

Abstract
A layer of endothelial cells attached to their underlying matrices by complex transmembrane structures termed focal adhesion (FA) proteins maintains the barrier property of microvascular endothelium. FAs sense the physical properties of the extracellular matrix (ECM) and organize the cytoskeleton accordingly. The close association of ahderens junction (A) protein, cadherin, with the cytoskeleton is known to be essential in coordinating the appropriate mechanical properties to cell-cell contacts. Recently, it has become clear that a crosstalk exists between focal adhesion kinase (FAK) and cadherin that regulates signaling at intercellular endothelial junctions. This review discusses recent advances in our understanding of the dynamic regulation of the molecular connections between FAK and the cadherin complex and cadherin-catenins-actin interaction-dependent changes as well as the role of small GTPases in endothelial barrier regulation. This review also discusses how a signaling network regulates a range of cellular processes important for barrier function and diseases.
Keywords: Barrier function, focal adhesion (FA), Focal adhesion kinase(FAK), Adherens junction (AJ), cadherin
INRODUCTION
Cell adhesion relies upon specialized transmembrane adhesion proteins, the cell adhesion molecules, through which cell-cell and cell-matrix interactions are mediated. These families of molecules act through receptor-ligand interactions that usually extend from the intracellular space to the extracellular space where they bind to other cell membranes (cell-cell) or to the cell-matrix. Adhesion sites that defined structural contact between cells and the ECM were initially described in studies using interference-reflection microscopy and electron microscopy (Abercrombie and Dunn, 1975; Abercrombie et al., 1971; Izzard and Lochner, 1976; Izzard and Lochner, 1980). These studies revealed that matrix adhesion occurs at many specialized, elongated small regions along the ventral plasma membrane tightly connected with the substrate. Moreover, these sites termed FAs, are associated with actin microfilaments at their cytoplasmic aspects and play an important role in the regulation of actin cytoskeleton organization, the adhesive interaction between integrins and their extracellular ligand, and the regulation of endothelial barrier integrity. FAK has emerged as a mediator of crosstalk between integrin-mediated FAs and intercellular adherens junction (AJs). FAK plays a central role in initiating and integrating various signaling pathways that ultimately affect barrier function. Evidence points to the importance of FAK activation in the regulation of microvascular barrier function (Holinstat et al., 2006; Quadri et al., 2003; Quadri and Bhattacharya, 2007). On one hand, FAK activation is essential in the maintenance of endothelial barrier properties, and inhibition of FAK activity leads to leaky microvessels (Holinstat et al., 2006; Quadri et al., 2003; Quadri and Bhattacharya, 2007). Conversely, FA assembly and activation serve as important signaling events in increasing endothelial permeability under stimulatory conditions, such as in the presence of angiogenic factors (Eliceiri et al., 2002; Zachary, 2003) and inflammatory mediators (Uehata et al., 1997).
Adhesion between cells is mediated by junctional proteins that constitute the intercellular junctional complex, which has an important role in defining the physiological function of a cell. Cadherins are plasma membrane proteins associated with AJs that make important contributions to barrier function, embryogenesis, and tissue homeostasis (Gumbiner, 2005; Halbleib and Nelson, 2006; Nishimura and Takeichi, 2009). AJs are characterized ultrastructurally as plasma membrane associated organelles comprised of opposing dense plaques at cell-cell contacts. The extracellular domains of cadherins are involved in homotypic interactions required for the formation of AJs; the cytoplasmic domain associates with catenins that link AJs to the actin cytoskeleton for junctional stabilization (Hirokawa and Heuser, 1981; Miyaguchi, 2000). The close association of the cadherin molecules with the cytoskeleton is known to be essential in coordinating the appropriate mechanical properties to cell-cell contacts. How adhesive interactions between cells generate and maintain the endothelial barrier remains one of the most challenging questions in understanding the basis of endothelial barrier function. AJs and the cadherin-catenin complex are therefore the subjects of intense research. Recent work has greatly advanced our understanding of the molecular organization of AJs and how cadherin-catenin complexes engage actin.
We reviewed the molecular structure and function of FAK (Sunita Bhattacharya, 2005) and cadherins (Parthasarathi, 2009). The review addresses FAK and VE/E-cadherin signaling in endothelial barrier regulation. This review also addresses cadherin-actin based adhesion, specifically the association between intracellular VE/E-cadherin molecules and the actin cytoskeleton. In addition to their adhesive function, cell adhesion molecules modulate signal transduction pathways by interacting with receptor tyrosine kinases and Rho-family GTPases for example (Braga, 2002; Noren et al., 2003; Yap and Kovacs, 2003). Hence, changes in the expression of cell adhesion molecules affect not only the adhesive properties, but also the signal transduction status of a cell. Conversely, signaling pathways modulate the function of cell adhesion molecules, altering the interactions between cells and their environment. This leads to changes in cell-cell and cell-matrix interactions, hence, microvascular endothelial barrier regulation. The combined application of new approaches, such as live cell imaging with molecular manipulation by DNA, protein transfection, and gene silencing will continue to provide excellent tools for FA and AJ regulation studies. Quantitative confocal and two photon microscopy methods that allow for simultaneous measurements of AJ protein dynamics and permeability in the intact microvessel provide a unique direction of future studies.
FAK LOCALIZATION AND FUNCTION
FAK is a major player in mediating signaling initiated at sites of cell-matrix attachment and at activated growth factor receptors, such as those for platelet-derived growth factor (PDGF), epidermal growth factor (EGF), hepatocyte growth factor (HGF), and vascular endothelial growth factor (VEGF) (Chen and Chen, 2006; Garces et al., 2006; Sieg et al., 2000). FAK is most commonly found at the cell membrane, in FAs, in smaller focal contacts, or in nascent spreading adhesions (de Hoog et al., 2004; Serrels et al., 2007). In epithelial cell-cell junctions the FAK NH2-terminal domain targeted to the nuclei and intercellular junction (Stewart et al., 2002), raises the possibility that the generation of NH2-terminal FAK fragments by post-translational processing may provide a novel mechanism for modulating cell junction. Different FAK domains may be postulated to play distinct cell type specific roles. FAK NH2-terminal fragments are generated during apoptosis (Lobo and Zachary, 2000). FAK resides in the nucleus, implying that it may ‘travel’ between subcellular locations. The role of nuclear localization of the FAK NH2-terminal domain is not known.
Protein interactions with the FAK carboxy-terminal, the focal adhesion targeting sequence (Hildebrand et al., 1993), are thought to determine its subcellular localization. FAK localization to focal adhesions is mediated primarily by the COOH-terminal focal adhesion targeting (FAT, residues 840–1052) domain (Hildebrand et al., 1993). The COOH-terminal domain of FAK is expressed in some tissues as an alternative transcript encoding a 41–43kDa protein called FRNK (for FAK-related non-kinase) (Schaller et al., 1993), and this domain antagonises FAK signaling by competing for binding to focal contacts (Taylor et al., 2001). Evidence shows (Holinstat et al., 2006) that inhibition of FAK by adenoviral expression of FRNK (a dominant negative FAK construct) in monolayer prevented p190RhoGAP phosphorylation, increased RhoA activity, induced actin stress fiber formation, and produced an irreversible increase in endothelial permeability in response to thrombin. Expression of FRNK in lung microvessel endothelia increased vascular permeability. RhoA is known to increase endothelial monolayer permeability by disrupting adherens junctions and reorganizing the cell-ECM attachment sites (Carbajal et al., 2000).
FAK activity is necessary for barrier enhancement (Holinstat et al., 2006; Quadri et al., 2003; Quadri and Bhattacharya, 2007) and controls diverse cellular processes, as well as biological properties associated with barrier function (Mehta et al., 2002; Quadri et al., 2003) and disease, such as vascular development, cardiomyocyte-induced hypertrophy, fibrosis, and epithelial cancer (Chishti et al., 1998; Lim et al., 2008; Luo and Guan, 2010; van Nimwegen and van de Water, 2007; Zhao and Guan, 2009). This range of functions is evidence that FAK performs fundamentally important roles in cells, the details of which continue to be uncovered. Indeed, at the cell cortex, FAK regulates integrin-dependent cell-matrix interactions, promoting dynamic actin and adhesion changes at the membrane and signaling to proliferation and survival pathways. Although FAK associated with cadherin, but does not have any binding site for cell-cell junctional protein for example cadherin /catenin, hence FAK does not interact directly with junctional protein. FAK transmit signals to junctional protein through intermediate molecules. It is not yet clear how the combined scaffold and kinase functions of FAK integrate signaling outputs that coordinate cell adhesion and barrier regulation.
