

Abstract
We used electron cryotomography of mitochondrial membranes from wild-type and mutant Saccharomyces cerevisiae to investigate the structure and organization of ATP synthase dimers in situ. Subtomogram averaging of the dimers to 3.7 nm resolution revealed a V-shaped structure of twofold symmetry, with an angle of 86° between monomers. The central and peripheral stalks are well resolved. The monomers interact within the membrane at the base of the peripheral stalks. In wild-type mitochondria ATP synthase dimers are found in rows along the highly curved cristae ridges, and appear to be crucial for membrane morphology. Strains deficient in the dimer-specific subunits e and g or the first transmembrane helix of subunit 4 lack both dimers and lamellar cristae. Instead, cristae are either absent or balloon-shaped, with ATP synthase monomers distributed randomly in the membrane. Computer simulations indicate that isolated dimers induce a plastic deformation in the lipid bilayer, which is partially relieved by their side-by-side association. We propose that the assembly of ATP synthase dimer rows is driven by the reduction in the membrane elastic energy, rather than by direct protein contacts, and that the dimer rows enable the formation of highly curved ridges in mitochondrial cristae.
Keywords: membrane-protein oligomerization, membrane deformation, molecular dynamics simulations, bioenergetics, ATP synthesis
The F1Fo ATP synthase is a highly conserved molecular machine that catalyses the production of ATP from ADP and Pi in energy-converting membranes of eukaryotes and bacteria. ATP synthesis is powered by an electrochemical transmembrane gradient. The mitochondrial ATP synthase is located in the inner membrane cristae (1, 2), where it forms dimers (3). These dimers are organized in rows, located along the highly curved ridges of the cristae (4–6). The ATP synthase of yeast (Saccharomyces cerevisiae) has a molecular mass of approximately 600 kDa and consists of thirteen different core subunits (α3, β3, γ, δ, ε, OSCP, 4, 6, 8, 910, d, f, and h) (7). The basic structure of the F1Fo ATP synthase as determined by electron cryomicroscopy (8, 9) is the same in all eukaryotes and bacteria. The F1Fo ATP synthase can be divided into four structural units, all of which are functionally essential: The catalytic head group [(αβ)3], the central stalk (γ, δ, ε), the peripheral stalk (4, d, h), and the membrane region (910, 6, 8, f). The first two units are often referred to as the F1 subcomplex and the last two as the Fo subcomplex. The oligomycin sensitivity conferral protein (OSCP) subunit connects the peripheral stalk to the catalytic domain. The complete enzyme works by rotary catalysis (reviewed in ref. 10). The atomic structures for both the yeast and bovine F1/rotor ring assemblies (11–13) have been determined by X-ray crystallography, as has part of the bovine peripheral stalk subcomplex (14, 15).
In addition to the 13 different core subunits, the yeast F1Fo ATP synthase contains four other protein subunits known as e, g, i, and k. Of these, e, g, k were first discovered in the ATP synthase dimer (3). Subunits e and g, which also occur in mammals, are small, integral membrane proteins with a single predicted transmembrane helix featuring a GXXXG motif. Subunit g has a small N-terminal matrix domain that can be cross-linked to subunit 4 (16), and subunit e has a short C-terminal domain with a predicted coiled-coil motif exposed to the cristae space. Initial characterization of these subunits suggested they were involved in ATP synthase dimer formation, as no dimers could be extracted from mitochondria lacking either subunit e or g (3, 17). However, in subsequent work small amounts of ATP synthase dimers were reported in digitonin extracts of these mutants (18). Biochemical cross-linking (19–21) and FRET (fluorescence resonance energy transfer) studies (22) likewise suggested dimers of the mitochondrial ATP synthase without subunits e and g, casting further doubt on the role of these subunits in dimer formation.
By electron cryotomography of mitochondria and mitochondrial membranes from six different species (5, 6) we found that the ATP synthase forms extensive rows of dimers along the highly curved cristae ridges in mammals, plants, and fungi. While the two F1 subcomplexes within each dimer were consistently 28 nm apart, the distance between adjacent dimers along the rows was variable, indicating that the dimers do not interact directly. Here, we report a three-dimensional map of the ATP synthase dimer in wild-type mitochondrial membranes, obtained by subtomogram averaging. At a resolution of 3.7 nm, the map clearly reveals the two catalytic heads in the dimer, with the central and peripheral stalks linking them to the membrane. The map indicates a constant angle of 86° between the monomers, and enables us to pinpoint the location of the dimer interface. To clarify the role of subunits e and g in the dimer, we obtained tomographic volumes of mitochondrial membranes from yeast mutants lacking these subunits or the first transmembrane helix of subunit 4 (su4TM1). We find that in these mutants the ATP synthase fails to form dimers or any higher oligomeric assemblies. Lastly, we show by large-scale molecular dynamics simulations that the energy of membrane deformation is sufficient to drive the formation of ATP synthase dimer rows without the need for specific protein-protein interactions.
Results
Subtomogram Averaging Reveals Architecture of the ATP Synthase Dimer In Situ.
