2.0) and then immediately neutralized with 10 M NaOH. Proteins in the buffer were exchanged, concentrated in PBS at 4°C using Centriprep YM-30 (Millipore, MA), and saved in aliquots at −80°C.
Limulus Amoebocyte Lysate (LAL) assay
The LAL assay was performed as previously described (17) to verify the endotoxin level to be lower than 0.01EU/mg in HMGB1, purified hemoglobin and hemopexin before use in cell cultures.
Preparation of macrophages
Bone marrow-derived macrophages (BMDMs) were prepared from mice, as described (18) with minor modifications (16;19). BMDMs were seeded in wells of 96-well culture plates at a density of 4.0 × 105 cells/cm2 (1.28 × 105/well) and were incubated at 37°C under humidified 5% CO2 to allow cells to adhere before use in assays.
Macrophage culture and cytokine assays
BMDMs were washed 3 times in serum-free medium, followed by incubation overnight with HMGB1, with or without hemoglobin, or with different TLR agonists as desired at indicated concentrations. Purified mouse or human hemopexin was added to the culture in some experiments as noted. Concentrations of TNF and IL-6 in the supernatants were quantitated by enzyme-linked immunosorbent assay (ELISA) (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
Statistics
Except where indicated, representative data from at least three experiments are presented in the figures. Data are expressed as means, and error bars represent SE. The data were analyzed by GraphPad Prism 5 (GraphPad software, La Jolla, CA). We used t tests to compare conditions with and without hemoglobin or with and without hemopexin. One-way ANOVA was used to analyze the data for the dose-dependent synergy between hemoglobin and HMGB1, the dose-dependent effect of hemopexin, and the dose dependent effect of piceacetannol. Values of p < 0.05 were considered to be statistically significant.
RESULTS
Hemoglobin strongly synergizes with HMGB1 to induce TNF and IL-6 from macrophages
Different concentrations of HMGB1 were incubated with a predetermined optimized concentration of mouse hemoglobin in cell culture with BMDMs, and concentrations of TNF and IL-6 were measured in the culture supernatants. As expected, treatment with HMGB1 significantly induced TNF and IL-6 release by macrophages (Figure 1). Hemoglobin alone did not induce detectable TNF at concentrations up to 1,000 μg/ml (8), but significantly enhanced TNF and IL-6 production by BMDMs induced by different concentrations of HMGB1 (Figure 1A, B). The effect of hemoglobin on TNF and IL-6 production was dose dependent (Figure 1C, D). Human hemoglobin also synergized with HMGB1 to induce high concentrations of TNF and IL-6 (data not shown). Together, these results indicate that hemoglobin synergistically increases HMGB1-dependent macrophage activation. We also found that free heme at low concentrations (< 1μM) synergizes with HMGB1 to induce increased TNF and IL-6 production from macrophages (data not shown).
Figure 1. Hemoglobin synergizes with HMGB1.

Bone marrow-derived macrophages (BMDMs) from C57BL/6 mice were washed 3 times with serum-free medium and then cultured overnight with HMGB1 at different concentrations alone or with mouse hemoglobin (30 μg/ml) (A, B), or HMGB1 at 4 μg/ml with mouse hemoglobin at different concentrations (C,D). Concentrations of TNF (A, C) or IL-6 (B, D) in the supernatants were determined by ELISA. The results represent mean ± s.e. and are representative of more than three independent experiments. * P < .05, ** P < .01, compared between cells treated with and without hemoglobin.