FAK AND ACTIN SIGNALING
FAK influences adhesion by its direct or indirect effects on actin and adhesion regulators, such as the RhoGTPases (Noren et al., 2003). Neural Wiskott–Aldrich syndrome protein (n-WASP), which is an effector for the RhoGTPase CDC42 (Wu et al., 2004) is a binding partner and substrate of FAK. WASP transduces extracellular signals into reorganization of the cytoskeleton and regulates actin-related protein (Arp2/3). Phosphorylation of n-WASP on Tyr256 affects its nuclear localization and promotes cell migration (Wu et al., 2004). In addition to n-WASP, the FAK-FERM domain (N-terminal) also binds directly to the Arp3, which induces an activating conformational change in the Arp2/3 complex (Fig. 1). This promotes nucleation by bringing an actin monomer to Arp2/3 via WASP-homology 2 domain, enhancing actin polymerization (Serrels et al., 2007), and stabilizing the newly formed actin (Winder, 2003). FAK is needed for proper assembly of nascent integrin adhesions. Arp3 is located at the tip; of nascent adhesion structures as they form (Serrels et al., 2007). The FAK FERM–Arp3 interaction is an example that directly links integrin signaling with actin polymerization machinery in the vicinity of nascent adhesions (Fig. 1).
Fig. 1. Model of FAK and the cadherin complex function in actin polymerization.
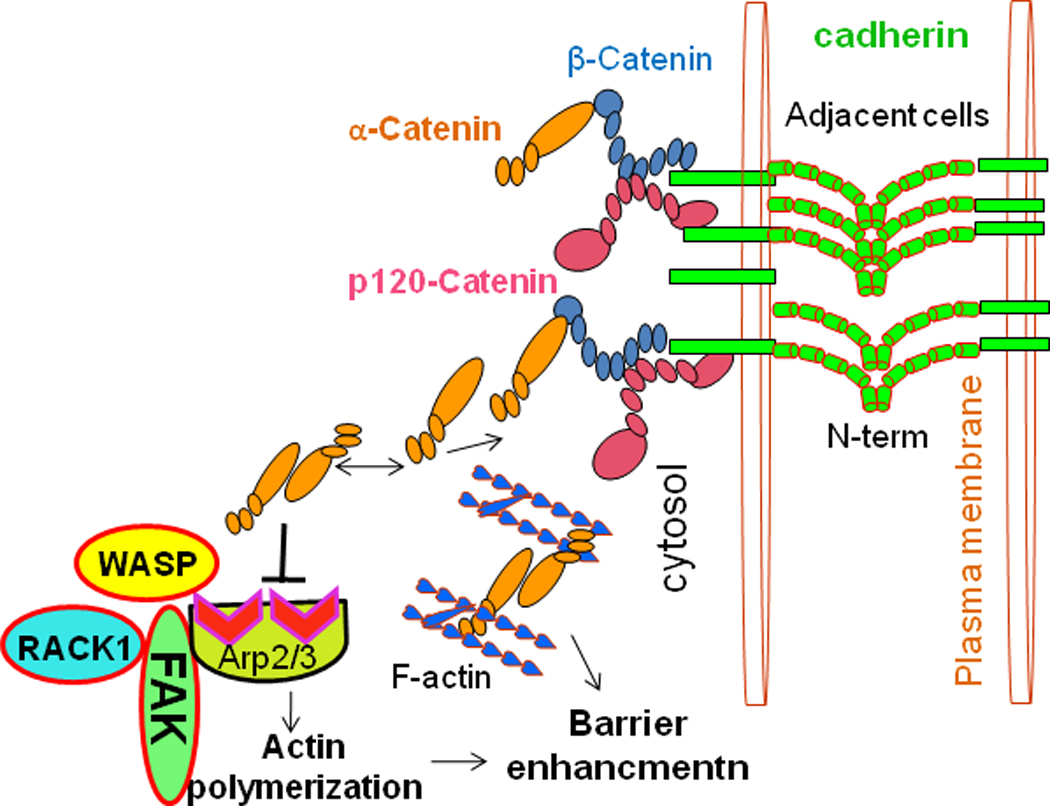
FAK signals to Arp2/3 in modulating cell adhesion and actin polymerization. The FAK N-terminal domain binds directly to Arp3, which promotes nucleation by bringing an actin monomer to Arp2/3 via the WASP-homology 2 domain, thereby enhancing actin polymerization. The α-catenin isoform binds to actin preferentially as a dimer, and the α-catenin monomer binds to β-catenin; therefore α-catenin does not bind to β-catenin and actin simultaneously.
Receptor for activated kinase C1 (RACK1) is found in nascent integrin adhesions but not in mature focal adhesion structures (de Hoog et al., 2004; Serrels et al., 2007). RACK1 binds to the FERM domain of FAK, at different FERM sequences to ARP3 (Fig. 1), and this complex is also located at nascent adhesions (Serrels et al., 2010). The FAK FERM–RACK1 interaction enables directional responses, and this may contribute to FAK’s role in an invasive cancer phenotype (Lahlou et al., 2007; Luo and Guan, 2010; McLean et al., 2004). Although this interaction between two molecular scaffolds (FAK and RACK1) is important physiologically, it is not clear whether RACK1 binding to the FERM domain activates FAK kinase activity (Serrels et al., 2010). Key effecter substrates of FAK in different functions have not widely been identified. The relative importance of the adaptor and kinase functions in all of FAK’s biological activities and endothelial barrier regulation remains unknown.
CADHERIN ORGANIZATION AND FUNCTION
The mechanisms of cellular signaling and adhesion are thought to be closely connected, such that adhesion components have double (or more) functions and interconnect in a signaling structural network (Pece and Gutkind, 2000). AJs function as clusters during zonula adherens assembly and dynamic cell–cell interactions. Cadherins are the principal components of AJs and clusters at sites of cell–cell contact (Parthasarathi, 2009). The cadherin family consists of classical cadherins, which are the main mediators of calcium-dependent cell–cell adhesion, and non-classical cadherins, which include desmosomal cadherins and the recently discovered large subfamily of protocadherins, which are implicated in neuronal plasticity. Classical cadherins are a family of single-span, transmembrane-domain glycoproteins that function specifically as cell–cell adhesion molecules. The classical cadherins are further subdivided into types I and II on the basis of sequence homology. These are three major cadherins found: vascular endothelial (VE), epithelial cadherin (E), and neuronal cadherins (N) (Corada et al., 1999; Liaw et al., 1990) in the vascular endothelium. VE-cadherin (also cadherin-5) is located at intercellular junctions of all endothelial types, and its expression has been confirmed both in vitro and in vivo (Dejana et al., 1999; Liaw et al., 1990). In the intact pulmonary vasculature, large vessels primarily express VE-cadherin (Gao et al., 2000; Parker et al., 2006; Safdar et al., 2003). Evidence show strong VE-cadherin expression in arteries, arterioles, and capillaries but almost no expression in veins and venules, suggesting vessel type-specific expression of VE-cadherin in regular human lung tissue, independent of age or sex (Herwig et al., 2008). Rat pulmonary microvessels express E-cadherin (Godzich et al., 2006; Ofori-Acquah et al., 2008; Parker et al., 2006; Quadri et al., 2003). VE-cadherin belongs to the type II sub group; only 23% of its sequence is identical with the classical cadherins, E-, and N-cadherins from the type I sub group (Breier et al., 1996). N-cadherin is not clustered at cell–cell junctions, but distributed diffusely in the cell membrane (Salomon et al., 1992). A morphological and functional endothelial heterogeneity has been proven for micro- and macrovascular endothelial cells of different organs, different species, and different compartments of the same organ (Cines et al., 1998; Volk and Kox, 2000).
Crystal structure studies show that cadherins contain a N-terminal extracellular region, a transmembrane anchor, and a cytoplasmic intracellular region (Fig. 1). The monitoring of cell aggregation by the binding of cells to immobilized cadherin ectodomains, or the binding of beads coated with purified cadherins, have led to the concept that cadherins function as homotypic cell adhesion molecules (Gumbiner, 2005). Cadherin molecules form homodimers on the cell surface; homotypic adhesion forms zipper-like adhesion, which may progress to extensive multimer formation (Boggon et al., 2002; Gumbiner, 2000). Functional features of homotypic adhesion might provide the barrier properties; for example, in mouse lung endothelial cells, a mutant of VE-cadherin lacking the extracellular domain, ΔEXD, increases vascular permeability (Broman et al., 2006).
Several groups have determined the three dimensional structures of the type I cadherin extracellular domain (Fig. 2) (Haussinger et al., 2004; Nagar et al., 1996; Pertz et al., 1999; Shapiro et al., 1995; Tamura et al., 1998). The extracellular domain consists of five ectodomains with immunoglobulin-like topology, ranging from the membrane-distal EC1 domain to the membrane-proximal EC5 domain (Boggon et al., 2002; Gumbiner, 2000; Harrison et al., 2010) Structural studies (Al-Amoudi and Frangakis, 2008; Nose et al., 1990; Shan et al., 1999), binding affinity measurements (Parisini et al., 2007), sequence analysis (Kitagawa et al., 2000), and molecular simulations (May et al., 2005; Tamura et al., 1998) have provided a detailed picture of the trans dimerization process that mediates cell-cell interaction. Trans dimerization is mediated by an interface formed between two cadherin molecules from opposing cells that swap the N-terminal β-strands of their EC1 domains, anchored by binding of the highly conserved Trp2 (Fig. 2). Cadherins show an exquisite specificity in their homophilic interactions by almost exclusively binding the same type of cadherin on the adjacent adherence cell. Binding between cadherin extracellular domains is relatively weak, but cell-cell adhesion may be strengthened by lateral clustering of cadherins mediated by protein linkages between the cadherin cytoplasmic domain and the actin cytoskeleton (Jamora and Fuchs, 2002). Thus, intracellular faces of these contacts are associated with the actin cytoskeleton in AJs (Fig. 1).
