

Abstract
The Multiple Chamber Aerosol Chemical Aging Study (MUCHACHAS) tested the hypothesis that hydroxyl radical (OH) aging significantly increases the concentration of first-generation biogenic secondary organic aerosol (SOA). OH is the dominant atmospheric oxidant, and MUCHACHAS employed environmental chambers of very different designs, using multiple OH sources to explore a range of chemical conditions and potential sources of systematic error. We isolated the effect of OH aging, confirming our hypothesis while observing corresponding changes in SOA properties. The mass increases are consistent with an existing gap between global SOA sources and those predicted in models, and can be described by a mechanism suitable for implementation in those models.
Keywords: atmospheric chemistry, biosphere–atmosphere interactions
Organic aerosol (OA) comprises a large fraction of fine-particle mass (PM2.5) (1). In the developed world, 1–2% of deaths are blamed on inhalation of PM2.5 (2), and the leading uncertainty in climate forcing is the interplay between the number of fine particles large enough to nucleate cloud droplets and the amount of sunlight reflected by those clouds (3). Oxidation and condensation of organics play a major but uncertain role in both phenomena.
Traditional models treat most OA as nonvolatile primary OA (POA), augmented by secondary OA (SOA) (4), and they underpredict OA concentrations by a factor of 3–10 (5). α-Pinene is a major biogenic SOA source, sometimes used to represent all SOA in global models (4, 6). However, less than 20% of the carbon from fresh α-pinene oxidation condenses in chambers at room temperature; (7) the remainder is gaseous (Fig. 1A). This “chamber” SOA is modestly oxidized, with an oxygen to carbon ratio (O∶C) < 0.4 (7). It is unambiguously semivolatile: Yields rise with increasing SOA mass loading (8, 9) and decreasing temperature (10), and the SOA evaporates upon heating (11–13) and after isothermal dilution (14).
Fig. 1.

(A) Volatility distribution of 1st-generation α-pinene ozonolysis products vs saturation concentration (C∗). Filled green bars indicate the condensed-phase and white bars the vapor-phase concentrations. The green arrow shows the total condensed-phase concentration (C∗ = COA = 66 μg m-3). Compounds with this saturation concentration will be 50-50 partitioned. Colored symbols show C∗ values for important species. 3-methyl-1,2,3-butanetricarboxylic acid (MBTCA) is a second-generation aging product. The large pool of vapors is the major target of MUCHACHAS. (B) Design and model predictions for MUCHACHAS experiments, showing mass concentration C vs. time in an idealized chamber with no surface or dilution losses. SOA formation from complete ozonolysis of α-pinene based on the C∗ product distribution in (A) is shown in the gray region (t < 0). Top curves model 200 μg m-3 α-pinene, as in (A), bottom curves model 50 μg m-3 α-pinene. After fresh SOA is stable, OH production commences at t = 0 (white region). Model predictions based on constant 107 OH cm-3 (about 5× global-average ambient daytime) suggest a rapid increase in OA, followed by a gradual decline over a period equivalent to nearly 1 w of oxidation under ambient conditions. Maximum enhancements are a factor of 2.5 for the top curve and a factor of 4 for the lower curve. The initial mass yield (gray region, t < 0) for the lower curve is 13%, a value commonly used in global models as a constant yield. The solid green curves show model predictions with no heterogeneous oxidation of particles by OH, while the dashed green curves show upper-limit predictions with heterogeneous oxidation roughly a factor 5 slower than gas-phase OH reactions. The dot-dash blue curve is a model for the lower case without fragmentation or heterogeneous oxidation, resulting in extreme added SOA upon OH aging.
In contrast, ambient OA is highly oxidized (0.5 ≤ O∶C ≤ 1.0) (1, 15) and not very volatile (16). Ambient SOA is much less volatile than ambient POA (16). Consequently, “chamber” SOA does not represent the atmosphere. Our hypothesis is that homogeneous gas-phase aging by OH is a major missing process connecting chamber studies to the atmosphere. Considerable attention has been paid to heterogeneous uptake of oxidants to particles (17, 18), and recently gas-phase oxidation of semivolatile primary emissions (19), but the degree to which gas-phase oxidation can age chamber SOA is uncertain (1, 4, 18, 20).
OA resides in the atmosphere for about one week (21), while the gas-phase lifetimes of major semivolatile SOA constituents are far shorter. Typical α-pinene products pinonaldehyde, cis-pinonic acid, and pinic acid all have lifetimes of only a few hours for summertime conditions (22). Without question, oxidation of semivolatile SOA vapors will perturb the equilibrium phase partitioning of these constituents. Because almost all of the first-generation products are less volatile than α-pinene (Fig. 1A), many second-generation products should be even less volatile, leading to increased SOA mass. Aging could thus rectify the shortfall in model OA predictions (5). However, SOA concentrations alone are not the only property of interest; the “target” for OA chemistry includes high oxidation state and low volatility (1, 15).