Synergistic induction of pro-inflammatory cytokines by HMGB1 and hemoglobin are partially dependent on TLR2, TLR4, RAGE, MyD88 and TRIF
It has been proposed that the cytokine release induced by HMGB1 may be mediated by several receptors including TLR2, TLR4 and the Receptor for Advanced Glycation Endproducts (RAGE) (12). Thus we next assessed the role of TLR2, TLR4, and RAGE signaling pathways in the synergy between hemoglobin with HMGB1. We compared cytokine responses of BMDMs from wild-type and knockout mice incubated with HMGB1 in the absence or presence of hemoglobin. Responses of BMDMs from TLR2 and RAGE knockout mice were compared with those of BMDMs from control wild type C57/BL6 mice, and responses of BMDMs from TLR4 deficient (C3H/HeJ) mice were compared with those of BMDMs from control (C3H/HeN) mice (Figure 2). TNF induced by HMGB1 alone was variably decreased in the TLR2 knockout cells, the C3H/HeJ cells, and the RAGE knockout cells compared to the control wild-type cells (data not shown). However, the synergy between HMGB1 and hemoglobin was still detected, despite the diminished responses in mice deficient in TLR2 (Figure 2A) or TLR4 (Figure 2B) or RAGE (Figure 2C), although in some cases there was a small percentage decrease in the synergy. These findings suggest that TLR2, TLR4, or RAGE each could be partially involved, but are not required for the synergy. We next studied cells obtained from myeloid differentiation primary response gene 88 (MyD88) knockout mice and TIR-domain-containing adapter-inducing interferon-β (TRIF) knockout mice to investigate if the adaptor proteins MyD88 and TRIF were involved in the synergy. Although as expected, BMDMs from TRIF and MyD88 knockout mice produced much less TNF (only ~ 5%) as compared to the wild type cells, there was still detectable synergy between HMGB1 and hemoglobin in cytokine production by both MyD88 and TRIF knockout BMDMs (Figure 2D). Together, these data suggest that TLR signaling may be involved but is not essential for the synergy to be present.
Figure 2. Synergy of hemoglobin with HMGB1 is not completely dependent on TLR2 or TLR4 or RAGE or MyD88 or TRIF.

BMDMs from (A) C57BL/6 or TLR2 knockout mice (TLR2KO) or (B) C3H/HeN mice or C3H/HeJ mice with deficient TLR4 or (C) C57BL/6 or RAGE knockout mice (RAGE KO) or (D) C57BL/6 or MyD88 knockout (MyD88 KO) or TRIF knockout (TRIF KO) mice were washed 3 times with serum-free medium and then cultured overnight with HMGB1 (4 μg/ml) with PBS or mouse hemoglobin (30 μg/ml). Concentrations of TNF in the supernatants were determined by ELISA. Each graph represents the percentage of cytokine production when cytokine production by HMGB1 with PBS on wild-type cells is assigned to 100%. Results represent mean ± s.e. and are representative of four independent experiments. * P < .05, ** P < .01, compared between cells treated with and without hemoglobin.
Involvement of induction of ROS and activation of spleen tyrosine kinase in the mechanisms of synergy
We used pharmacological inhibitors to investigate the role of reactive oxygen species (ROS) in the synergy between hemoglobin and HMGB1. As shown in figure 3A, the anti-oxidant NAC significantly decreased, and the flavoprotein inhibitor DPI partially decreased TNF that was synergistically induced by hemoglobin and HMGB1, whereas the xanthine oxidase inhibitor allopurinol and NO synthase inhibitor L-NAME did not decrease the synergy (figure 3A). NAC also decreased TNF induced by HMGB1 in the absence of hemoglobin. It has been reported that the synergy of heme with LPS is mediated through spleen tyrosine kinase (Syk)-dependent ROS generation (7). To evaluate the role of Syk in the hemoglobin-HMGB1 synergy, the selective Syk inhibitor piceatannol was used to pre-treat the macrophages. As shown in figure 3B, the synergy was significantly reduced by piceatannol in a dose dependent manner. Piceatannol also decreased the signal in cells that received HMGB1 alone. These results suggest that Syk-dependent ROS generation plays an important role in the induction of TNF by HMGB1, as well as in the synergy of HMGB1 with hemoglobin.
Figure 3. Involvement of induction of ROS and activation of Syk in the mechanisms of synergy.