Fig. 2. Structure of strand-swapping in wild-type E-cadherin fragments (trans-dimer).
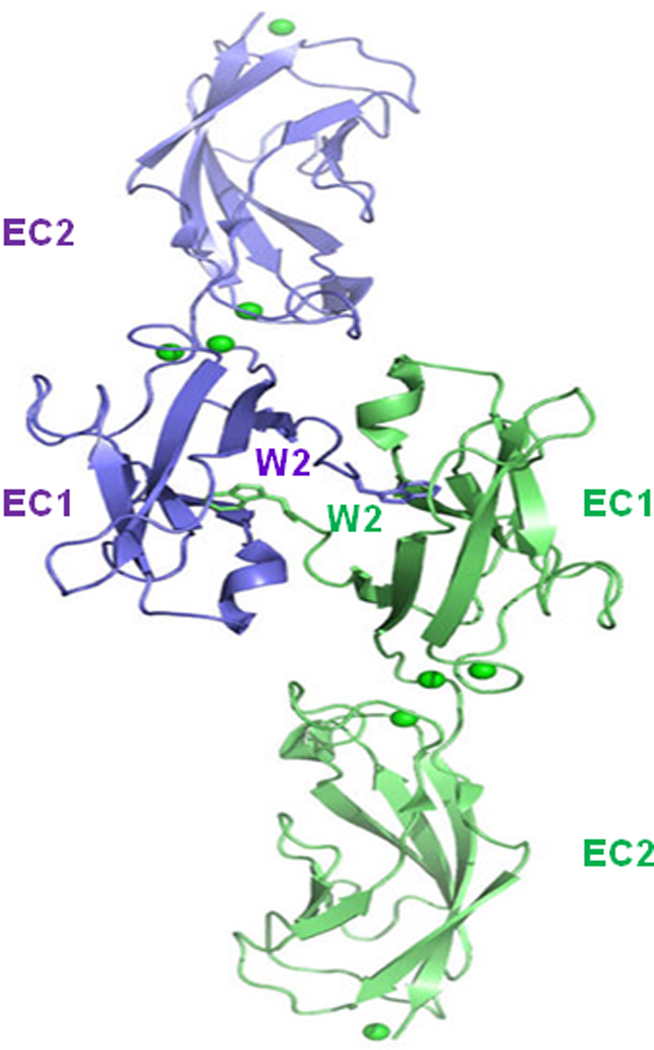
Ribbon diagram shows the strand-swapped dimer formed between protomers of wild-type E-cadherin EC1 in the crystal. Side chain atoms are shown for Trp2 residues (W2) and calcium ions are displayed as green spheres (reproduced with permission of the Nature Structure Molecular Biology Harrison, 2010).
CADHERIN-CATENIN-ACTIN INTERACTION
The intracellular domain of classical cadherins, which is lacking in non-classical cadherins and protocadherins, interacts with various catenin proteins to form the cytoplasmic adhesion complex (Parthasarathi, 2009). In mice and humans, 5 type I cadherins and 13 type II cadherins have been described (Posy et al., 2008). Type I and type II cadherins share several common structural features. They both contain an ectodomain region, which is composed of five tandem extracellular cadherin domains, each of about 110 amino acids (Boggon et al., 2002; Nagar et al., 1996). Classical cadherins are anchored by a single-transmembrane region and have a short cytoplasmic domain with conserved binding sites for β/γ-catenins and p120 catenins (Ishiyama et al., 2010; Lampugnani et al., 1995), which help to mediate attachment to the cytoskeleton and to control cadherin trafficking (Liu et al., 2007; Reynolds and Carnahan, 2004). The cytoplasmic C-terminus of cadherin binds to intracellular proteins; β-catenin and p120 catenin (Type I cadherin) or γ-catenin (Typ II cadherin). β-Catenin binds to both the C-terminus of the cadherin intracellular domain and the N-terminus of α-catenin (Fig. 1). The E-cadherin cytoplasmic domain forms a high affinity, 1:1 complex with β-catenin, which binds with lower affinity to α-catenin (Aberle et al., 1994; Hinck et al., 1994; Huber and Weis, 2001; Pokutta and Weis, 2000). α-Catenin binds directly to F-actin of the cytoskeleton (Fig. 1) also through number of actin binding proteins, such as α-actinin and vinculin. Absence of α- or β-catenin results in defective cell adhesion and failure of cadherin-catenin complexes to associate with the actin cytoskeleton. The Nelson group has challenged this view and suggested alternative roles for α-catenin in the junction (Drees et al., 2005; Yamada et al., 2005). They demonstrated with purified recombinant proteins that α-catenin cannot bind to β-catenin and actin simultaneously (Fig. 1), even in the presence of actin binding proteins. In fluorescence recovery after photobleaching (FRAP) experiments, E-cadherin, β-catenin, and α-catenin displayed very similar diffusional behaviors on the membrane, whereas actin associated with cell-cell contacts diffused more rapidly and was more mobile (Yamada et al., 2005). Moreover, deletion of the cadherin cytoplasmic domain or the actin-binding domain of α-catenin, which would break the link to actin, did not significantly alter the dynamics of the cadherin-catenin complex (Yamada et al., 2005). This evidence independently confirms the lack of a stable linkage between the cadherin-catenin complex and actin through α-catenin at cell-cell contacts. The Nelson group also has shown that the mammalian α-catenin isoform binds to actin preferentially as a dimer, and the α-catenin monomer binds to β-catenin (Drees et al., 2005). Thus, α-catenin may not be able to bind β-catenin and actin at the same time (Fig. 1), suggesting that α-catenin is not directly involved in the linkage between the E-cadherin–β-catenin complex and actin filaments (Drees et al., 2005). How cadherin cytoplasmic domains contribute to the stabilization of cell adhesion and endothelial barrier regulation is not clear.
REGULATION OF CADHERIN COMPLEX THROUGH SIGNALING MOLECULES
Cadherins are considered structural proteins, but there is evidence that cadherins are targets for signaling pathways that regulate adhesion, but also signaling molecules may themselves that regulate basic cellular processes, such as migration, proliferation, apoptosis and cell differentiation (Barth et al., 1997; Hulsken et al., 1994; Morin et al., 1997). Cadherin does not exhibit any enzymatic activity; therefore, their ability to function as signal transducing receptors depends on their physical interactions with other effectors. For example, phosphoinositide 3'-kinase (PI3K) is recruited to cell-cell contacts (Singleton et al., 2005; Sovova et al., 2004), activated by cadherin (Singleton et al., 2005; Sovova et al., 2004) and E-cadherin interacts with receptor tyrosine kinases, such as epidermal growth factor receptor (Andl and Rustgi, 2005).
Role of receptor tyrosine kinase (RTK) signaling
Tyrosine phosphorylation has been implicated in the regulation of cadherin function resulting in the disassembly of the cytoplasmic adhesion complex and, subsequently, the disruption of cadherin-mediated cell-cell adhesion. This includes phosphorylation of receptor tyrosine kinases, RTKs, which are frequently activated in cancer cells: epidermal growth factor receptor, hepatocyte growth factor receptor (c-MET), and fibroblast growth factor receptor. Converse to the regulation of E-cadherin function by RTKs, functional adhesion junctions can also affect the RTKs activity. For example, E-cadherin-mediated, cell-cell adhesion has been shown to repress EGF-induced epidermal growth factor receptor activation (Takahashi and Suzuki, 1996). Ligated E-cadherin also recruits epidermal growth factor receptor and induces its ligand-independent activation, leading to the activation of signal transduction cascades, including the PI3K and mitogen activated protein kinase (MAPK) pathways and to tumor cell survival (Kovacs et al., 2002; Pece and Gutkind, 2000). E-cadherin-mediated cell adhesion also induces the activation and phosphorylation of the RTK, resulting in the repression of cell-matrix adhesion (Zantek et al., 1999). However, the functional implication of this mechanism in barrier function is not known.