Tomograms of mitochondrial membranes from the yeast Saccharomyces cerevisiae were assessed for the distribution of ATP synthase, which appears as 10 nm spherical densities positioned 5 nm above the membrane (5, 6). Pairs of particles along highly curved membrane ridges were extracted and averaged. The resulting subtomogram average was generated from 121 subvolumes and shows two ATP synthase monomers forming a V-shaped dimer, with their long axes crossing at an angle of 86° (Fig. 1). The peripheral and central stalks are clearly resolved. The dimer interface is located in the membrane between the two peripheral stalks. The two ATP synthase complexes can be superimposed onto each other by a rotation of 180° about an axis normal to the membrane. Fourier shell correlation [FSC (23)] between two randomly-selected halves of the data indicated a resolution of 3.7 nm at 50% correlation (Fig. S1).
Fig. 1.

Subtomogram average of the ATP synthase dimer from yeast mitochondria. Two ATP synthase monomers form a V-shaped dimer with an angle of 86° between their long axes. The central and peripheral stalks are clearly resolved. The dimer interface is located in the membrane between the two peripheral stalks. Threshold levels: mesh, 1σ; light grey, 2σ; dark grey, 3σ. See also Movie S1.
Fit of Subcomplex Structures.
In the subtomogram average of the dimer, each ATP synthase monomer resembles the published cryo-EM maps of the bovine and yeast mitochondrial ATP synthase obtained by single-particle analysis [Fig. S2 (8, 9)]. To determine whether our map is also consistent with the results of high-resolution X-ray crystallography, the X-ray structures of the yeast F1/rotor ring assembly [PDB entry 2WPD; (12)] and the bovine peripheral stalk fragment [2CLY; (14)] were fitted as rigid bodies (Fig. 2). Alternatively, the X-ray structure of the bovine F1/peripheral stalk assembly [2WSS; (15)] was placed into the subtomogram average (Fig. S3).
Fig. 2.

Fitted atomic models of ATP synthase dimer. (A) side view of subtomogram average 3D volume with fitted atomic models. Blue-purple, F1/rotor ring assembly [PDB: 2WPD (12)]; green, OSCP [PDB:2B05 (25)]; yellow-red, peripheral stalk fragment [PDB: 2CLY (14)] with additional residues from [PDB: 2WSS (15)]. Arrow indicates the N-terminal ends of the rotor ring, which account for the bulge protruding into the cristae space; red arrowhead indicates subunit h. (B) contour map of projected density slice between the dashed red lines in (A). (C) same as (B) but with fitted atomic models. The F1 density has near-sixfold symmetry with three stronger density peaks (black arrowheads) alternating with weaker peaks. The β subunits (deep blue) were positioned in the strong densities with the peripheral stalk complex adjacent to a non-catalytic interface. See Movie S1.
In both cases, the catalytic F1 unit fitted the main globular density very well, and the γ, δ, and ε subunits filled the density of the central stalk. As in the single particle maps (8, 9), the F1 region in the subtomogram average has pseudo-sixfold symmetry with alternating short and long edges (Fig. 2B). These edges correspond to the catalytic and noncatalytic α/β interfaces of the F1 subcomplex (24), which allowed us to optimize the fit of the F1/rotor ring assembly. The resulting fit positioned the β subunits in the three stronger of the six densities, with a noncatalytic α/β interface roughly parallel to the peripheral stalk density (9, 15) (Fig. 2C). The N-terminal ends of the rotor ring subunits in the F1/rotor ring assembly [2WPD; (12)] filled the bulge of density on the intra-cristae side of the membrane (Fig. 2A).
In the alternative fit of the F1/peripheral stalk assembly (2WSS), the peripheral stalk subunits did not sit fully in the corresponding density of the subtomogram average (Fig. S3 A and C), whereas the structure of the bovine peripheral stalk fragment (2CLY) fitted this density almost perfectly (Fig. 2, and Fig. S3 B and D). This finding indicates that the position of the peripheral stalk in the bovine crystal structure (2WSS) is different from that in yeast ATP synthase dimers. To account for a small volume above F1, we extended the 2CLY fragment to include the C-terminal residues of the b subunit from the 2WSS structure. Finally, we fitted the N-terminal part of the OSCP subunit to the map region immediately above an α subunit, with which it is known to interact (15, 25). The fit as shown in Fig. 2 was now excellent overall, with the b-subunit (homologous to subunit 4 in yeast) extending from the membrane to the top of the F1 subcomplex. Note that the twist of the peripheral stalk seen in the crystal structure (2CLY) is also evident in the subtomogram average.
Mutants Lacking Specific ATP Synthase Subunits Do Not Have Dimers or Lamellar Cristae.