POA sources emit compounds over a wide range of volatility, and oxidation of those vapors dramatically transforms OA properties. After emissions are diluted to ambient concentrations (evaporating semivolatile compounds), OH exposure results in large increases in aerosol mass and oxygen content (23). However the aging mechanism driving this enhancement is uncertain. The first few hours of chamber data on POA aging can be reproduced with a simple mechanism that irreversibly drives semivolatile organics to lower volatility (19). Extended to aging SOA, this mechanism improves model agreement in both mass and O∶C in short-term box model simulations of Mexico City (24), but for biogenic SOA it leads to a severe over-prediction of rural OA concentrations in the Southeastern United States. (25). Consequently, the effects of aging on biogenic SOA remain highly uncertain despite its immense importance.
Experimental Procedures
MUCHACHAS was designed to separate biogenic SOA formation from aging by OH, and to constrain the effects of that aging. The design is presented as a model simulation in Fig. 1B. The model employs a mechanism based on a priori consideration of how oxidation influences volatility. By employing multiple state-of-the-art chambers, MUCHACHAS was able to separate systematic effects such as wall losses and radical sources from the fundamental OH aging process occurring in all of the chambers and by extension the atmosphere.
To reduce systematic issues associated with wall losses, light sources, and chamber size, we conducted parallel experiments in four very different chambers described in the Materials and Methods section. Chamber attributes include Teflon (SAPHIR, PSI, CMU) and aluminum (AIDA) walls with large (SAPHIR, AIDA) and medium (PSI, CMU) volumes. OH was produced by HONO photolysis (SAPHIR, PSI, CMU), alkene ozonolysis (AIDA, PSI, CMU), and HOOH photolysis (CMU). Photolysis was driven by sunlight (SAPHIR), quasi-solar lamps (PSI), and 350 nm UV lamps (CMU). The critical design feature permitting isolation of the aging effect was the near stepwise introduction of OH after the initial SOA formation had reached completion.
Wall and dilution losses impede chamber studies, which are mass balances (26); they must be modeled. Particle wall losses are high in small chambers (CMU = 0.4 h-1, PSI = 0.17 h-1) and low in large chambers (AIDA = 0.014 h-1, SAPHIR = 0.04–0.16 h-1 depending on fan and roof state). Dilution is significant in SAPHIR (0.034 h-1). Any loss of condensible vapors other than the target chemical loss or condensation to suspended particles is an unseen part of the mass balance. Direct vapor wall losses are high only in aluminum-walled AIDA (0.36 h-1) (10, 27). For the teflon chambers (SAPHIR, PSI, and especially CMU, where 2/3 of the mass is on the walls at t = 0), vapor condensation to particles after deposition to the walls is a major uncertainty.
Model
The mechanism behind Fig. 1B is built on the 2D volatility basis set (2D-VBS) framework. The 2D-VBS classifies organics based on volatility (C∗) and oxidation extent (1, 28–30). To construct a lumped mechanism, described in detail in the SI Text, we discretize this 2D space into bins and consider the changes to C∗ and O∶C caused by OH oxidation. Reaction with OH transforms source organics in one bin to products distributed among many other bins. Here vapors react with OH at 3 × 10-11 cm3 molec-1 s-1, while heterogeneous OH uptake is either five times slower (with the same product distribution) or negligibly slow. While the gas-phase OH rate constants are uncertain, there is no doubt that the organic vapors will react with OH rapidly, and there is also no doubt that heterogeneous OH uptake is much slower.
For the full product distribution we describe two processes via generalized oxidation “kernels” (Fig. S1): Functionalization, where the carbon number is conserved and products move to lower volatility and higher oxidation state, and fragmentation, where products are spread widely over lower carbon numbers and thus (mostly) higher volatility and oxidation state (30, 31).
Functionalization always forms products with lower volatility than the reactant; one generation of chemistry in our model makes products with C∗ decreased by 1–6 (mostly 2–4) orders of magnitude, consistent with average addition of one ─OH and one ═O group per oxidation step (28). Fragmentation generates products over a wide volatility range. Fragmentation is a two-step process: Initial fragments are distributed between the C∗ of the reactant and the most volatile C∗ in the model, but half of these fragments are radicals that immediately functionalize to form lower C∗, more oxygenated stable products. Some fragmentation products are thus less volatile than the precursor. In addition, fragments can also functionalize in a subsequent oxidation step; for both of these reasons fragmentation products must be tracked because they can contribute to SOA formation. These two processes are reasonably well constrained for hydrocarbons (32, 33); we assume this extends to more oxidized organics.