BMDMs from C57BL/6 mice were washed 3 times and pre-treated with (A) Serum free media (SFM), or NAC (10mM) or DPI (10 μM) or allopurinol (10 μM) or L-NAME (100 μM) or (B) piceacetannol at indicated concentrations for 30 min. Cells were then further treated overnight with serum-free medium or with 4 μg/ml of HMGB1 in the absence or presence of 30 μg/ml mouse hemoglobin (Hb). Concentrations of TNF in the supernatants were determined by ELISA. The results denote the mean ± s.e and are representative of more than four independent experiments. * P < .05, ** P < .01, compared between cells treated with and without hemoglobin in the presence of serum free media or the same type of inhibitor. In panel A, † P < .001, compared between cells treated with HMGB1 and hemoglobin in the absence and presence of DPI. In panel B, P < .001 for the dose effect of piceacetannol on cells treated with HMGB1 and hemoglobin.
Hemopexin blocks the synergistic induction of pro-inflammatory cytokines from macrophages by HMGB1 and hemoglobin
We have shown that, hemopexin, the major heme scavenger in the plasma (10;20), significantly blocks the synergy of hemoglobin with LPS (8). In addition, hemopexin has some immunomodulatory activities in that it modestly downregulates pro-inflammatory cytokines from macrophages (16) and functions as an anti-inflammatory component of serum HDL in atherosclerosis (21). It was therefore of interest to assess if hemopexin would also affect the synergistic induction of pro-inflammatory cytokines that were induced by hemoglobin with HMGB1. We observed that addition of hemopexin significantly suppressed the synergistic induction of TNF in a dose dependent manner (Figure 4A). Similar results were obtained with IL-6 (Figure 4B). Addition of equal concentrations of endotoxin-free albumin (Sigma) under the same conditions did not suppress the response (data not shown).
Figure 4. Hemopexin suppresses hemoglobin synergy with HMGB1 on BMDMs.

BMDMs from C57BL/6 mice were washed 3 times with serum-free medium and then cultured overnight with 4 μg/ml of HMGB1 in the absence or presence of 30 μg/mL mouse hemoglobin (Hb) and indicated concentrations of hemopexin (Hx). Concentrations of TNF (A) and IL-6 (B) in the supernatants were determined by ELISA. The results denote the mean ± s.e and are representative of more than four independent experiments. ** P < .01, compared between cells treated with and without hemopexin in the presence of both HMGB1 and hemoglobin.
TLR2 and TLR4 agonists, HMGB1, and hemoglobin synergize to induce macrophages to release high concentrations of TNF and IL-6 that are suppressed by hemopexin
Because it is common that tissue damage, necrosis and bleeding coexist in some settings of infection, we next assessed the synergy from three types of stimuli including HMGB1, hemoglobin and the microbial derived TLR agonist, LPS. Very low concentrations of LPS (250 pg/ml), HMGB1(2 μg/ml) and hemoglobin (10 μg/ml) were used to stimulate BMDMs. Significantly higher concentrations of TNF and IL-6 (Figure 5A, B) were observed in the culture after overnight incubation, compared to the cultures with the stimulus alone or any combinations of two stimuli. Similar results were observed using the TLR2 agonist Pam3Cys (1 ng/ml) instead of LPS (Figure 5C, D). The addition of hemopexin to the culture resulted in remarkable suppression in synergy between the three types of stimuli (Figure 6A, B). These data suggest that synergy between HMGB1, hemoglobin and microbial products influences the magnitude of the innate immune response to injury and invasion, and that hemopexin may play a role in limiting this response.
Figure 5. Synergistic inflammation induced by hemoglobin, HMGB1 and TLR agonist LPS or P3C.

BMDMs from C57BL/6 mice were washed 3 times with serum-free medium and then cultured overnight with hemoglobin (Hb) (10 μg/ml), HMGB1 (2 μg/ml), LPS (250 pg/ml) (A) or Pam3Cys (P3C) (1 ng/ml) (B) alone or combinations of two or three of the stimuli. Concentrations of TNF in the supernatants were determined by ELISA. The results represent mean ± s.e. and are representative of three independent experiments. ** P < .01, compared between cells treated with three stimuli and with one or two stimuli.