The ATP synthase components that have been proposed to be involved in dimer formation include subunits h, i, 6, e, g, and 4 (16, 19–21, 26, 27). Our map indicates that the interface between protomers is located in the membrane between the peripheral stalks. Subunit h is positioned at the top of the peripheral stalk, near the OSCP subunit, and therefore cannot be part of the dimer interface (Fig. 2A). The remaining five subunits are all membrane-associated (16, 17, 28–30), and are thus candidates for dimer contacts in the membrane. Among these subunits, only subunits e, g, and su4TM1 can be deleted without affecting the assembly or function of the monomeric enzyme (3, 16). To assess whether these subunits are involved in dimer formation, we imaged mitochondria from mutant yeast strains lacking either subunit e, g, or su4TM1 by electron cryotomography.
The ATP synthase in the mutant mitochondria appeared monomeric and the mitochondria lacked the flat lamellar cristae with tightly curved ridges that are typical of wild type. Instead the inner mitochondrial membrane formed a number of separate vesicles with few or no cristae (Fig. 3 and Fig. S4). When cristae were present, they tended to be balloon-shaped with smooth, gently curving surfaces. Isolated membranes obtained from these mutant mitochondria were likewise spherical, with randomly distributed ATP synthase monomers (Fig. 4A), instead of being tubular or disk-shaped with rows of dimers as in wild type (Fig. 4 C and D). Some of the mutant membrane vesicles were accidentally flattened on the EM grid, resulting in highly curved edges. However, these edges were always devoid of ATP synthase particles (Fig. 4B).
Fig. 3.

Morphology of mitochondria from wild-type and mutant yeast strains. Surface-rendered volume of a mitochondrion from (A) wild-type and (B) the su4ΔTM1 yeast strain. Wild-type mitochondria have lamellar cristae with highly curved edges whereas mitochondria from mutants lacking either subunit e, g, or su4TM1 contain a number of separate inner membrane vesicles but few or no cristae. When cristae were present, they tended to be balloon-shaped with smooth, gently curving surfaces. Light grey-outer membrane, sky blue-inner membrane. (Scale bar, 200 nm). See also Fig. S4.
Fig. 4.

ATP synthase distribution in isolated mitochondrial membranes. Surface-rendered volumes of mitochondrial membranes from yeast strains lacking subunit g (A, B), and wild type (C, D). In the mutants, the ATP synthase complexes are monomeric, and randomly distributed over flat or gently curving membrane regions (A, B). By contrast ATP synthase from wild-type mitochondria form rows of dimers along the highly curved ridges of tubular (D) or disk-shaped (C) cristae vesicles.
We also imaged mitochondria from a yeast strain lacking subunit k, which is believed to stabilize the ATP synthase dimers (31). Tomograms of mitochondria and subtomogram averages of ATP synthase dimers from this mutant were indistinguishable from wild type (Fig. S5). We therefore conclude that (i) subunits e, g, and 4, but not k, are essential for the formation of the yeast ATP synthase dimer; (ii) monomeric ATP synthase does not by itself converge on highly curved membrane regions; (iii) rows of dimers are a prerequisite for the formation of lamellar cristae with tightly curved ridges that are typical of normal, wild-type yeast mitochondria.
Membrane Deformation Drives Assembly of Dimer Rows.
The assembly of ATP synthase dimers into rows has been implied to result from two different dimer geometries, one with an angle of approximately 35° and the other with an angle of approximately 90° (32) between monomers. It has been postulated that these two different dimers are formed by interaction of either subunits e and g or of subunits 4, i, a, and h (18–22). We show here that subunits e, g, and 4 are each essential for dimer formation. However, we only find one type of dimer in our tomograms. The distances and angles between dimers in a row are highly variable (Fig. 4 C and D), indicating that the organization of ATP synthase dimers into rows is not due to direct dimer-dimer contacts mediated by interstitial proteins.
The self-assembly of ATP synthase dimers into rows might instead be due to the deformation imposed by individual dimers on the surrounding lipid bilayer. To examine this perturbation and its potential role in the formation of dimer rows, we carried out a series of coarse-grained molecular dynamics simulations of single and multiple ATP synthase dimers in a model phospholipid membrane. We found that an individual dimer causes a pronounced convex curvature (as seen from F1) along the direction connecting the two Fo domains, which persists over a distance of 15–20 nm from the dimer x-axis, (Fig. 5 A and B and Fig. S6A). However, the dimer also enforces a concave, or negative membrane curvature along the perpendicular y-axis, which persists over a similar distance (Fig. 5 A and C, and Fig. S6B). We considered that if two ATP synthase dimers approached one another by random lateral diffusion in the membrane, the curvature deformation in the y direction would be partially relieved if their x-axes became aligned to be parallel, thus stabilizing the arrangement observed in our tomograms. To quantify this effect, we carried out additional simulations of a membrane containing a dimer of dimers, and a tetramer of dimers (Fig. 5 D–F), and compared the overall curvature deformation of these membranes with that of the single-dimer system (Fig. S6). These calculations indeed indicate that a side-by-side association of ATP synthase dimers, as seen in wild type mitochondria, reduces the elastic energy of the surrounding membrane to the order of several kBT for each dimer-dimer interface (Fig. S6). This energy reduction occurs because the curvature of the membrane along the direction of the row (y-axis) is significantly reduced between adjacent dimers (Fig. 5E), while that in the perpendicular direction (x-axis) is largely unchanged. Although other factors may also contribute, we propose that this net gain in membrane elastic energy is a major driving force for the self-assembly of ATP synthase dimers into rows.