Role of nonreceptor tyrosine kinase (Src)
Disassembly of cadherin includes phosphorylation of the non-RTK, Src, which phosphorylates E-cadherin, neuronal (N)-cadherin, β-catenin, γ-catenin and p120-catenin (Fig. 3), resulting in the disruption of cadherin-mediated cell–cell adhesion (Behrens et al., 1993; Fujita et al., 2002; Hamaguchi et al., 1993). Cadherin molecules are not stably exposed at the cell surface; rather, they cycle on and off the plasma membrane in a highly dynamic fashion by exo- and endocytic events (Akhtar and Hotchin, 2001; Xiao et al., 2005). Internalization of E-cadherin from AJs is initiated by the Src-mediated tyrosine phosphorylation of E-cadherin (McLachlan et al., 2007; Papkoff, 1997). This posttranslational modification induces the dissociation of p120 from E-cadherin (Fig. 3), and the binding of the cbl-like ubiquitin-ligase, Hakai, which results in the ubiquitination of E-cadherin and internalization within clathrin-coated endosomes (Fujita et al., 2002; Palacios et al., 2005; Pece and Gutkind, 2002). p120 is a Src substrate and member of the catenin family (Anastasiadis and Reynolds, 2000) that binds to the juxtamembrane domain of E-cadherin (Fig. 3). p120 is involved in the maintenance of E-cadherin at the plasma membrane (Anastasiadis and Reynolds, 2000), (Ireton et al., 2002; Xiao et al., 2005). In an E-cadherin-bound state, p120 prevents the internalization of E-cadherin (Fujita et al., 2002; Pece and Gutkind, 2002). How the endocytic machinery regulates adherens junction formation or opening is an issue that requires further investigation.
Fig. 3. Interactions between FAK and the cadherin complex.
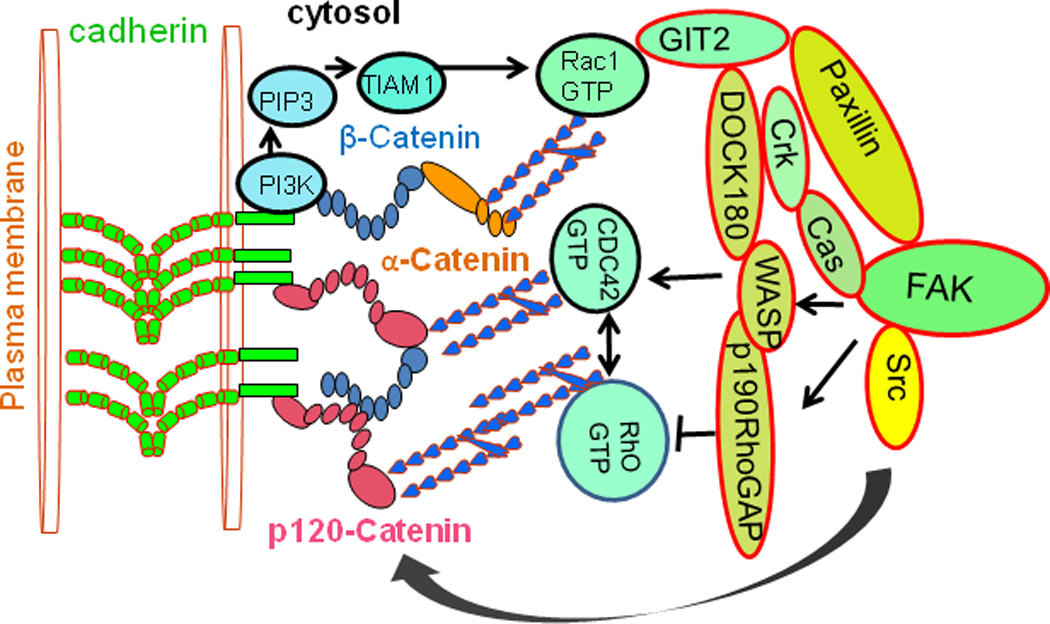
Signal from FAK to Rac1 plays a role in modulating cell adhesion and actin polymerization. PI3K is recruited to the membrane by intact E-cadherin AJs, where it generates PIP3, resulting in the activation of the Rho-GEF, TIAM1 and subsequently of Rac1 and CDC42. Activated FAK enhances Rac1 activity via a Cas/CrkII/DOCK180 complex. Paxillin binds to GIT2, participate in Rac- and Rho-mediated signaling events at FAs. Association of β-catenin with paxillin is depends on Rac and Cdc42 activities. Src family Activates p190 Rho-GAP, activated p190 Rho-GAP suppresses Rho activity. p120 is a Src substrate and is involved in the maintenance of cadherin at the plasma membrane.
Role of small GTPases
E-cadherin, once engaged in cell-cell adhesion, suppresses Rho activity by activating p190 Rho-GAP, probably through Src-family kinases, indicating that active signals are induced by the formation of cell junctions (Fig. 3). In addition to interacting with RhoGTPases through p190 Rho-GAP and p120-catenin, cadherin also communicates with these molecules through PI3K signaling. Ligation of cadherin molecules between two neighboring cells recruits PI3K to the cytoplasmic adhesion complex (Fig. 3), thereby generating phosphatidylinositol-(3,4,5)-triphosphate (PIP3) at the plasma membrane. Guanine nucleotide exchange factors (GEFs) that contain phosphatidylinositol-(3,4,5)-triphosphate (PIP3) binding pleckstrin-homology domains, such as TIAM1, are then recruited to the membrane, activating Rac1, and possibly activateing CDC42 (Fig. 3). Disrupting Rac1 or Rho interrupts AJ assembly (Braga, 2002; Yap and Kovacs, 2003), whereas CDC42 seems to regulate AJ maintenance (Kouklis et al., 2004). The Malik group demonstrated in mouse lung endothelial cells that the mutant of VE-cadherin lacking the extracellular domain (ΔEXD) increases vascular permeability, and coexpression of dominant-negative CDC42 (N17CDC42) prevents the increase of permeability induced by ΔEXD (Broman et al., 2006). This was attributed to inhibition of the α-catenin association with the ΔEXD-β-catenin complex, suggesting that CDC42 regulates AJ permeability by controlling the binding of α-catenin with β-catenin and the consequent interaction of the VE-cadherin/catenin complex with the actin cytoskeleton. Rho GTPases RhoA, Rac1, and CDC42 are important in regulating AJ assembly (Fukata and Kaibuchi, 2001; Zigmond, 2004). Cadherin-catenin interactions specifically activate Rac1, as seen in response to cadherin-based cell-cell adhesion (Lampugnani et al., 2002; Noren et al., 2001) and in cells binding to cadherin-coated substrates (Kovacs et al., 2002; Noren et al., 2001). Rac1 and CDC42 may support E-cadherin function. There seems to be a fine balance between Rac1 and Rho activity during AJ assembly. As cells make contact, Rac1 activation occurs at cell-cell contacts, whereas Rho acts at later contractile cables (Yamada and Nelson, 2007). Crosstalk between Rac1 and Rho helps the actin reconfiguration during AJ assembly.
Unfortunately, the overall picture of Rho proteins and barrier function is still not clear. For example, changing the composition of the ECM changed the function of Rac1 from a proadhesive to an anti-adhesive molecule (Sander et al., 1998). Rho family GTPases are certainly involved in many different aspects of the various stages of cell-cell adhesion formation; however, details of their actual functional roles remain to be determined.
Role of the WNT signaling pathway
Assembly of the cadherin, β-catenin, and γ-catenin complex, which mediates cell adhesion, also has important functions in the canonical WNT signaling pathway (Bienz and Clevers, 2000) (Fig. 4). Non-sequestered, free β-catenin and γ-catenin are rapidly phosphorylated by glycogen synthase kinase 3β (GSK-3β) in the adenomatous polyposis coli (APC)-axin-GSK-3β complex and are subsequently degraded by the ubiquitin-proteasome pathway. If the tumor suppressor APC is non-functional, as in many colon cancer cells, or if the activated WNT-signaling pathway blocks GSK-3β activity, β-catenin accumulates at high levels in the cytoplasm (Fig. 4). The WNT ligand ultimately results in the stabilization of cytoplasmic β-catenin, which is then free to enter the nucleus. Subsequently, β-catenin translocates to the nucleus, where it binds to members of the transcription factor TCF/LEF1 and modulates the expression of their target genes. This dual function of β-catenin raised the question of whether the loss of cadherin function would subsequently lead to the activation of the WNT signaling pathway. In various cellular systems, it has been demonstrated that sequestration of β-catenin by E-cadherin competes with the β-catenin/TCF-mediated transcriptional activity of the canonical WNT signaling pathway (Fig. 4). The fact that E-cadherin does not completely deplete cytoplasmic catenin indicates that β-catenin exists in different functional pools (Gottardi et al., 2001; Orsulic et al., 1999; Stockinger et al., 2001). Since the activation of transcription factor Kruppel-like factor (KLF4), regulates VE cadherin expression (Cowan et al. 2010), and also interacts with C-terminal domain of β-catenin (Evans et al. 2010), might inhibits Wnt signaling (Fig. 4). Hence maintains the integrity of AJs, preventing vascular leakage in response to inflammatory stimuli.
Fig. 4. Signaling pathways affected by loss of cadherin function.