Figure 6. Hemopexin suppresses the synergy induced by hemoglobin, HMGB1 and TLR agonist LPS or P3C.
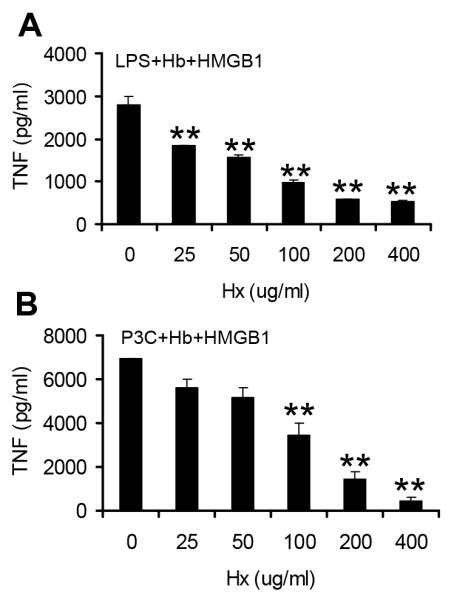
BMDMs from C57BL/6 mice were washed 3 times with serum free medium and then cultured overnight with hemoglobin (Hb) (10 μg/ml), HMGB1 (2 μg/ml), LPS (250 pg/ml) (A) or Pam3Cys (P3C) (1ng/ml) (B) in the absence or presence of hemopexin (Hx) at indicated concentrations. Concentrations of TNF in the supernatants were determined by ELISA. The results denote the mean ± s.e and are representative of three independent experiments. ** P < .01, compared between cells treated with and without hemopexin.
DISCUSSION
Damage to blood vessel walls with release of blood into tissues occurs frequently in diseases and after medical procedures. Red blood cells are either re-absorbed without rupture or undergo secondary degradation of cellular membranes with the release of hemoglobin into the extracellular space. Often there is an associated primary or secondary local inflammation, with generation and release of HMGB1, an intracellular protein that is integral in causing and increasing inflammation through active secretion after activation of NF-kB, or when released by permeabilized cell membranes during necrosis. We describe here that very low concentrations of hemoglobin synergize with HMGB1 to increase pro-inflammatory cytokines from macrophages. The results suggest that breakdown products of red blood cells that are released into tissues likely play an important role in amplifying sterile inflammation that is present when blood is released into tissues.
This finding extends an observation made many years ago that hemoglobin synergizes with LPS in vitro and in vivo to increase release of TNF (1-3). There is an extensive literature on the role of HMGB1 in promoting inflammation in tissues in which degrading blood cells would be present (11). While difficult to obtain a numerical value for the concentration of hemoglobin in this complex tissue microenvironment, synergy with HMGB1 was observed at a hemoglobin concentration of 3 μg/ml, which is 1/50,000th of the total hemoglobin concentration in whole blood. Hemoglobin concentrations in areas of necrosis or other inflamed fluids almost certainly exceed this.
Several earlier studies indicate that LPS and hemoglobin physically interact (3;22;23). However, the finding that hemoglobin leads to synergistic increases in cytokine production with multiple other ligands, including HMGB1, suggests that the synergy may occur at the cellular level. Synergy of hemoglobin with LPS is due to the heme moiety (6), and synergy of heme with LPS appears to be mediated through reactive oxygen species generation that is dependent upon spleen tyrosine kinase (7). The mechanisms for the synergy of hemoglobin with HMGB1 may involve similar pathways. Our results using cells from several types of knockout mice suggest that TLR2, TLR4 and RAGE signaling might contribute to, but is not essential, to the synergy. Our studies on ROS and Syk using inhibitors are consistent with the concept that Syk-dependent ROS generation is important, but not specific, to the synergy. In our studies the stimulation of macrophages after removal of several of the receptors reported to be involved in HMGB1 signaling, or after removal of adaptor signaling pathways, or with pharmacological inhibition of ROS or Syk resulted in suppressed responses to different extents with HMGB1 alone, confounding interpretation of the specificity of these mechanisms on the synergy. Further studies on additional receptors including CD163 for hemoglobin-bound haptoglobin and CD91 for heme-bound hemopexin may provide more understanding of the synergistic mechanisms.