Fig. 5.

Membrane curvature induced by ATP synthase dimers. (A) perspective view of a simulated membrane patch with an ATP synthase dimer distorting the planar lipid bilayer. The simulation is based on a coarse-grained representation of the dimer structure and its environment (see Methods); for clarity, the solvent is omitted. (B, C) cross sections through the membrane patch in (A) showing the curvature profile of the lipid bilayer in x and y direction. (D) and (E), curvature profiles as in (C) for membranes with two or four ATP synthase dimers, side by side. Note how the membrane deformation in y direction is relieved when two or more dimers assemble into a row. (F) perspective view of the row of four dimers shown in (E). 2D curvature maps are compared quantitatively in Fig. S6.
Discussion
Mitochondria are the powerhouse of eukaryotic cells and the main site of ATP synthesis by aerobic respiration. The inner membrane of a typical mitochondrion has numerous deep invaginations called cristae. The cristae are generally presumed to increase the membrane area available for oxidative phosphorylation, and have been proposed to play an important role in optimizing ATP synthesis (5, 6). The formation of cristae results in extreme local membrane curvature, which, as seen from the matrix, is either concave (negative) at the point of membrane invagination or convex (positive) at the cristae ridges. In our previous work we have shown that the ATP synthase dimers are located along the highly curved ridges of lamellar cristae in mitochondria from six different species (5). In this work, we present the structure of the yeast ATP synthase dimer in cristae membranes and examine the formation of both the ATP synthase dimer and the dimer rows, and ask how they affect mitochondrial morphology.
The Yeast ATP Synthase Dimer.
Up to now, all structural studies of the yeast or mammalian F1Fo ATP synthase dimers have been carried out with complexes extracted from the membrane by detergent (8, 9, 32–34). Attempts to determine the detailed structure of isolated ATP synthase dimers have been hindered by the instability of the dimeric complex outside the membrane. Dudkina et al. reported two types of detergent-solubilized yeast ATP synthase dimers with angles of 35° or 90° (32). Others found that the detergent-extracted dimers were heterogenous, with angles ranging from 55° to 140° (33, 34). In one study (33), the predominant dimer angle was consistent with ours, whereas another reported a predominant angle of 36–48° (34). A 3D map (34) showed the peripheral stalks to be offset from the dimer interface by an angle of 30°. Our extensive analysis of tomographic volumes of cristae membranes revealed no evidence of ATP synthase dimers with angles other than approximately 90°. Therefore dimers with more acute angles (34) are most likely the result of nonspecific, hydrophobic interaction of the Fo domains upon detergent solubilization. Evidently, the membrane environment is essential for the stability of the native yeast ATP synthase dimer.
ATP synthase dimers from some organisms other than yeast or vertebrates appear to be more stable when extracted from the membrane. One such example is the mitochondrial F1Fo ATP synthase dimer from Polytomella, a chlorophyll-less green alga. The Polytomella dimer has been examined by single-particle analysis (35) and subtomogram averaging (36). The two reported structures differ in terms of dimer angle (50° or 70°) and the proposed interaction of peripheral stalks at the dimer interface. The yeast ATP synthase dimer is clearly different from both. The yeast dimer angle is wider, the peripheral stalk less thick, and the monomers interact only in the membrane. Above all, the organization of dimers into rows is different. In Polytomella, subtomogram averaging revealed a constant offset of +9° between successive dimers (36). This offset would lead to a helical arrangement of the rows spiralling around tubular cristae, as observed in Polytomella (32) and Paramecium (4), a protist. By contrast, dimer rows in yeast and vertebrates run along ridges of lamellar cristae (5, 6) and are never helical. This pronounced structural dissimilarity most likely reflects substantial differences in F1Fo ATP synthase subunit composition (37, 38).
Dimer Interactions and Row Formation.
The formation of ATP synthase dimer rows has been assumed to be protein-driven and considerable effort has been invested into finding the proteins responsible (16, 18–21, 26, 27). Subunits e and g have been widely assumed to provide the protein contacts between dimers (19–22). We demonstrate here that mutants lacking subunits e or g do not have ATP synthase dimers in situ, and that subunits e, g, and su4TM1 are all essential for dimer formation in the membrane. Deletion of either subunit e or su4TM1 results in the loss of subunit g (3, 16), which suggests that all three subunits are in close proximity at the dimer interface. As mitochondrial membranes contain only one type of dimer, there is also only one dimer interface. Subunits e and g are therefore unlikely to be involved both in dimer formation and in contacts between dimers along rows. Rather, the variable distance between neighboring dimers suggests a more general, protein-independent mechanism of row formation.
Dimer Rows and Membrane Curvature.