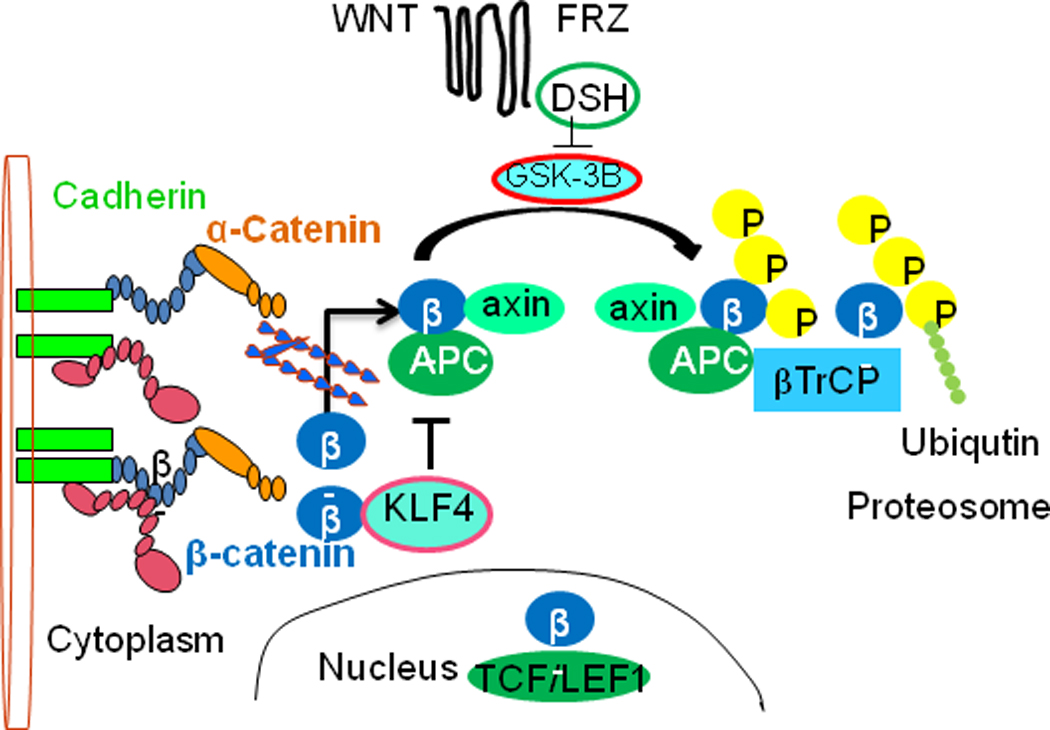
Upon disassembly of the cytoplasmic cell-adhesion complex, catenins are released and accumulate in the cytoplasm. β-Catenin (β) is then sequestered by the adenomatous polyposis coli (APC)–axin–glycogen synthase kinase 3β (GSK-3β) complex and phosphorylated by GSK-3β. Phosphorylated β-catenin is specifically bound by βTrCP, a subunit of the E3 ubiquitin-ligase complex, which ubiquitylates β-catenin and thereby marks it for rapid proteosomal degradation. However, on activation of the WNT signalling pathway, GSK-3β is repressed, and β-catenin is no longer phosphorylated. β-catenin translocates to the nucleus where, together with the TCF/LEF1 transcription factors, it modulates the expression of several target genes. Transcriton factor KLF4 interacts with β-catenin and inhibits Wnt signaling pathway.
CROSS TALK BETWEEN FAK AND CADHERIN-MEDIATED ADHESIONS
FAK is a critical bidirectional linkage between the actin cytoskeleton and the cell-matrix interface, thus providing stability that maintains endothelial cell barrier integrity. FAK activation and enhancement of AJs associated with RhoGTPase (Birukov et al., 2002; Shikata et al., 2003). RhoGTPase activity is subject to regulation by GEFs, guanine nucleotide dissociation inhibitors (GDIs), or GTPase activating proteins (GAPs). RhoGDI-1 (RhoGDIα) represses RhoA activation and thus protects endothelial cell junctions from disassembly (Gorovoy et al., 2007). RhoA activity also inhibited through the activation of p190RhoGAP (Holinstat et al., 2006). FAK activates p190RhoGAP after thrombin stimulation to inhibit the increase in permeability facilitated by RhoA (Fig. 3) and reassembles disrupted endothelial cell junctions, (Holinstat et al., 2006).
In a barrier protective effect, cAMP directly activates Epac, a Rap1GEF, (de Rooij et al., 1998) enhancing VE-cadherin junctional integrity and actin reorganization to decrease endothelial permeability (Kooistra et al., 2005). Rap1 decreases basal endothelial permeability by enhancing distribution of both AJs and tight junctions (Cullere et al., 2005; Kooistra et al., 2005), and Rap1 antagonizes thrombin-induced increased permeability by inhibiting activation of RhoA (Cullere et al., 2005; Kooistra et al., 2005). Since activation of FAK and CDC42 (Fig. 3) also parallels the time course of re-formation of AJs and endothelial barrier protection following thrombin challenge (Kouklis et al., 2004; Schilling et al., 1992), it is likely that Rap-1, FAK, and CDC42 act in concert to down regulate RhoA activity and to promote the reformation of endothelial cell junctions.
The effect of FAK, barrier strengthening or weakening, varies depending on the nature of stimuli and the physical or chemical states of surrounding matrices. For example, inhibiting signaling through FAK or decreasing FAK expression can promote assembly or disassembly of cadherin-mediated cell-cell adhesions, respectively, depending on cell context and cadherin type (Avizienyte et al., 2002; Yano et al., 2004). In one case, loss of FAK or paxillin from HeLa cells leads to increased peripheral Rac1 activity and deregulation of N-cadherin-mediated cell–cell adhesion. In contrast, it is reported that integrin-induced activation of FAK can also result in activation of Rac1 via a p130Cas/CrkII/DOCK180 complex with DOCK180 acting as a Rac1 GEF (Cheresh et al., 1999; Hsia et al., 2003) (Fig. 3). This suggests that FAK can signal to Rac1 via different effectors and that these signaling pathways may have distinct, and probably localized, biological consequences.
From our studies using rat lung microvascular endothelial cells, it is evident that E-cadherin acts as a switch to either increase or decrease barrier strength through FAK signaling, which in turn regulates cadherin accumulation or clustering (Quadri and Bhattacharya, 2007). Moreover, H2O2 exposure induces an immediate loss of surface E-cadherin that then progressively increases with time. This response may be due to focal adhesions driving E-cadherin toward the surface. Thus, inhibition of FAK activation may block the signal for E-cadherin translocation to the surface, thereby compromising the integrity of the microvascular barrier. This suggests that in ECs, FAK activation is required for proper localization of E-cadherin to the cell periphery and for consequent strengthening of the endothelial cell barrier (Quadri and Bhattacharya, 2007). By contrast in other cell types, as in KM12C colon cancer cells, Src induced deregulation of E-cadherin requires αv/β1 integrin and Src-dependent tyrosine phosphorylation of FAK, suggesting Src–FAK has a negative influence on cadherin-mediated intercellular adhesion in motile phenotypes (Avizienyte et al., 2002). Since p120 is a Src substrate (Anastasiadis and Reynolds, 2000), cytoplasmic p120 binds the Vav2 exchange factor (Fig. 5) and regulates the activity of the small G-proteins Rac1, CDC42 and RhoA (Noren et al., 2001); could explain the cadherin-mediated intercellular deregulation, but E-cadherin-bound p120 prevents the internalization of E-cadherin (Fujita et al., 2002; Pece and Gutkind, 2002). This diversity of responses of FAK to Src could be due to some cell- or context-dependent signaling from FAK to RAC1 and on other upstream signaling inputs, such as Src activity. These findings suggest that FAK’s activation induced signaling positively regulates intercellular adhesion; however, the Src-induced signaling pathway negatively regulates cell–cell adhesion.
Fig 5. Cross talk between FAK and cadherin.

Since β-catenin does not contain FAK or paxillin binding sites, FAK signals to Rac, activated Rac1 and CDC42 sequester the GTPase-activating protein IQGAP1, and prevent its binding to β-catenin, thereby stabilizing cadherin-mediated cell adhesion. Otherwise in free form IQGAP1 binds to β-catenin, thereby displacing α-catenin from the cytoplasmic adhesion complex and disrupting the anchoring of the cytoplasmic complex to the cytoskeleton. Cytoplasmic p120-catenin (p120) activates the Rho-family GTPases Rac1 and CDC42 through the VAV2 (Rho-GEF), and represses Rho by an unknown mechanism.