We found that the induction of the pro-inflammatory cytokines TNF and IL-6 as a result of synergy between hemoglobin and HMGB1 was further increased by the presence of low concentrations of microbial ligands such as LPS or Pam3Cys that activate TLR4 or TLR2 respectively. This increase was not only additive but synergistic, leading to a “3-way synergy” in which three compounds were much more potent than any two of the other compounds. Countless studies have utilized single TLR agonists such as LPS alone in model systems as reagents to probe immune interactions. However, isolated activation by any single TLR agonist ligand is unlikely to occur in nature (24), and synergy between hemoglobin, HMGB1, and microbial ligands is likely important in sepsis syndrome (7;14;25). Our finding confirms and extends prior studies reporting synergistic activation of signaling pathways and subsequent induction of pro-inflammatory cytokines and chemokines triggered by different TLR agonists (26-32).
Although our findings seem of particular importance to extracellular hemoglobin in tissues outside of the vasculature, in some situations a similar synergy between extracellular hemoglobin and HMGB1 may occur in the bloodstream itself, such as in sickle cell crisis, hemolysis caused by infection including hemorrhagic fevers and malaria, ischemic-perfusion syndrome, cardiopulmonary bypass, severe sepsis, and after transfusions in which there is liberation of hemoglobin into blood (33-37).
Hemopexin strongly suppressed the synergistic inflammation of HMGB1 with hemoglobin. Hemopexin has been considered primarily as a protein that binds heme rather than hemoglobin (10). Our findings here are consistent with the concept that hemopexin may transfer or “steal” heme from degrading hemoglobin, as has been described by Hrkal (9). Sequestration of a low amount of heme would lead to a potent effect because low concentrations of hemoglobin greatly amplify cytokine release. Transfer of heme out of hemoglobin would also generate heme-free globin, which itself has an anti-inflammatory effect on macrophages (6). Other mechanisms for the suppression are also possible, including downstream induction of heme-oxygenase 1 with secondary anti-inflammatory effects (38).
Hemopexin concentrations in blood are decreased in some clinical situations, including during intravascular hemolysis (39), in premature infants (40), and during severe sepsis (25). It is possible that low concentrations could reflect a decrease in heme buffering capacity, in which case measurement of the concentrations of hemopexin might be helpful as a biomarker, alone or perhaps in combination with other inflammatory markers including HMGB1. It was recently reported that infused hemopexin decreases organ failure and mortality in a cecal ligation and puncture model in mice (25), a finding that raises the possibility of administering hemopexin as a therapy to treat sepsis, as well as other infections in which hemoglobin is liberated such hemorrhagic fevers or malaria. The findings here raise the possibility that infusion of hemopexin might likewise be helpful in settings where hemoglobin and HMGB1 are both present in the absence of infection, such as in sterile inflammation or necrosis.
Acknowledgments
This work was funded by the Shriners Hospital for Crippled Children (8720), the National Institute of Health (AI059010, GM59694 to HSW, GM62508 to KJT, and GM098446 to HY, and DARPA grant W911NF-10-1-0271 (to HSW).
Footnotes
Conflicts of interest: In accordance with institutional policy, HSW has reported the use of hemopexin as potential anti-inflammatory agent, and patent protection has been applied for. No other author reports and potential conflicts.