A key element of the mechanism is the branching ratio between functionalization and fragmentation for lumped species in a given 2D-VBS bin. Here the effect of increasing oxygenation can be described. This branching ratio is uncertain, and so we rely on an empirical parameterization. Early models had no fragmentation (19, 24), but data reported by Kroll, et al. (34) showed that fragmentation increases dramatically as O∶C rises from 0 to roughly 0.5. Jimenez et al. (1) parameterized the branching ratio with O∶C1/6. Because the kernels are based on mechanistic insight from VOC oxidation, we limit adjustment to this single ad hoc parameter (Fig. S2), with O∶C1/4 giving the best model-measurement agreement. The important feature is the rapid increase in fragmentation for functionalized compounds. Our simulations are most sensitive to branching at the O∶C of first-generation products (80% fragmentation at O∶C = 0.4).
The mechanism is bounded by several constraints. First-generation volatility distributions (Fig. 1A) are well constrained by chamber-SOA formation data. It is very likely that second-generation products will be, on average, less volatile and thus SOA mass yields will rise with continued OH oxidation (Fig. 1B). However, complete oxidation to CO2 includes complete volatilization; eventually mass yields must fall, and particles do shrink after intense OH exposure (34). The mechanism thus interpolates between the (observed) rising mass yields between the first and second generations and the inevitably decreasing yields over longer timescales. The uncertainties are the magnitude and timing of the maximum in Fig. 1B and how this varies with photochemical conditions (i.e., VOC∶NOx, UV light, etc.).
Results
In all experiments a chamber was prepared by thorough cleaning and then filled with clean air and at least one tracer to follow OH radical production. We then added ozone and α-pinene and allowed the reaction to proceed to completion. This generated a collection of first-generation reaction products in both the vapor and condensed phases as in Fig. 1A. This dark interval (the shaded region in Figs. 1B and 2) served as a control and continued long enough to constrain the wall-loss parameters. After the dark interval we turned on an OH-radical source and monitored changes in the SOA.
Fig. 2.

A common experiment in multiple chambers: representative data and model curves. All experiments include a “dark” period with ozone present (indicated with dark gray shading) followed by an OH aging period (indicated with white shading). Each box shows suspended SOA concentration vs time: (A) SAPHIR (HONO photolysis—ambient light, approximately 1.6 × 106 OH cm-3); (B) AIDA (TME + O3, approximately 3.5 × 106 OH cm-3); (C) PSI (HONO photolysis—quasi-solar Xe light, approximately 5 × 106 OH cm-3); (D) CMU (TME + O3, approximately 2 × 106 OH cm-3). Observations are the dark green circles. The solid green curve in each panel is the same model shown in Fig. 1, with no heterogeneous oxidation but added terms for wall losses, while the dashed green curve omits the OH aging. Overall, for the complete MUCHACHAS dataset the model generally somewhat underpredicts aging-induced SOA growth even though the model without wall losses (or heterogeneous oxidation) describes a nearly three-fold increase, as shown in Fig. 1B.
Representative results from each chamber for similar OA concentrations are shown in Fig. 2. OH exposure almost always led to a significant increase in SOA concentrations. The model used in Fig. 1B, with additional treatment of wall losses, is shown with solid green curves in Fig. 2, while the dashed curves show model runs with no OH aging. Wall losses caused the observed mass concentration increase from OH aging to be much less than the twofold increase intrinsic to the mechanism (upper solid curve in Fig. 1B) because we assumed that wall-bound particles behave like suspended particles and thus consume some of the condensible vapors (Figs. S3–S5). The step function in OH makes the effect of aging evident even in experiments with low OH and high wall losses.
For the complete MUCHACHAS dataset, the different chambers and OH sources spanned a wide range of conditions. Dark OH production from TME ozonolysis (35) was used in AIDA (exclusively) and also at PSI and CMU. HONO photolysis at PSI (Fig. 2C) and CMU (Fig. S5) produced more OH and sharper initial SOA concentration increases. The natural photolysis in SAPHIR (36) produced the lowest OH concentrations (typical of ambient OH) and consequently the smallest net SOA concentration increase.
For temperature-dependent experiments in AIDA we controlled the α-pinene concentrations to keep the fresh SOA level constant. At 253 K, more than half of the carbon was in the condensed phase, while at 313 K less than 10% was condensed. We observed greater aging mass increases at elevated temperatures, consistent with the larger pool of vapors available for OH oxidation. Our model also predicts larger fractional increases for lower SOA concentrations at 300 K, because a greater fraction of the carbon is in the gas phase (lower curves in Fig. 1B).
The different chamber loss mechanisms taken together constitute an experimental advantage. For example, we clearly observed rapid loss of organic acid vapors to the AIDA walls for directly injected cis-pinonic acid (37). The mass loss for AIDA experiments (Fig. 2B) was because of semivolatile vapor loss, consistent with other isothermal observations (14, 38). At PSI and CMU, however, particle diameters were stable after SOA formation, and mass losses were associated with particle number loss via deposition to the walls.