Although lipids can form curved bilayers by themselves, biological membranes are mostly shaped by proteins e.g., BAR (Bin1/amphiphysin/Rvs167) domain proteins, clathrins, and epsins (39). In mitochondria, the ATP synthase dimer has been shown to play a major role in shaping the cristae (40). On the basis of our computer simulations we propose that the curvature induced by the ATP synthase dimer in a simple lipid membrane is sufficient to drive the formation of long dimer rows, which would then give rise to highly curved membrane ridges and lamellar cristae. Although the lipid composition of the membrane may also play a role (41), the fact that a tetramer of dimers can bend a lipid bilayer composed of only POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) into a ridge suggests that the dimer alone is perfectly adequate to drive its own self-association without the need for additional protein-protein or protein-lipid interactions.
Considering that the rows of ATP synthase dimers are a highly conserved, fundamental feature of all normal mitochondria, the advantage of a membrane-driven self-assembly over a protein-mediated interaction becomes clear when the energy balance for remodelling is considered. The energetic cost of remodelling the dimer rows, according to our calculations, requires only a few kBT per dimer. However, if the interaction between dimers were mediated by protein, much more energy would be needed to disrupt each interaction, as can be deduced for example from the measured free energy of association for a glycophorin A homodimer, which is 15 kBT (42). The mitochondrion is a highly dynamic organelle, which undergoes constant changes in its shape in the course of fusion and fission events, organelle migration, or large-scale changes in cell morphology during division or contraction. As the mitochondria provide the energy for these processes, it is essential that they can remodel their inner membrane quickly without unnecessary energy consumption, or the need for additional, costly protein factors. The self-organization of ATP synthase dimers into rows by means of random diffusion and minimization of the elastic membrane energy meets these requirements exactly.
Conclusion
We used electron cryotomography and subtomogram averaging to determine the structure of the mitochondrial F1Fo ATP synthase dimer in situ at an estimated resolution of 3.7 nm, and examined the formation of dimers and dimer rows in mitochondrial membranes by molecular dynamics simulations. We conclude that: (i) two ATP synthase monomers interact at the base of the peripheral stalks in the membrane, thereby forming a symmetrical V-shaped dimer with the central stalks including an angle of 86°. (ii) Subunits e, g, and 4 are part of this dimer interface, and are each essential for dimer formation. (iii) Only one type of ATP synthase dimer exists in the cristae membranes of yeast mitochondria. This dimer is similar to that in other fungi and in vertebrates, but different from that in Polytomella and Paramecium. (iv) Molecular dynamics simulations indicate that an individual ATP synthase dimer causes a marked deformation of the surrounding lipid bilayer, inducing a positive, convex curvature in one direction and a concave curvature in the perpendicular direction. (v) The side-by-side association of multiple dimers into a row reduces the overall cost in elastic energy of membrane deformation. We propose that this effect is sufficient to drive the formation of rows of ATP synthase dimers in mitochondria, without the need for specific protein or lipid-mediated interactions between dimers. The dimers are thus themselves responsible for the formation of the highly curved cristae ridges, and an essential element of normal mitochondrial morphology.
Methods
Saccharomyces cerevisiae Strains.
Yeast strains W303, BY4742, Δe in W303, Δe in BY4742, 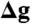 in W303,
in W303,  in BY4742, Δk in W303 and 4ΔTM1 in D273-10B [EUROSCAF,(3, 16)] were grown under nonfermentable conditions as described (43). Mitochondria were isolated by enzymatic digestion of the cell wall, followed by differential centrifugation as described (44). Whole mitochondria were fragmented by successive freeze-thaw cycles at -20 °C.
in BY4742, Δk in W303 and 4ΔTM1 in D273-10B [EUROSCAF,(3, 16)] were grown under nonfermentable conditions as described (43). Mitochondria were isolated by enzymatic digestion of the cell wall, followed by differential centrifugation as described (44). Whole mitochondria were fragmented by successive freeze-thaw cycles at -20 °C.
Electron Cryotomography.
Mitochondria samples were washed with trehalose buffer (250 mM trehalose, 10 mM Tris-HCl pH 7.4) and mixed 1∶1 with fiducial gold markers (6 nm gold particles conjugated to protein A, Aurion) immediately before plunge-freezing in liquid ethane. Both single and dual tilt image series (± 60°, step size 1.5°) were collected on an FEI Polara microscope operating at 300 kV equipped with a post-column energy filter and a 2 × 2 k CCD camera (GIF Tridiem 863, Gatan). Images were recorded at a nominal magnification of 41,000×, corresponding to a pixel size of 0.576 nm. The defocus was 7 μm. Tomograms were aligned using the gold fiducials and tomographic volumes were reconstructed using the IMOD package (45). Tomograms were filtered by nonlinear anisotropic diffusion to increase contrast (46), and manually segmented with the program AMIRA (Mercury Systems).
Subtomogram Averaging and Fitting of X-Ray Structures.