REFERENCES
- 1.Roth RI, Kaca W, Levin J. Hemoglobin: a newly recognized binding protein for bacterial endotoxins (LPS) Prog.Clin.Biol.Res. 1994;388:161–172. [PubMed] [Google Scholar]
- 2.Su D, Roth RI, Yoshida M, Levin J. Hemoglobin increases mortality from bacterial endotoxin. Infect.Immun. 1997;65:1258–1266. doi: 10.1128/iai.65.4.1258-1266.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaca W, Roth RI, Levin J. Hemoglobin, a newly recognized lipopolysaccharide (LPS)-binding protein that enhances LPS biological activity. J.Biol.Chem. 1994;269:25078–25084. [PubMed] [Google Scholar]
- 4.Bodet C, Chandad F, Grenier D. Hemoglobin and LPS act in synergy to amplify the inflammatory response. J.Dent.Res. 2007;86:878–882. doi: 10.1177/154405910708600914. [DOI] [PubMed] [Google Scholar]
- 5.Howe J, Richter W, Hawkins L, Rossle M, Alexander C, Fournier K, Mach JP, Waelli T, Gorczynski RM, Ulmer AJ, Brade H, Zamyatina A, Kosma P, Rietschel ET, Brandenburg K. Hemoglobin enhances the biological activity of synthetic and natural bacterial (endotoxic) virulence factors: a general principle. Med.Chem. 2008;4:520–525. doi: 10.2174/157340608786242089. [DOI] [PubMed] [Google Scholar]
- 6.Yang H, Wang H, Bernik TR, Ivanova S, Wang H, Ulloa L, Roth J, Eaton JW, Tracey KJ. Globin attenuates the innate immune response to endotoxin. Shock. 2002;17:485–490. doi: 10.1097/00024382-200206000-00008. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez PL, Dutra FF, Alves L, Figueiredo RT, Mourao-Sa D, Fortes GB, Bergstrand S, Lonn D, Cevallos RR, Pereira RM, Lopes UG, Travassos LH, Paiva CN, Bozza MT. Heme amplifies the innate immune response to microbial molecules through spleen tyrosine kinase (Syk)-dependent reactive oxygen species generation. J.Biol.Chem. 2010;85:32844–32851. doi: 10.1074/jbc.M110.146076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin T, Kwak YH, Sammy F, He P, Thundivalappil S, Sun G, Chao W, Warren HS. Synergistic inflammation is induced by blood degradation products with microbial Toll-like receptor agonists and is blocked by hemopexin. J.Infect.Dis. 2010;202:624–632. doi: 10.1086/654929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hrkal Z, Vodrazka Z, Kalousek I. Transfer of heme from ferrihemoglobin and ferrihemoglobin isolated chains to hemopexin. Eur.J.Biochem. 1974;43:73–78. doi: 10.1111/j.1432-1033.1974.tb03386.x. [DOI] [PubMed] [Google Scholar]
- 10.Tolosano E, Altruda F. Hemopexin: structure, function, and regulation. DNA Cell Biol. 2002;21:297–306. doi: 10.1089/104454902753759717. [DOI] [PubMed] [Google Scholar]
- 11.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu.Rev.Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al Abed Y, Akira S, Bierhaus A, Erlandsson-Harris H, Andersson U, Tracey KJ. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc.Natl.Acad.Sci.U.S.A. 2010;107:11942–11947. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Wang H, Mason JM, Levine J, Yu M, Ulloa L, Czura CJ, Tracey KJ, Yang H. Recombinant HMGB1 with cytokine-stimulating activity. J.Immunol.Methods. 2004;289:211–223. doi: 10.1016/j.jim.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 14.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 15.Andrade CT, Barros LA, Lima MC, Azero EG. Purification and characterization of human hemoglobin: effect of the hemolysis conditions. Int.J.Biol.Macromol. 2004;34:233–240. doi: 10.1016/j.ijbiomac.