We observed much more than SOA concentration changes. The oxygen to carbon ratio (O∶C) clearly increased after OH production [Fig. 3A, measured with a high-resolution aerosol mass spectrometer (HR-AMS) (1)]. Individual fragment ions showed more nuanced behavior. Despite the nearly constant SOA mass late in the dark period, the AMS spectra evolved steadily, at a rate independent of external parameters (ozone concentrations, OH concentrations, etc.). We call this evolution “ripening” to distinguish it from OH-driven chemical aging. During ripening, f44 (the fraction of the organic signal at m/z = 44, predominantly 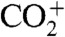 ) rose, f43 (mostly CH3CO+) fell, but O∶C remained roughly constant (Fig. S6).
) rose, f43 (mostly CH3CO+) fell, but O∶C remained roughly constant (Fig. S6).
Fig. 3.

Additional changes to SOA properties and constituents during OH aging experiments. (A) Typical evolution of molar oxygen to carbon ratio (O∶C, magenta circles) modeled with the 2D-VBS (magenta curve). The model reproduces the increase in O∶C for the first 1.5 h well. Afterwards model and measurements diverge, either because oxygenation in the model is too aggressive or because OH levels (and consequently aging) declined later in the experiment. The sharp early drop in the model before t = - 3 h arises from fresh SOA growth; the least volatile constituents have the highest O∶C. (B) Volatility data combine a steady decrease in volatility (increase in volume fraction remaining) unrelated to OH exposure with a sharp increase in volatility immediately after OH exposure, suggesting that fresh semi-volatile SOA addition dominates over aging of existing semi-volatile SOA. (C) Gas-phase fraction of cis-pinonic acid (magenta circles) and levels of MBTCA (red triangles) vs. temperature in AIDA. Separate experiments with pure cis-pinonic acid showed conclusively that MBTCA is formed from cis-pinonic acid oxidation by OH in the gas phase. These results show that as the cis-pinonic acid is sequestered in the condensed phase at low temperatures, the MBTCA formation is reduced proportionally.
We observed cloud condensation nucleus (CCN) activity (39), volatility via tandem differential mobility analysis (V-TDMA) (12, 13), and subsaturated water uptake via hygroscopicity TDMA (H-TDMA) (12). The CCN activity of the organics was largely unaffected by either ripening or OH oxidation (39). Volatility (Fig. 3B) showed a consistent temporal pattern in AIDA, PSI, and SAPHIR, responding to ripening and to OH aging (12, 13). Volatility measured by the volume fraction remaining at a fixed elevated temperature decreased progressively during ripening, increased sharply with the introduction of OH, and then reverted to a more gradual progressive decrease after roughly 1 h of OH aging. Though the average volatility of the SOA increased with OH aging, this masked a concurrent broadening of the volatility distribution. This broadening suggests that while the newly added mass formed from OH aging had fairly high volatility, this aging also formed a significant amount of very low volatility material. During the ripening period the subsaturated hygroscopicity observed with H-TDMA remained roughly constant (12).
Production of low volatility organics via OH aging was confirmed by measurement of individual products. In AIDA, organic acids were measured with atmospheric pressure chemical ionization mass spectrometry (APCI-MS) (37). We observed the phase partitioning of acids shown in Fig. 1 (e.g., the cis-pinonic acid partitioning in Fig. 3C), and established conclusively that the triacid MBTCA is produced from gas-phase oxidation of cis-pinonic acid by OH. MBTCA in SOA increased sharply after OH formation. Production was correlated with the fraction of cis-pinonic acid in the gas phase (Fig. 3C), suggesting that this triacid is effectively a first-generation product of cis-pinonic acid. We subsequently introduced a mixture of pinic and cis-pinonic acids into AIDA; only when cis-pinonic acid vapors were being actively added to the chamber while OH was also being generated did the MBTCA signal rise (37).
Discussion
MUCHACHAS experiments were carefully designed to separate initial SOA formation from subsequent exposure to OH radicals in an aging step, and to ensure that substantial concentrations of OH were produced during that step. The results confirm dramatic aging effects and strongly suggest that the major effect of gas-phase OH reactions is to enhance SOA concentrations by a factor of 2–4 (Fig. 1B). The fundamental result is that gas-phase reactions of semivolatile SOA vapors are clearly an important part of OA evolution, and these experiments substantially extend the timescale of the chamber studies.
Model-measurement agreement varies between experiments. It is best for the AIDA and PSI experiments, with the highest and most intense OH exposure relative to wall losses. The fresh SOA prediction (t ≤ 0 in Fig. 2) is based on the scavenger-free SOA mass yields in Fig. 1A (40). SOA production is known to vary with conditions [VOC∶NOx, UV light, etc. (4)] that were not kept constant in different experiments. We did not attempt to model those differences because the essential and invariant feature is that a large pool of vapors can react with OH radicals during aging. Consequently, we adjusted the initial α-pinene by ≤ 10% to match the SOA level at t = 0 so that the aging data are easy to compare with the model in Fig. 2.