Subvolumes containing an ATP synthase dimer were extracted from single-tilt tomograms prior to contrast enhancement, prealigned according to the position of the F1 subcomplex to the membrane and averaged in IMOD (45). The initial alignment was optimized in PEET (47) using the preliminary average as a reference. The final volume was twofold averaged and filtered to 30 Å using a Fermi filter. A total of 121 out of an initial 138 subvolumes were used to calculate the final map. All features described were present after filtering to 50 Å. Fourier shell calculations were performed in PEET. Atomic models were docked in CHIMERA using the sequential fit routine (48). Replacement of the subvolumes into the original tomograms was carried out with the AMIRA EM toolbox (49).
Molecular Simulations.
Molecular dynamics simulations were carried out with GROMACS 4.5.3 (50) and the coarse-grained (CG) MARTINI 2.1 forcefield (51) at constant temperature (298 K) and pressure (1 bar). The simulation system with a single F1Fo dimer comprised approximately 1,728,000 CG particles (protein, POPC lipids, water, Na+ and Cl- ions), enclosed in a periodic box of dimensions 85.3 × 85.3 × 27.8 nm. The two-dimer simulation system was a duplicate of the single-dimer system, and thus included approximately 3,456,000 CG pseudoatoms in a box of 85.0 × 170.0 × 27.9 nm. Lastly, the tetramer-of-dimers system is 84.8 × 159.6 × 27.9 nm in size, and comprised approximately 3,260,000 CG particles. Each system was simulated for 850 ns, split in two trajectories of 750 and 100 ns. The computing core-hours used by these simulations were approximately 200,000 (single-dimer), approximately 450,000 (dimer-of-dimers) and approximately 550,000 (tetramer-of-dimers). Further details are provided in SI Methods.
Supplementary Material
ACKNOWLEDGMENTS.
We thank Marie-France Giraud and Rosemary A. Stuart for the kind gift of the su4ΔTM1 and Δe, 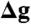 , and Δk yeast strains respectively, Deryck Mills for maintenance of the electron microscopy facility, and Maximilian Mattil for excellent technical assistance. This work was supported by the Max Planck Society (K.M.D., C.A., J.D.F-G, and W.K.), the Cluster of Excellence Frankfurt “Macromolecular Complexes” (W.K., and J.D.F-G) and Project Z1 of the SFB 815 “Redox Proteomics” (I.W.) funded by the Deutsche Forschungsgemeinschaft and the Bundesministerium für Bildung und Forschung BMBF 01GM1113B; mitoNET—Deutsches Netzwerk für mitochondriale Erkrankungen (I.W.).
, and Δk yeast strains respectively, Deryck Mills for maintenance of the electron microscopy facility, and Maximilian Mattil for excellent technical assistance. This work was supported by the Max Planck Society (K.M.D., C.A., J.D.F-G, and W.K.), the Cluster of Excellence Frankfurt “Macromolecular Complexes” (W.K., and J.D.F-G) and Project Z1 of the SFB 815 “Redox Proteomics” (I.W.) funded by the Deutsche Forschungsgemeinschaft and the Bundesministerium für Bildung und Forschung BMBF 01GM1113B; mitoNET—Deutsches Netzwerk für mitochondriale Erkrankungen (I.W.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The map and fitted model reported in this paper have been deposited in the Electron Microscopy Data Bank (accession no. 2161) and the Protein Data Bank (accession no. 4b2q), respectively.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1204593109/-/DCSupplemental.
References
- 1.Kagawa Y, Racker E. Partial resolution of the enzymes catalyzing oxidative phosphorylation. X. Correlation of morphology and function in submitochondrial particles. J Biol Chem. 1966;241:2475–2482. [PubMed] [Google Scholar]
- 2.Gilkerson RW, Selker JM, Capaldi RA. The cristal membrane of mitochondria is the principal site of oxidative phosphorylation. FEBS Lett. 2003;546:355–358. doi: 10.1016/s0014-5793(03)00633-1. [DOI] [PubMed] [Google Scholar]
- 3.Arnold I, Pfeiffer K, Neupert W, Stuart RA, Schagger H. Yeast mitochondrial F1F0-ATP synthase exists as a dimer: identification of three dimer-specific subunits. EMBO J. 1998;17:7170–7178. doi: 10.1093/emboj/17.24.7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen R, Schroeder C, Fok A. An investigation of mitochondrial inner membranes by rapid-freeze deep-etch techniques. J Cell Biol. 1989;108:2233–2240. doi: 10.1083/jcb.108.6.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies KM, et al. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc Natl Acad Sci USA. 2011;108:14121–14126. doi: 10.1073/pnas.1103621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strauss M, Hofhaus G, Schröder RR, Kühlbrandt W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 2008;27:1154–1160. doi: 10.1038/emboj.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Velours J, Arselin G. The Saccharomyces cerevisiae ATP synthase. J Bioenerg Biomembr. 2000;32:383–390. doi: 10.1023/a:1005580020547. [DOI] [PubMed] [Google Scholar]