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 16.Liang X, Lin T, Sun G, Beasley-Topliffe L, Cavaillon JM, Warren HS. Hemopexin down-regulates LPS-induced proinflammatory cytokines from macrophages. J.Leukoc.Biol. 2009;86:229–235. doi: 10.1189/jlb.1208742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Novitsky TJ, Roslansky PF, Siber GR, Warren HS. A turbidometric method for quantifying serum inhibition of limulus amoebocyte lysate response. J.Clin.Micro. 1985;20:211–216. doi: 10.1128/jcm.21.2.211-216.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schilling D, Thomas K, Nixdorff K, Vogel SN, Fenton MJ. Toll-like receptor 4 and Toll-IL-1 receptor domain-containing adapter protein (TIRAP)/myeloid differentiation protein 88 adapter-like (Mal) contribute to maximal IL-6 expression in macrophages. J.Immunol. 2002;169:5874–5880. doi: 10.4049/jimmunol.169.10.5874. [DOI] [PubMed] [Google Scholar]
- 19.Bagchi A, Herrup EA, Warren HS, Trigilio J, Shin HS, Valentine C, Hellman J. MyD88-Dependent and MyD88-Independent Pathways in Synergy, Priming, and Tolerance between TLR Agonists. J.Immunol. 2007;178:1164–1171. doi: 10.4049/jimmunol.178.2.1164. [DOI] [PubMed] [Google Scholar]
- 20.Paoli M, Anderson BF, Baker HM, Morgan WT, Smith A, Baker EN. Crystal structure of hemopexin reveals a novel high-affinity heme site formed between two beta-propeller domains. Nat.Struct.Biol. 1999;6:926–931. doi: 10.1038/13294. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe J, Grijalva V, Hama S, Barbour K, Berger FG, Navab M, Fogelman AM, Reddy ST. Hemoglobin and Its Scavenger Protein Haptoglobin Associate with ApoA-1-containing Particles and Influence the Inflammatory Properties and Function of High Density Lipoprotein. J.Biol.Chem. 2009;284:18292–18301. doi: 10.1074/jbc.M109.017202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howe J, Garidel P, Roessle M, Richter W, Alexander C, Fournier K, Mach JP, Waelli T, Gorczynski RM, Ulmer AJ, Zahringer U, Hartmann A, Rietschel ET, Brandenburg K. Structural investigations into the interaction of hemoglobin and part structures with bacterial endotoxins. Innate.Immun. 2008;14:39–49. doi: 10.1177/1753425907087257. [DOI] [PubMed] [Google Scholar]
- 23.Bahl N, Du R, Winarsih I, Ho B, Tucker-Kellogg L, Tidor B, Ding JL. Delineation of LPS-binding sites on Hemoglobin - from in silico predictions to biophysical characterization. J.Biol.Chem. 2011 doi: 10.1074/jbc.M111.245472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warren HS. Toll-like receptors. Crit Care Med. 2005;33:S457–S459. doi: 10.1097/01.ccm.0000185504.39347.5d. [DOI] [PubMed] [Google Scholar]
- 25.Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, Bonaparte D, Cavalcante MM, Chora A, Ferreira A, Marguti I, Cardoso S, Sepulveda N, Smith A, Soares MP. A central role for free heme in the pathogenesis of severe sepsis. Sci.Transl.Med. 2010;2:51ra71. doi: 10.1126/scitranslmed.3001118. [DOI] [PubMed] [Google Scholar]
- 26.Gouwy M, Struyf S, Verbeke H, Put W, Proost P, Opdenakker G, Van Damme J. CC chemokine ligand-2 synergizes with the nonchemokine G protein-coupled receptor ligand fMLP in monocyte chemotaxis, and it cooperates with the TLR ligand LPS via induction of CXCL8. J.Leukoc.Biol. 2009 doi: 10.1189/jlb.1008638. [DOI] [PubMed] [Google Scholar]