We could have adjusted chamber-specific parameters further in order to match experimental data. As an example, the observed aging growth in the CMU chamber (Fig. 2D) is larger than the model; this could be due to the large modeled flux of condensible vapors to particles on the wall, but we lack sufficient evidence to “tweak” this parameter. Such adjustments would be as likely to mask chamber-specific systematic errors as to provide new insight. Instead we applied a common model that reproduces the general features of all the chamber experiments; this makes a stronger case for our main conclusion regarding OH-initiated aging of biogenic SOA. The differences between the model and measurements reflect real uncertainties that will not be reduced by model- and chamber-dependent parameter tuning.
The MUCHACHAS findings also highlight significant remaining uncertainties that must be addressed before a complete aging mechanism can be described. One is the degree of fragmentation. Without fragmentation the 2D-VBS rapidly converts vapors to SOA with approximately 180% mass yields because of added oxygen (blue dash-dot curve, Fig. 1B). The preferred mechanism peaks at ≲50% (lower green curves, Fig. 1B). Low fragmentation may be suitable for high molecular weight POA vapors (19), but not for lighter and more oxygenated biogenic SOA. The empirical O∶C1/4 relation for the fragmentation branching ratio captures this difference, but it is quantitatively uncertain. As shown in the SI Text, different numerical values of this parameter result in different SOA enhancements from aging, but the overall conclusion that OH oxidation results in a substantial SOA mass increase is robust.
Another uncertainty is the nature and effect of ripening, which may well be oligomerization (20). Some properties evolve, such as volatility, while others remain roughly constant, such as organic mass and CCN activity. Furthermore, the MUCHACHAS experiments were carried out at medium to low relative humidity and thus avoided (by design) potential aqueous-phase aging reactions, which could well be important in the atmosphere.
UV photolysis may also affect the oxidized SOA. Experiments at CMU showed a strong UV effect; in the extreme case, with weak OH formation (approximately 106 cm-3) via HOOH photolysis using 360 nm UV lights, SOA mass diminished monotonically during aging. For HONO photolysis the SOA initially rose, consistent with the PSI results in Fig. 2C, but fell again after OH concentrations declined from approximately 107 to approximately 106 cm-3. With TME + ozone OH production, SOA concentrations rose on aging with OH production in the dark (as in Fig. 2D), but the rise halted if the UV lights were turned on (41). The same effect may be evident in the SAPHIR data, as shown in Fig. 2A; OA concentrations initially rose upon aging but ultimately failed to increase as rapidly as our model predicted, again when OH production was relatively weak in the presence of UV light (in this case sunlight).
A final uncertainty is the effect of heterogeneous OH uptake, with limiting behavior indicated by the solid and dashed curves in Fig. 1B. The differences between the curves are substantial; both accommodation coefficients for OH and the resulting mechanisms are highly uncertain for atmospheric aerosols. OH uptake clearly oxidizes organic particles (34), causing the oxidation state to rise and carbon numbers to decrease (30). For the model producing the dashed green curves in Fig. 1B we presume that the homogeneous gas-phase and heterogeneous mechanisms are the same despite the different rates, which is probably not true. Because of gas-phase diffusion limitations heterogeneous oxidation is almost certainly a factor of 5–20 slower than the corresponding homogeneous oxidation (42), but the heterogeneous oxidation timescale is still shorter than the aerosol residence timescale in the atmosphere (21). If the mechanism including heterogeneous oxidation is valid, as shown with the dashed curves in Fig. 1B this could partly counteract the mass increases of aging, though the average concentrations would still be elevated and variable. Furthermore, the residual aerosol would be highly oxidized and distributed down to very low volatility, consistent with observations in remote locations (15, 43).
Conclusions
OH aging strongly influences biogenic SOA properties and concentrations throughout the aerosol life cycle in the atmosphere. Aging can significantly increase SOA concentrations beyond first-generation mass yields, by a factor similar to the disagreement between some models and observations (4, 5). The interval in Fig. 1B spans approximately 1 w in the atmosphere, yet many photochemical models currently only describe the gray interval (t ≤ 0), or even simply assume a constant mass yield at t = 0 by parameterizing SOA formation as a fraction of the terpene flux from the biosphere. Based on the MUCHACHAS results, those current assumptions are clearly inadequate, and the mechanism presented here provides at least a partial solution to a more accurate representation.
An ongoing challenge is to isolate the various effects influencing OA over the week or so average residence time in the atmosphere. OA comprises thousands of molecules, making complete elucidation a formidable challenge; a hypothesis of MUCHACHAS was that meaningful average behaviors could be described and applied to atmospheric simulations. This first step demonstrates the influence of OH aging chemistry on biogenic SOA, and the potential application of OH aging in regional and global models to accurately simulate organic aerosol mass concentrations and properties.
Materials and Methods
Chambers.
Carnegie Mellon University (CMU) 12 m3 teflon chamber (44); the Paul Scherrer Institute (PSI) 27 m3 teflon chamber (45); the Karlsruhe Institute of Technology 84.5 m3 Aerosol Interaction and Dynamics in the Atmosphere (AIDA) aluminum chamber (46); and the Forschungszentrum Jülich 270 m3 Simulation of Atmospheric Photochemistry in a large Reaction chamber (SAPHIR) double-walled teflon outdoor facility (36).