Downstream of FAK, paxillin is also important for endothelial barrier regulation. Paxillin is a multidomain adapter, FA protein that functions as a molecular scaffold for protein recruitment to FAs and thereby facilitates protein networking and efficient signal transmission (Turner, 2000; Turner and Brown, 2001). Evidence shows that in mouse lungs and in HUVEC cells, loss of VE-cadherin junctional assembly in microvessels causes permeability and the reversal of the loss of barrier function after VE-cadherin junctions were reannealed in Ca2+ switch assay in the intact mouse lung (Gao et al., 2000). Reported findings has shown that human pulmonary endothelial cells undergo S1P-induced enhancement of VE-cadherin and association of β-catenin with paxillin (Fig. 3), which is critically dependent on Rac and CDC42 activities (Birukova et al., 2007) and is abolished by pharmacological or small interfering RNA (siRNA)-mediated inhibition of Rac and CDC42. It is also showed that enhancement of the VE-cadherin interaction with α-catenin and β-catenin was associated with the increased formation of FAK-β-catenin complexes. Depletion of β-catenin by siRNA resulted in loss of S1P-mediated, VE-cadherin association with FAK as well as paxillin rearrangement (Birukova et al., 2007) (Sun et al., 2009). Since β-catenin does not contain FAK or paxillin binding sites, possibly FAK and paxillin indirectly interacts with β-catenin and VE cadherin. Paxillin interacts with signaling proteins Crk, p60Src-kinase, FAK, (Fig. 3), and structural FA-associated proteins such as vinculin, actopaxin, and tubulin (Turner, 2000; Turner and Brown, 2001). Paxillin also binds to paxillin kinase linker (PKL/GIT2). GIT2 is a member of ADP-ribosylation factor GTPase activation factors (ARF GAP) family, and participate in Rac- and Rho-mediated signaling events at FAs (Mazaki et al., 2001; Turner, 2000; Turner and Brown, 2001). Enhancement of cadherin and association of β-catenin with paxillin is critically dependent on Rac and CDC42 activities (Fig. 3). These findings suggested that Rac and CDC42 GTPases have been implicated in the assembly of these complexes.
In addition to the effects of cadherin-mediated adhesion on Rho GTPase activity, cytoskeleton-associated signaling proteins also have an effect on the stability of the cytoplasmic adhesion complex. GTPase-activating protein, IQGAP1, a downstream effector of Rac1 and CDC42 (Fig. 5) is known to negatively regulate E-cadherin mediated cell-cell adhesion by interacting with β-catenin and displacing α-catenin from the cytoplasmic adhesion complex (Kuroda et al., 1998). Activated GTP-bound forms of Rac1 and CDC42 sequester IQGAP1 and prevent its binding to β-catenin, thereby stabilizing cadherin-mediated cell adhesion (Fukata et al., 1999). Indeed, IQGAP1 expression or function has been observed during tumor progression in gastric cancer cells, for example (Takemoto et al., 2001). However, it remains to be determined whether IQGAP1-mediated disruption of cadherin function is a general process in barrier disruption. The understanding that the linkage between the cadherin-catenin complex and the actin cytoskeleton (Conacci-Sorrell et al., 2002; Gumbiner, 1996) is important for barrier regulation comes from findings that barrier-deteriorating stimuli deplete both the cadherin-catenin complex (Rabiet et al., 1996) and actin (Ehringer et al., 1999) from the cell periphery, thereby raising the possibility that FAK and cadherin-mediated, cell-cell contacts communicate with each other.
SUMMARY
As described above, FAK and VE/E-cadherin are able to associate with actin and signal transduction pathways by interacting with molecules such as receptor tyrosine kinases, Rho-family GTPases and components of the WNT signaling pathway. The expression of FAK and cadherin affect not only the adhesive properties of a cell, but also the signal transduction status. Conversely, signaling pathways can modulate the function of FAK and cadherin, altering the interactions between cells and their environment. Although many different examples of signaling mediated by FAK and cadherin have been reported, the functional implications of signaling molecules between FAK and cadherin crosstalk will certainly be a key focus of future research.
FUTURE PERSPECTIVE
In this review, I have discussed topics that appear to be crucial for understanding the structure and function of adhesions, including the molecular complexity of these sites, their heterogeneity, and their dynamics. The molecular complexity of FAs is probably considerably greater since many of these components are still unknown and others can be post-translationally modified or proteolytically processed, undergoing conformational changes. To provide an insight into the local molecular architecture of adhesion sites, advanced ‘multi-dimensional microscopy’ is needed; this will allow the simultaneous localization of multiple components at a high spatial and temporal resolution. Imaging of molecular interactions using fluorescence resonance energy transfer will be needed for studies of these complex molecular interactions in situ. Such approaches may help uncover not only the molecular architecture of adhesion sites but also the ways in which they function in matrix rearrangement, adhesion-mediated signaling, and endothelial barrier regulation.
Highlights.
FAK and cadherin signaling coordinates appropriate changes at the cell-cell contact.
Cadherin organization and endothelial barrier function.
Cadherin-catenins-actin interactions are in question.
The role of small GTpases in the FAK and cadherin mediated cross talk.
The signaling molecules between FAK and cadherin cross talk will be a key focus of future.
ACKNOWLEDGEMENTS
I am grateful to Dr. Bhattacharya for helpful advice. Dr. Sunita Bhattacharya read the manuscript. I am thankful to Tara Guclu for critical reading of this manuscript and Feroze Hakim (8th grade) for drawing figures. Supported by NIH by HL 36024 (PI: Jahar Bhattacharya).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abercrombie M, Dunn GA. Adhesions of fibroblasts to substratum during contact inhibition observed by interference reflection microscopy. Exp Cell Res. 1975;92:57–62. doi: 10.1016/0014-4827(75)90636-9. [DOI] [PubMed] [Google Scholar]
- Abercrombie M, et al. The locomotion of fibroblasts in culture. IV. Electron microscopy of the leading lamella. Exp Cell Res. 1971;67:359–367. doi: 10.1016/0014-4827(71)90420-4. [DOI] [PubMed] [Google Scholar]
- Aberle H, et al. Assembly of the cadherin-catenin complex in vitro with recombinant proteins. J Cell Sci. 1994;107(Pt 12):3655–3663. doi: 10.1242/jcs.107.12.3655. [DOI] [PubMed] [Google Scholar]
- Al-Amoudi A, Frangakis AS. Structural studies on desmosomes. Biochem Soc Trans. 2008;36:181–187. doi: 10.1042/BST0360181. [DOI] [PubMed] [Google Scholar]
- Anastasiadis PZ, Reynolds AB. The p120 catenin family: complex roles in adhesion, signaling and cancer. J Cell Sci. 2000;113(Pt 8):1319–1334. doi: 10.1242/jcs.113.8.1319. [DOI] [PubMed] [Google Scholar]
- Andl CD, Rustgi AK. No one-way street: cross-talk between e-cadherin and receptor tyrosine kinase (RTK) signaling: a mechanism to regulate RTK activity. Cancer Biol Ther. 2005;4:28–31. doi: 10.4161/cbt.4.1.1431. [DOI] [PubMed] [Google Scholar]
- Avizienyte E, et al. Src-induced de-regulation of E-cadherin in colon cancer cells requires integrin signalling. Nat Cell Biol. 2002;4:632–638. doi: 10.1038/ncb829. [DOI] [PubMed] [Google Scholar]
- Barth AI, et al. Cadherins, catenins and APC protein: interplay between cytoskeletal complexes and signaling pathways. Curr Opin Cell Biol. 1997;9:683–690. doi: 10.1016/s0955-0674(97)80122-6. [DOI] [PubMed] [Google Scholar]
- Behrens J, et al. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol. 1993;120:757–766. doi: 10.1083/jcb.120.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311–320. doi: 10.1016/s0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- Birukov KG, et al. Shear stress-mediated cytoskeletal remodeling and cortactin translocation in pulmonary endothelial cells. Am J Respir Cell Mol Biol. 2002;26:453–464. doi: 10.1165/ajrcmb.26.4.4725. [DOI] [PubMed] [Google Scholar]
- Boggon TJ, et al. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 2002;296:1308–1313. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- Birukova AA, et al. Paxillin-beta-catenin interactions are involved in Rac/Cdc42-mediated endothelial barrier-protective response to oxidized phospholipids. Am J Physiol Lung Cell Mol Physiol. 2007;293:L199–L211. doi: 10.1152/ajplung.00020.2007. [DOI] [PubMed] [Google Scholar]
- Boggon TJ, et al. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 2002;296:1308–1313. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- Braga VM. Cell-cell adhesion and signalling. Curr Opin Cell Biol. 2002;14:546–556. doi: 10.1016/s0955-0674(02)00373-3. [DOI] [PubMed] [Google Scholar]
- Breier G, et al. Molecular cloning and expression of murine vascular endothelial-cadherin in early stage development of cardiovascular system. Blood. 1996;87:630–641. [PubMed] [Google Scholar]