- 8.Rubinstein JL, Walker JE, Henderson R. Structure of the mitochondrial ATP synthase by electron cryomicroscopy. EMBO J. 2003;22:6182–6192. doi: 10.1093/emboj/cdg608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lau WC, Baker LA, Rubinstein JL. Cryo-EM structure of the yeast ATP synthase. J Mol Biol. 2008;382:1256–1264. doi: 10.1016/j.jmb.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 10.Meier T, Faraldo-Gomez J, Börsch B. ATP synthase—a paradigmatic molecular machine. In: Frank J, editor. Molecular Machines in Biology. Cambridge: Cambridge Univ Press; 2011. pp. 208–238. [Google Scholar]
- 11.Stock D, Leslie AG, Walker JE. Molecular architecture of the rotary motor in ATP synthase. Science. 1999;286:1700–1705. doi: 10.1126/science.286.5445.1700. [DOI] [PubMed] [Google Scholar]
- 12.Dautant A, Velours J, Giraud MF. Crystal structure of the Mg.ADP-inhibited state of the yeast F1c10-ATP synthase. J Biol Chem. 2010;285:29502–29510. doi: 10.1074/jbc.M110.124529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watt IN, Montgomery MG, Runswick MJ, Leslie AG, Walker JE. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci USA. 2010;107:16823–16827. doi: 10.1073/pnas.1011099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dickson VK, Silvester JA, Fearnley IM, Leslie AG, Walker JE. On the structure of the stator of the mitochondrial ATP synthase. EMBO J. 2006;25:2911–2918. doi: 10.1038/sj.emboj.7601177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rees DM, Leslie AG, Walker JE. The structure of the membrane extrinsic region of bovine ATP synthase. Proc Natl Acad Sci USA. 2009;106:21597–21601. doi: 10.1073/pnas.0910365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soubannier V, et al. In the absence of the first membrane-spanning segment of subunit 4(b), the yeast ATP synthase is functional but does not dimerize or oligomerize. J Biol Chem. 2002;277:10739–10745. doi: 10.1074/jbc.M111882200. [DOI] [PubMed] [Google Scholar]
- 17.Arnold I, Pfeiffer K, Neupert W, Stuart RA, Schagger H. ATP synthase of yeast mitochondria. Isolation of subunit j and disruption of the ATP18 gene. J Biol Chem. 1999;274:36–40. doi: 10.1074/jbc.274.1.36. [DOI] [PubMed] [Google Scholar]
- 18.Wittig I, Velours J, Stuart R, Schagger H. Characterization of domain interfaces in monomeric and dimeric ATP synthase. Mol Cell Proteomics. 2008;7:995–1004. doi: 10.1074/mcp.M700465-MCP200. [DOI] [PubMed] [Google Scholar]
- 19.Paumard P, et al. Two ATP synthases can be linked through subunits i in the inner mitochondrial membrane of Saccharomyces cerevisiae. Biochemistry. 2002;41:10390–10396. doi: 10.1021/bi025923g. [DOI] [PubMed] [Google Scholar]
- 20.Spannagel C, et al. Evidence of a subunit 4 (subunit b) dimer in favor of the proximity of ATP synthase complexes in yeast inner mitochondrial membrane. Biochim Biophys Acta. 1998;1414:260–264. doi: 10.1016/s0005-2736(98)00174-6. [DOI] [PubMed] [Google Scholar]
- 21.Fronzes R, Weimann T, Vaillier J, Velours J, Brèthes D. The peripheral stalk participates in the yeast ATP synthase dimerization independently of e and g subunits. Biochemistry. 2006;45:6715–6723. doi: 10.1021/bi0601407. [DOI] [PubMed] [Google Scholar]
- 22.Gavin P, Prescott M, Devenish R. F1F0-ATP synthase complex interactions in vivo can occur in the absence of the dimer specific subunit e. J Bioenerg Biomembr. 2005;37:55–66. doi: 10.1007/s10863-005-4128-8. [DOI] [PubMed] [Google Scholar]
- 23.Saxton WO, Baumeister W. The correlation averaging of a regularly arranged bacterial cell envelope protein. J Microsc. 1982;127:127–138. doi: 10.1111/j.1365-2818.1982.tb00405.x. [DOI] [PubMed] [Google Scholar]
- 24.Abrahams JP, Leslie AG, Lutter R, Walker JE. Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature. 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 25.Carbajo RJ, et al. Structure of the F1-binding domain of the stator of bovine F1F0-ATPase and how it binds an alpha-subunit. J Mol Biol. 2005;351:824–838. doi: 10.1016/j.jmb.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 26.Velours J, et al. Organization of the yeast ATP synthase F(0): a study based on cysteine mutants, thiol modification and cross-linking reagents. Biochim Biophys Acta. 2000;1458:443–456. doi: 10.1016/s0005-2728(00)00093-1. [DOI] [PubMed] [Google Scholar]
- 27.Velours J, et al. Evidence of the proximity of ATP synthase subunits 6(a) in the inner mitochondrial membrane and in the supramolecular forms of Saccharomyces cerevisiae ATP synthase. J Biol Chem. 2011;286:35477–35484. doi: 10.1074/jbc.M111.275776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arnold I, Bauer MF, Brunner M, Neupert W, Stuart RA. Yeast mitochondrial F1F0-ATPase: the novel subunit e is identical to Tim11. FEBS Lett. 1997;411:195–200. doi: 10.1016/s0014-5793(97)00691-1. [DOI] [PubMed] [Google Scholar]