The CMU (44) and PSI (45) chambers are similar. However, the CMU chamber relies on 360 nm UV lights for illumination, while the PSI chamber uses filtered Xe lamps to produce quasi solar light. For MUCHACHAS each chamber was held near 298 K. The AIDA chamber has temperature and pressure control over a very wide range (188–333 K, 0.01–1100 hPa) (46); SOA experiments were conducted at 253, 273, 293, and 313 K and 1,013 mb, while the cis-pinonic acid oxidation experiments were conducted at 283 K. Finally, the SAPHIR chamber is housed within a completely retractible outdoor enclosure. Illumination is solar, and conditions are nearly ambient (it is held slightly over atmospheric pressure to ensure that any leakage goes out of the chamber, and it is typically slightly warmer than ambient due to radiational heating).
Instrumentation.
All experiments included a proton-transfer reaction mass spectrometer (44, 47), a scanning mobility particle sizer , an Aerodyne aerosol mass spectrometer (48, 49), and either a thermodenuder (11) or a volatility tandem differential mobility analyzer (12, 13). Most experiments measured SOA hygroscopicity, either with tandem differential mobility (12) measurements below 100% RH or with cloud condensation nucleus counting (39, 50) above 100% humidity or both.
At AIDA we employed an atmospheric pressure ionization mass spectrometer (37) and filter solvent extraction GC/MS to measure condensed-phase organic acids, while at PSI ion chromotography was employed to measure selected acids in both phases. At SAPHIR, OH and HO2 radicals were measured directly using Laser Induced Fluorescence (51).
OH Sources.
We used several different OH radical sources. These were the dark reaction of ozone and tetramethyl ethylene (35) (CMU, AIDA, PSI), UV photolysis of hydrogen peroxide at 360 nm (52, 53) (CMU), and photolysis of nitrous acid in the near UV (CMU, PSI, SAPHIR). The different sources result in different balances between OH, HO2 and organic peroxy radicals, different peroxy radical oxidation pathways (especially under high and low NOx conditions) and different amounts of photolysis of oxygenated organic oxidation products.
Supplementary Material
ACKNOWLEDGMENTS.
This work was funded by the EU project EUROCHAMP, the Swiss National Science Foundation, the Swedish Research Council, and the US EPA STAR program.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1115186109/-/DCSupplemental.
References
- 1.Jimenez JL, et al. Evolution of organic aerosols in the atmosphere: A new framework connecting measurements to models. Science. 2009;326:1525–1529. doi: 10.1126/science.1180353. [DOI] [PubMed] [Google Scholar]
- 2.Pope CA, Ezzati M, Dockery DW. Fine-particulate air pollution and life expectancy in the United States. N Engl J Med. 2009;360:376–386. doi: 10.1056/NEJMsa0805646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Intergovernmental Panel on Climate Change. Climate Change 2007: The Scientific Basis. Cambridge, UK: Cambridge Univ Press; 2007. [Google Scholar]
- 4.Hallquist M, et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos Chem Phys. 2009;10:5155–5236. [Google Scholar]
- 5.Spracklen DV, et al. Aerosol mass spectrometer constraint on the global secondary organic aerosol budget. Atmos Chem Phys. 2011;11:12109–12136. [Google Scholar]
- 6.Kanakidou M, et al. Organic aerosol and global climate modelling: a review. Atmos Chem Phys. 2005;5:1053–1123. [Google Scholar]
- 7.Shilling JE, et al. Loading-dependent elemental composition of α-pinene SOA particles. Atmos Chem Phys. 2009;9:771–782. [Google Scholar]
- 8.Odum J, et al. Gas/particle partitioning and secondary organic aerosol yields. Environ Sci Technol. 1996;30:2580–2585. [Google Scholar]
- 9.Presto AA, Donahue NM. Investigation of α-pinene plus ozone secondary organic aerosol formation at low total aerosol mass. Environ Sci Technol. 2006;40:3536–3543. doi: 10.1021/es052203z. [DOI] [PubMed] [Google Scholar]
- 10.Saathoff H, et al. Temperature dependence of yields of secondary organic aerosols from the ozonolysis of α-pinene and limonene. Atmos Chem Phys. 2009;9:1551–1577. [Google Scholar]
- 11.An WJ, Pathak RK, Lee BH, Pandis SN. Aerosol volatility measurement using an improved thermodenuder: Application to secondary organic aerosol. J Aerosol Sci. 2007;38:305–314. [Google Scholar]