- Broman MT, et al. Cdc42 regulates adherens junction stability and endothelial permeability by inducing alpha-catenin interaction with the vascular endothelial cadherin complex. Circ Res. 2006;98:73–80. doi: 10.1161/01.RES.0000198387.44395.e9. [DOI] [PubMed] [Google Scholar]
- Carbajal JM, et al. ROCK mediates thrombin's endothelial barrier dysfunction. Am J Physiol Cell Physiol. 2000;279:C195–C204. doi: 10.1152/ajpcell.2000.279.1.C195. [DOI] [PubMed] [Google Scholar]
- Chen SY, Chen HC. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol Cell Biol. 2006;26:5155–5167. doi: 10.1128/MCB.02186-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheresh DA, et al. Regulation of cell contraction and membrane ruffling by distinct signals in migratory cells. J Cell Biol. 1999;146:1107–1116. doi: 10.1083/jcb.146.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chishti AH, et al. The FERM domain: a unique module involved in the linkage of cytoplasmic proteins to the membrane. Trends Biochem Sci. 1998;23:281–282. doi: 10.1016/s0968-0004(98)01237-7. [DOI] [PubMed] [Google Scholar]
- Cines DB, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–3561. [PubMed] [Google Scholar]
- Conacci-Sorrell M, et al. The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest. 2002;109:987–991. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corada M, et al. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci U S A. 1999;96:9815–9820. doi: 10.1073/pnas.96.17.9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan CE, et al. Kruppel-like factor-4 transcriptionally regulates VE-cadherin expression and endothelial barrier function. Circ Res. 2010;107:959–966. doi: 10.1161/CIRCRESAHA.110.219592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullere X, et al. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood. 2005;105:1950–1955. doi: 10.1182/blood-2004-05-1987. [DOI] [PubMed] [Google Scholar]
- de Hoog CL, et al. RNA and RNA binding proteins participate in early stages of cell spreading through spreading initiation centers. Cell. 2004;117:649–662. doi: 10.1016/s0092-8674(04)00456-8. [DOI] [PubMed] [Google Scholar]
- de Rooij J, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- Dejana E, et al. Vascular endothelial (VE)-cadherin: only an intercellular glue? Exp Cell Res. 1999;252:13–19. doi: 10.1006/excr.1999.4601. [DOI] [PubMed] [Google Scholar]
- Drees F, et al. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell. 2005;123:903–915. doi: 10.1016/j.cell.2005.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- Ehringer WD, et al. Quantitative image analysis of F-actin in endothelial cells. Microcirculation. 1999;6:291–303. [PubMed] [Google Scholar]
- Eliceiri BP, et al. Src-mediated coupling of focal adhesion kinase to integrin alpha(v)beta5 in vascular endothelial growth factor signaling. J Cell Biol. 2002;157:149–160. doi: 10.1083/jcb.200109079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PM, et al. KLF4 interacts with beta-catenin/TCF4 and blocks p300/CBP recruitment by beta-catenin. Mol Cell Biol. 2010;30:372–381. doi: 10.1128/MCB.00063-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, et al. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol. 2002;4:222–231. doi: 10.1038/ncb758. [DOI] [PubMed] [Google Scholar]
- Fukata M, Kaibuchi K. Rho-family GTPases in cadherin-mediated cell-cell adhesion. Nat Rev Mol Cell Biol. 2001;2:887–897. doi: 10.1038/35103068. [DOI] [PubMed] [Google Scholar]
- Fukata M, et al. Cdc42 and Rac1 regulate the interaction of IQGAP1 with beta-catenin. J Biol Chem. 1999;274:26044–26050. doi: 10.1074/jbc.274.37.26044. [DOI] [PubMed] [Google Scholar]
- Gao X, et al. Reversibility of increased microvessel permeability in response to VE-cadherin disassembly. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1218–L1225. doi: 10.1152/ajplung.2000.279.6.L1218. [DOI] [PubMed] [Google Scholar]
- Garces CA, et al. Vascular endothelial growth factor receptor-3 and focal adhesion kinase bind and suppress apoptosis in breast cancer cells. Cancer Res. 2006;66:1446–1454. doi: 10.1158/0008-5472.CAN-05-1661. [DOI] [PubMed] [Google Scholar]
- Godzich M, et al. Activation of the stress protein response prevents the development of pulmonary edema by inhibiting VEGF cell signaling in a model of lung ischemia-reperfusion injury in rats. FASEB J. 2006;20:1519–1521. doi: 10.1096/fj.05-4708fje. [DOI] [PubMed] [Google Scholar]
- Gorovoy M, et al. RhoGDI-1 modulation of the activity of monomeric RhoGTPase RhoA regulates endothelial barrier function in mouse lungs. Circ Res. 2007;101:50–58. doi: 10.1161/CIRCRESAHA.106.145847. [DOI] [PubMed] [Google Scholar]
- Gottardi CJ, et al. E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J Cell Biol. 2001;153:1049–1060. doi: 10.1083/jcb.153.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM. Regulation of cadherin adhesive activity. J Cell Biol. 2000;148:399–404. doi: 10.1083/jcb.148.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- Halbleib JM, Nelson WJ. Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes Dev. 2006;20:3199–3214. doi: 10.1101/gad.1486806. [DOI] [PubMed] [Google Scholar]
- Hamaguchi M, et al. p60v-src causes tyrosine phosphorylation and inactivation of the N-cadherin-catenin cell adhesion system. EMBO J. 1993;12:307–314. doi: 10.1002/j.1460-2075.1993.tb05658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison OJ, et al. Two-step adhesive binding by classical cadherins. Nat Struct Mol Biol. 2010;17:348–357. doi: 10.1038/nsmb.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussinger D, et al. Proteolytic E-cadherin activation followed by solution NMR and X-ray crystallography. EMBO J. 2004;23:1699–1708. doi: 10.1038/sj.emboj.7600192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herwig MC, et al. Endothelial VE-cadherin expression in human lungs. Pathol Res Pract. 2008;204:725–730. doi: 10.1016/j.prp.2008.04.014. [DOI] [PubMed] [Google Scholar]
- Hildebrand JD, et al. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAK, to cellular focal adhesions. J Cell Biol. 1993;123:993–1005. doi: 10.1083/jcb.123.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinck L, et al. Dynamics of cadherin/catenin complex formation: novel protein interactions and pathways of complex assembly. J Cell Biol. 1994;125:1327–1340. doi: 10.1083/jcb.125.6.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Heuser JE. Quick-freeze, deep-etch visualization of the cytoskeleton beneath surface differentiations of intestinal epithelial cells. J Cell Biol. 1981;91:399–409. doi: 10.1083/jcb.91.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holinstat M, et al. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J Biol Chem. 2006;281:2296–2305. doi: 10.1074/jbc.M511248200. [DOI] [PubMed] [Google Scholar]
- Hsia DA, et al. Differential regulation of cell motility and invasion by FAK. J Cell Biol. 2003;160:753–767. doi: 10.1083/jcb.200212114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AH, Weis WI. The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell. 2001;105:391–402. doi: 10.1016/s0092-8674(01)00330-0. [DOI] [PubMed] [Google Scholar]
- Hulsken J, et al. E-cadherin and APC compete for the interaction with beta-catenin and the cytoskeleton. J Cell Biol. 1994;127:2061–2069. doi: 10.1083/jcb.127.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireton RC, et al. A novel role for p120 catenin in E-cadherin function. J Cell Biol. 2002;159:465–476. doi: 10.1083/jcb.200205115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama N, et al. Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell. 2010;141:117–128. doi: 10.1016/j.cell.2010.01.017. [DOI] [PubMed] [Google Scholar]
- Izzard CS, Lochner LR. Cell-to-substrate contacts in living fibroblasts: an interference reflexion study with an evaluation of the technique. J Cell Sci. 1976;21:129–159. doi: 10.1242/jcs.21.1.129. [DOI] [PubMed] [Google Scholar]
- Izzard CS, Lochner LR. Formation of cell-to-substrate contacts during fibroblast motility: an interference-reflexion study. J Cell Sci. 1980;42:81–116. doi: 10.1242/jcs.42.1.81. [DOI] [PubMed] [Google Scholar]
- Jamora C, Fuchs E. Intercellular adhesion, signalling and the cytoskeleton. Nat Cell Biol. 2002;4:E101–E108. doi: 10.1038/ncb0402-e101. [DOI] [PubMed] [Google Scholar]
- Kitagawa M, et al. Mutation analysis of cadherin-4 reveals amino acid residues of EC1 important for the structure and function. Biochem Biophys Res Commun. 2000;271:358–363. doi: 10.1006/bbrc.2000.2636. [DOI] [PubMed] [Google Scholar]
- Kooistra MR, et al. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005;579:4966–4972. doi: 10.1016/j.febslet.2005.07.080. [DOI] [PubMed] [Google Scholar]
- Kouklis P, et al. Cdc42 regulates the restoration of endothelial barrier function. Circ Res. 2004;94:159–166. doi: 10.1161/01.RES.0000110418.38500.31. [DOI] [PubMed] [Google Scholar]
- Kovacs EM, et al. E-cadherin homophilic ligation directly signals through Rac and phosphatidylinositol 3-kinase to regulate adhesive contacts. J Biol Chem. 2002;277:6708–6718. doi: 10.1074/jbc.M109640200. [DOI] [PubMed] [Google Scholar]