- 27.Lombardi V, Van Overtvelt L, Horiot S, Moingeon P. Human dendritic cells stimulated via TLR7 and/or TLR8 induce the sequential production of Il-10, IFN-gamma, and IL-17A by naive CD4+ T cells. J.Immunol. 2009;182:3372–3379. doi: 10.4049/jimmunol.0801969. [DOI] [PubMed] [Google Scholar]
- 28.Strandskog G, Skjaeveland I, Ellingsen T, Jorgensen JB. Double-stranded RNA- and CpG DNA-induced immune responses in Atlantic salmon: comparison and synergies. Vaccine. 2008;26:4704–4715. doi: 10.1016/j.vaccine.2008.06.054. [DOI] [PubMed] [Google Scholar]
- 29.Vanhoutte F, Paget C, Breuilh L, Fontaine J, Vendeville C, Goriely S, Ryffel B, Faveeuw C, Trottein F. Toll-like receptor (TLR)2 and TLR3 synergy and cross-inhibition in murine myeloid dendritic cells. Immunol.Lett. 2008;116:86–94. doi: 10.1016/j.imlet.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 30.He H, Genovese KJ, Nisbet DJ, Kogut MH. Synergy of CpG oligodeoxynucleotide and double-stranded RNA (poly I:C) on nitric oxide induction in chicken peripheral blood monocytes. Mol.Immunol. 2007;44:3234–3242. doi: 10.1016/j.molimm.2007.01.034. [DOI] [PubMed] [Google Scholar]
- 31.Roelofs MF, Joosten LA, Abdollahi-Roodsaz S, van Lieshout AW, Sprong T, van den Hoogen FH, van den Berg WB, Radstake TR. The expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis Rheum. 2005;52:2313–2322. doi: 10.1002/art.21278. [DOI] [PubMed] [Google Scholar]
- 32.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat.Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hebert PC, Wells G, Blajchman MA, Marshall J, Martin C, Pagliarello G, Tweeddale M, Schweitzer I, Yetisir E. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N.Engl.J.Med. 1999;340:409–417. doi: 10.1056/NEJM199902113400601. [DOI] [PubMed] [Google Scholar]
- 34.Koch CG, Li L, Sessler DI, Figueroa P, Hoeltge GA, Mihaljevic T, Blackstone EH. Duration of red-cell storage and complications after cardiac surgery. N.Engl.J.Med. 2008;358:1229–1239. doi: 10.1056/NEJMoa070403. [DOI] [PubMed] [Google Scholar]
- 35.Hod EA, Brittenham GM, Billote GB, Francis RO, Ginzburg YZ, Hendrickson JE, Jhang J, Schwartz J, Sharma S, Sheth S, Sireci AN, Stephens HL, Stotler BA, Wojczyk BS, Zimring JC, Spitalnik SL. Transfusion of human volunteers with older, stored red blood cells produces extravascular hemolysis and circulating non-transferrin-bound iron. Blood. 2011 doi: 10.1182/blood-2011-08-371849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toy P, Popovsky MA, Abraham E, Ambruso DR, Holness LG, Kopko PM, McFarland JG, Nathens AB, Silliman CC, Stroncek D. Transfusion-related acute lung injury: definition and review. Crit Care Med. 2005;33:721–726. doi: 10.1097/01.ccm.0000159849.94750.51. [DOI] [PubMed] [Google Scholar]
- 37.El Dib M, Narang S, Lee E, Massaro AN, Aly H. Red blood cell transfusion, feeding and necrotizing enterocolitis in preterm infants. J.Perinatol. 2011;31:183–187. doi: 10.1038/jp.2010.157. [DOI] [PubMed] [Google Scholar]
- 38.Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24:449–455. doi: 10.1016/s1471-4906(03)00181-9. [DOI] [PubMed] [Google Scholar]
- 39.Muller-Eberhard U, Javid J, Liem HH, Hanstein A, Hanna M. Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood. 1968;32:811–815. [PubMed] [Google Scholar]
- 40.Kanakoudi F, Drossou V, Tzimouli V, Diamanti E, Konstantinidis T, Germenis A, Kremenopoulos G. Serum concentrations of 10 acute-phase proteins in healthy term and preterm infants from birth to age 6 months. Clin.Chem. 1995;41:605–608. [PubMed] [Google Scholar]