- 12.Tritscher T, et al. Volatility and hygroscopicity of aging secondary organic aerosol in a smog chamber. Atmos Chem Phys. 2011;11:11477–11496. [Google Scholar]
- 13.Salo K, et al. Volatility of secondary organic aerosol during OH radical induced ageing. Atmos Chem Phys. 2011;11:11055–11067. [Google Scholar]
- 14.Grieshop AP, Donahue NM, Robinson AL. Is the gas-particle partitioning in α-pinene secondary organic aerosol reversible? Geophys Res Lett. 2007;34:L14810. [Google Scholar]
- 15.Ng NL, et al. Organic aerosol components observed in worldwide datasets from aerosol mass spectrometry. Atmos Chem Phys. 2010;9:4625–4641. [Google Scholar]
- 16.Huffman JA, et al. Chemically-resolved aerosol volatility measurements from two megacity field studies. Atmos Chem Phys. 2009;9:7161–7182. [Google Scholar]
- 17.Pöschl U, Rudich Y, Ammann M. Kinetic model framework for aerosol and cloud surface chemistry and gas-particle interactions—part 1: General equations, parameters, and terminology. Atmos Chem Phys. 2007;7:5989–6023. [Google Scholar]
- 18.Rudich Y, Donahue NM, Mentel TF. Aging of organic aerosol: Bridging the gap between laboratory and field studies. Annu Rev Phys Chem. 2007;58:321–352. doi: 10.1146/annurev.physchem.58.032806.104432. [DOI] [PubMed] [Google Scholar]
- 19.Robinson AL, et al. Rethinking organic aerosols: Semivolatile emissions and photochemical aging. Science. 2007;315:1259–1262. doi: 10.1126/science.1133061. [DOI] [PubMed] [Google Scholar]
- 20.Kalberer M, et al. Identification of polymers as major components of atmospheric organic aerosols. Science. 2004;303:1659–1662. doi: 10.1126/science.1092185. [DOI] [PubMed] [Google Scholar]
- 21.Wagstrom KM, Pandis SN. Determination of the age distribution of primary and secondary aerosol species using a chemical transport model. J Geophys Res. 2009;114:D011784. [Google Scholar]
- 22.Vereecken L, Peeters J. Enhanced H-atom abstraction from pinonaldehyde, pinonic acid, pinic acid, and related compounds: Theoretical study of C─H bond strengths. Phys Chem Chem Phys. 2002;4:467–472. [Google Scholar]
- 23.Robinson AL, Grieshop AP, Donahue NM, Hunt SW. Updating our conceptual model for fine particle mass emissions from combustion systems. J Am Waste Manage Assoc. 2010;60:1204–1222. doi: 10.3155/1047-3289.60.10.1204. [DOI] [PubMed] [Google Scholar]
- 24.Dzepina K, et al. Evaluation of new secondary organic aerosol models for a case study in Mexico City. Atmos Chem Phys. 2009;9:5681–5709. [Google Scholar]
- 25.Murphy BN, Pandis SN. Simulating the formation of semivolatile primary and secondary organic aerosol in a regional chemical transport model. Environ Sci Technol. 2009;43:4722–4728. doi: 10.1021/es803168a. [DOI] [PubMed] [Google Scholar]
- 26.Matsunaga A, Ziemann PJ. Gas-wall partitioning of organic compounds in a teflon film chamber and potential effects on reaction product and aerosol yield measurements. Aerosol Sci Technol. 2010;44:881–892. [Google Scholar]
- 27.Saathoff H, et al. Coating of soot and (NH4)2SO4 particles by ozonolysis products of α-pinene. J Aerosol Sci. 2003;34:1297–1321. [Google Scholar]
- 28.Donahue NM, Epstein SA, Pandis SN, Robinson AL. A 2-dimensional volatility basis set: 1. Organic mixing thermodynamics. Atmos Chem Phys. 2011;11:3303–3318. [Google Scholar]
- 29.Donahue NM, Kroll JH, Robinson AL, Pandis SN. A 2-dimensional volatility basis set: 2. Diagnostics of laboratory and ambient organic aerosol. Atmos Chem Phys. 2012;12:615–634. [Google Scholar]
- 30.Kroll JH, et al. Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol. Nat Chem. 2011;3:133–139. doi: 10.1038/nchem.948. [DOI] [PubMed] [Google Scholar]
- 31.Chacon-Madrid HJ, Presto AA, Donahue NM. Functionalization vs fragmentation: n-aldehyde oxidation mechanisms and secondary organic aerosol formation. Phys Chem Chem Phys. 2010;12:13975–13982. doi: 10.1039/c0cp00200c. [DOI] [PubMed] [Google Scholar]
- 32.Atkinson R. Gas phase tropospheric chemistry of organic compounds. J Phys Chem Ref Data. 1997;26:215–290. [Google Scholar]
- 33.Lim YB, Ziemann PJ. Effects of molecular structure on aerosol yields from oh radical-initiated reactions of linear, branched, and cyclic alkanes in the presence of nox. Environ Sci Technol. 2009;43:2328–2334. doi: 10.1021/es803389s. [DOI] [PubMed] [Google Scholar]