- 29.Boyle GM, Roucou X, Nagley P, Devenish RJ, Prescott M. Identification of subunit g of yeast mitochondrial F1F0-ATP synthase, a protein required for maximal activity of cytochrome c oxidase. Eur J Biochem. 1999;262:315–323. doi: 10.1046/j.1432-1327.1999.00345.x. [DOI] [PubMed] [Google Scholar]
- 30.Cox GB, Fimmel AL, Gibson F, Hatch L. The mechanism of ATP synthase: a reassessment of the functions of the b and a subunits. Biochim Biophys Acta. 1986;849:62–69. doi: 10.1016/0005-2728(86)90096-4. [DOI] [PubMed] [Google Scholar]
- 31.Wagner K, Perschil I, Fichter CD, van der Laan M. Stepwise assembly of dimeric F(1)F(o)-ATP synthase in mitochondria involves the small F(o)-subunits k and i. Mol Biol Cell. 2010;21:1494–1504. doi: 10.1091/mbc.E09-12-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dudkina NV, Sunderhaus S, Braun H-P, Boekema EJ. Characterization of dimeric ATP synthase and cristae membrane ultrastructure from Saccharomyces and Polytomella mitochondria. FEBS Lett. 2006;580:3427–3432. doi: 10.1016/j.febslet.2006.04.097. [DOI] [PubMed] [Google Scholar]
- 33.Thomas D, et al. Supramolecular organization of the yeast F1F0-ATP synthase. Biol Cell. 2008;100:591–601. doi: 10.1042/BC20080022. [DOI] [PubMed] [Google Scholar]
- 34.Couoh-Cardel SJ, Uribe-Carvajal S, Wilkens S, Garcia-Trejo JJ. Structure of dimeric F1F0-ATP synthase. J Biol Chem. 2010;285:36447–36455. doi: 10.1074/jbc.M110.144907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cano-Estrada A, et al. Subunit-subunit interactions and overall topology of the dimeric mitochondrial ATP synthase of Polytomella sp. Biochim Biophys Acta. 2010;1797:1439–1448. doi: 10.1016/j.bbabio.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 36.Dudkina NV, Oostergetel GT, Lewejohann D, Braun HP, Boekema EJ. Row-like organization of ATP synthase in intact mitochondria determined by cryo-electron tomography. Biochim Biophys Acta. 2010;1797:272–277. doi: 10.1016/j.bbabio.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 37.Vázquez-Acevedo M, et al. The mitochondrial ATP synthase of chlorophycean algae contains eight subunits of unknown origin involved in the formation of an atypical stator-stalk and in the dimerization of the complex. J Bioenerg Biomembr. 2006;38:271–282. doi: 10.1007/s10863-006-9046-x. [DOI] [PubMed] [Google Scholar]
- 38.Lapaille M, et al. Atypical subunit composition of the chlorophycean mitochondrial F1F0-ATP synthase and role of Asa7 protein in stability and oligomycin resistance of the enzyme. Mol Biol Evol. 2010;27:1630–1644. doi: 10.1093/molbev/msq049. [DOI] [PubMed] [Google Scholar]
- 39.McMahon HT, Gallop JL. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 2005;438:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- 40.Paumard P, et al. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002;21:221–230. doi: 10.1093/emboj/21.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Acehan D, et al. Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys J. 2011;100:2184–2192. doi: 10.1016/j.bpj.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fleming KG, Ackerman AL, Engelman DM. The effect of point mutations on the free energy of transmembrane alpha-helix dimerization. J Mol Biol. 1997;272:266–275. doi: 10.1006/jmbi.1997.1236. [DOI] [PubMed] [Google Scholar]
- 43.Rabl R, et al. Formation of cristae and crista junctions in mitochondria depends on antagonism between Fcj1 and Su e/g. J Cell Biol. 2009;185:1047–1063. doi: 10.1083/jcb.200811099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daum G, Bohni PC, Schatz G. Import of proteins into mitochondria. Cytochrome b2 and cytochrome c peroxidase are located in the intermembrane space of yeast mitochondria. J Biol Chem. 1982;257:13028–13033. [PubMed] [Google Scholar]
- 45.Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 46.Frangakis AS, Hegerl R. Noise reduction in electron tomographic reconstructions using nonlinear anisotropic diffusion. J Struct Biol. 2001;135:239–250. doi: 10.1006/jsbi.2001.4406. [DOI] [PubMed] [Google Scholar]
- 47.Nicastro D, et al. The molecular architecture of axonemes revealed by cryoelectron tomography. Science. 2006;313:944–948. doi: 10.1126/science.1128618. [DOI] [PubMed] [Google Scholar]
- 48.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 49.Pruggnaller S, Mayr M, Frangakis AS. A visualization and segmentation toolbox for electron microscopy. J Struct Biol. 2008;164:161–165. doi: 10.1016/j.jsb.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 50.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–337. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 51.Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH. The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.