- 34.Kroll JH, et al. Measurement of fragmentation and functionalization pathways in the heterogeneous oxidation of oxidized organic aerosol. Phys Chem Chem Phys. 2009;11:8005–8014. doi: 10.1039/b905289e. [DOI] [PubMed] [Google Scholar]
- 35.Lambe AT, Zhang J, Sage AM, Donahue NM. Controlled OH radical production via ozone-alkene reactions for use in aerosol aging studies. Environ Sci Technol. 2007;41:2357–2363. doi: 10.1021/es061878e. [DOI] [PubMed] [Google Scholar]
- 36.Rohrer F, et al. Characterization of the photolytic HONO-source in the atmosphere simulation chamber SAPHIR. Atmos Chem Phys. 2005;5:2189–2201. [Google Scholar]
- 37.Müller L, et al. Formation of 3-methyl-1,2,3-butanetricarboxylic acid via gas phase oxidation of pinonic acid—a mass spectrometric study of SOA aging. Atmos Chem Phys. 2012;12:1483–1496. [Google Scholar]
- 38.Vaden TD, Imre D, Beránek J, Shrivastava M, Zelenyuk A. Evaporation kinetics and phase of laboratory and ambient secondary organic aerosol. Proc Nati Acad Sci USA. 2011;108:2190–2195. doi: 10.1073/pnas.1013391108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frosch M, et al. Relating CCN activity and atomic O/C ratio of α-pinene secondary organic aerosol. J Geophys Res A. 2011;116:D22212. [Google Scholar]
- 40.Henry KM, Donahue NM. Effect of the OH radical scavenger hydrogen peroxide on secondary organic aerosol formation from α-pinene ozonolysis. Aerosol Sci Technol. 2011;45:686–690. [Google Scholar]
- 41.Henry KM, Donahue NM. Photochemical aging of α-pinene secondary organic aerosol: Effects of OH radical sources and photolysis. J Phys Chem. 2012;116:5932–5940. doi: 10.1021/jp210288s. [DOI] [PubMed] [Google Scholar]
- 42.Lambe AT, Miracolo MA, Hennigan CJ, Robinson AL, Donahue NM. Effective rate constants and uptake coefficients for the reactions of organic molecular markers (n-alkanes, hopanes and steranes) in motor oil and diesel primary organic aerosols with OH radicals. Environ Sci Technol. 2009;43:8794–8800. doi: 10.1021/es901745h. [DOI] [PubMed] [Google Scholar]
- 43.Hildebrandt L, et al. Formation of low-volatility oxygenated organic aerosol in the atmosphere: Insights from the Finokalia Aerosol Measurement Experiments. Geophys Res Lett. 2010;37:L23801. [Google Scholar]
- 44.Presto AA, Hartz KEH, Donahue NM. Secondary organic aerosol production from terpene ozonolysis. 1. effect of UV radiation. Environ Sci Technol. 2005;39:7036–7045. doi: 10.1021/es050174m. [DOI] [PubMed] [Google Scholar]
- 45.Paulsen D, et al. Secondary organic aerosol formation by irradiation of 1,3,5-trimethylbenzene-NOx-H2O in a new reaction chamber for atmospheric chemistry and physics. Environ Sci Technol. 2005;39:2668–2678. doi: 10.1021/es0489137. [DOI] [PubMed] [Google Scholar]
- 46.Saathoff H, et al. The AIDA soot aerosol characterisation campaign 1999. J Aerosol Sci. 2003;35:1277–1296. [Google Scholar]
- 47.Lindinger W, Hansel A, Jordan A. Online monitoring of volatile organic compounds at ppt levels by means of proton-transfer-reaction mass spectrometry (PTR-MS): Medical aplications, food control and environmental research. Int J Mass Spectrom Ion Procs. 1998;173:181–241. [Google Scholar]
- 48.Jayne JT, et al. Development of an aerosol mass spectrometer for size and composition analysis of submicron particles. Aerosol Sci Technol. 2000;33:49–70. [Google Scholar]
- 49.DeCarlo PF, et al. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal Chem. 2006;78:8281–8289. doi: 10.1021/ac061249n. [DOI] [PubMed] [Google Scholar]
- 50.Asa-Awuku A, Engelhart GJ, Lee BH, Pandis SN, Nenes A. Relating CCN activity, volatility, and droplet growth kinetics of beta-caryophyllene secondary organic aerosol. Atmos Chem Phys. 2009;9:795–812. [Google Scholar]
- 51.Hofzumahaus A, et al. Amplified trace gas removal in the troposphere. Science. 2009;324:1702–1704. doi: 10.1126/science.1164566. [DOI] [PubMed] [Google Scholar]
- 52.Kroll JH, Ng NL, Murphy SM, Flagan RC, Seinfeld JH. Secondary organic aerosol formation from isoprene photooxidation. Environ Sci Technol. 2006;40:1869–1877. doi: 10.1021/es0524301. [DOI] [PubMed] [Google Scholar]
- 53.Hildebrandt L, Donahue NM, Pandis SN. High formation of secondary organic aerosol from the photo-oxidation of toluene. Atmos Chem Phys. 2009;9:2973–2986. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.