- Kuroda S, et al. Role of IQGAP1, a target of the small GTPases Cdc42 and Rac1, in regulation of E-cadherin-mediated cell-cell adhesion. Science. 1998;281:832–835. doi: 10.1126/science.281.5378.832. [DOI] [PubMed] [Google Scholar]
- Lahlou H, et al. Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumor progression. Proc Natl Acad Sci U S A. 2007;104:20302–20307. doi: 10.1073/pnas.0710091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani MG, et al. The molecular organization of endothelial cell to cell junctions: differential association of plakoglobin, beta-catenin, and alpha-catenin with vascular endothelial cadherin (VE-cadherin) J Cell Biol. 1995;129:203–217. doi: 10.1083/jcb.129.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani MG, et al. VE-cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. Mol Biol Cell. 2002;13:1175–1189. doi: 10.1091/mbc.01-07-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw CW, et al. Identification and cloning of two species of cadherins in bovine endothelial cells. Embo J. 1990;9:2701–2708. doi: 10.1002/j.1460-2075.1990.tb07456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ST, et al. FERM control of FAK function: implications for cancer therapy. Cell Cycle. 2008;7:2306–2314. doi: 10.4161/cc.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, et al. Involvement of p120 carboxy-terminal domain in cadherin trafficking. Cell Struct Funct. 2007;32:127–137. doi: 10.1247/csf.07023. [DOI] [PubMed] [Google Scholar]
- Lobo M, Zachary I. Nuclear localization and apoptotic regulation of an amino-terminal domain focal adhesion kinase fragment in endothelial cells. Biochem Biophys Res Commun. 2000;276:1068–1074. doi: 10.1006/bbrc.2000.3547. [DOI] [PubMed] [Google Scholar]
- Luo M, Guan JL. Focal adhesion kinase: a prominent determinant in breast cancer initiation, progression and metastasis. Cancer Lett. 2010;289:127–139. doi: 10.1016/j.canlet.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May C, et al. Identification of a transiently exposed VE-cadherin epitope that allows for specific targeting of an antibody to the tumor neovasculature. Blood. 2005;105:4337–4344. doi: 10.1182/blood-2005-01-0010. [DOI] [PubMed] [Google Scholar]
- Mazaki Y, et al. An ADP-ribosylation factor GTPase-activating protein Git2-short/KIAA0148 is involved in subcellular localization of paxillin and actin cytoskeletal organization. Mol Biol Cell. 2001;12:645–662. doi: 10.1091/mbc.12.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan RW, et al. E-cadherin adhesion activates c-Src signaling at cell-cell contacts. Mol Biol Cell. 2007;18:3214–3223. doi: 10.1091/mbc.E06-12-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean GW, et al. Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev. 2004;18:2998–3003. doi: 10.1101/gad.316304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, et al. Modulatory role of focal adhesion kinase in regulating human pulmonary arterial endothelial barrier function. J Physiol. 2002;539:779–789. doi: 10.1113/jphysiol.2001.013289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaguchi K. Ultrastructure of the zonula adherens revealed by rapid-freeze deep-etching. J Struct Biol. 2000;132:169–178. doi: 10.1006/jsbi.2000.4244. [DOI] [PubMed] [Google Scholar]
- Morin PJ, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Nagar B, et al. Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature. 1996;380:360–364. doi: 10.1038/380360a0. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Takeichi M. Remodeling of the adherens junctions during morphogenesis. Curr Top Dev Biol. 2009;89:33–54. doi: 10.1016/S0070-2153(09)89002-9. [DOI] [PubMed] [Google Scholar]
- Noren NK, et al. Cadherin engagement inhibits RhoA via p190RhoGAP. J Biol Chem. 2003;278:13615–13618. doi: 10.1074/jbc.C200657200. [DOI] [PubMed] [Google Scholar]
- Noren NK, et al. Cadherin engagement regulates Rho family GTPases. J Biol Chem. 2001;276:33305–33308. doi: 10.1074/jbc.C100306200. [DOI] [PubMed] [Google Scholar]
- Nose A, et al. Localization of specificity determining sites in cadherin cell adhesion molecules. Cell. 1990;61:147–155. doi: 10.1016/0092-8674(90)90222-z. [DOI] [PubMed] [Google Scholar]
- Ofori-Acquah SF, et al. Heterogeneity of barrier function in the lung reflects diversity in endothelial cell junctions. Microvasc Res. 2008;75:391–402. doi: 10.1016/j.mvr.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsulic S, et al. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J Cell Sci. 1999;112(Pt 8):1237–1245. doi: 10.1242/jcs.112.8.1237. [DOI] [PubMed] [Google Scholar]
- Palacios F, et al. Lysosomal targeting of E-cadherin: a unique mechanism for the down-regulation of cell-cell adhesion during epithelial to mesenchymal transitions. Mol Cell Biol. 2005;25:389–402. doi: 10.1128/MCB.25.1.389-402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papkoff J. Regulation of complexed and free catenin pools by distinct mechanisms. Differential effects of Wnt-1 and v-Src. J Biol Chem. 1997;272:4536–4543. [PubMed] [Google Scholar]
- Parisini E, et al. The crystal structure of human E-cadherin domains 1 and 2, and comparison with other cadherins in the context of adhesion mechanism. J Mol Biol. 2007;373:401–411. doi: 10.1016/j.jmb.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JC, et al. Hydraulic conductance of pulmonary microvascular and macrovascular endothelial cell monolayers. Am J Physiol Lung Cell Mol Physiol. 2006;291:L30–L37. doi: 10.1152/ajplung.00317.2005. [DOI] [PubMed] [Google Scholar]
- Parthasarathi K, Q SK. Cadherin and connexin in Pulmonary EndothelialF Function. In: N FV, Rounds S, editors. The Pulmonary Endothelium Function in Health and Disease. Chichester, UK: John Wiley & Sons, Ltd.; 2009. [Google Scholar]
- Pece S, Gutkind JS. Signaling from E-cadherins to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell-cell contact formation. J Biol Chem. 2000;275:41227–41233. doi: 10.1074/jbc.M006578200. [DOI] [PubMed] [Google Scholar]
- Pece S, Gutkind JS. E-cadherin and Hakai: signalling, remodeling or destruction? Nat Cell Biol. 2002;4:E72–E74. doi: 10.1038/ncb0402-e72. [DOI] [PubMed] [Google Scholar]
- Pertz O, et al. A new crystal structure, Ca2+ dependence and mutational analysis reveal molecular details of E-cadherin homoassociation. EMBO J. 1999;18:1738–1747. doi: 10.1093/emboj/18.7.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokutta S, Weis WI. Structure of the dimerization and beta-catenin-binding region of alpha-catenin. Mol Cell. 2000;5:533–543. doi: 10.1016/s1097-2765(00)80447-5. [DOI] [PubMed] [Google Scholar]
- Posy S, et al. Sequence and structural determinants of strand swapping in cadherin domains: do all cadherins bind through the same adhesive interface? J Mol Biol. 2008;378:954–968. doi: 10.1016/j.jmb.2008.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri SK, et al. Endothelial barrier strengthening by activation of focal adhesion kinase. J Biol Chem. 2003;278:13342–13349. doi: 10.1074/jbc.M209922200. [DOI] [PubMed] [Google Scholar]
- Quadri SK, Bhattacharya J. Resealing of endothelial junctions by focal adhesion kinase. Am J Physiol Lung Cell Mol Physiol. 2007;292:L334–L342. doi: 10.1152/ajplung.00228.2006. [DOI] [PubMed] [Google Scholar]
- Rabiet MJ, et al. Thrombin-induced increase in endothelial permeability is associated with changes in cell-to-cell junction organization. Arterioscler Thromb Vasc Biol. 1996;16:488–496. doi: 10.1161/01.atv.16.3.488. [DOI] [PubMed] [Google Scholar]
- Reynolds AB, Carnahan RH. Regulation of cadherin stability and turnover by p120ctn: implications in disease and cancer. Semin Cell Dev Biol. 2004;15:657–663. doi: 10.1016/j.semcdb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Safdar Z, et al. Hyperosmolarity enhances the lung capillary barrier. J Clin Invest. 2003;112:1541–1549. doi: 10.1172/JCI18370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon D, et al. Extrajunctional distribution of N-cadherin in cultured human endothelial cells. J Cell Sci. 1992;102(Pt 1):7–17. doi: 10.1242/jcs.102.1.7. [DOI] [PubMed] [Google Scholar]
- Sander EE, et al. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J Cell Biol. 1998;143:1385–1398. doi: 10.1083/jcb.143.5.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, et al. Autonomous expression of a noncatalytic domain of the focal adhesion-associated protein tyrosine kinase pp125FAK. Mol Cell Biol. 1993;13:785–791. doi: 10.1128/mcb.13.2.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling WP, et al. Depletion of the inositol 1,4,5-trisphosphate-sensitive intracellular Ca2+ store in vascular endothelial cells activates the agonist-sensitive Ca(2+)-influx pathway. Biochem J. 1992;284(Pt 2):521–530. doi: 10.1042/bj2840521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrels B, et al. A complex between FAK, RACK1, and PDE4D5 controls spreading initiation and cancer cell polarity. Curr Biol. 2010;20:1086–1092. doi: 10.1016/j.cub.2010.04.042. [DOI] [PubMed] [Google Scholar]
- Serrels B, et al. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat Cell Biol. 2007;9:1046–1056. doi: 10.1038/ncb1626. [DOI] [PubMed] [Google Scholar]
- Shan WS, et al. The adhesive binding site of cadherins revisited. Biophys Chem. 1999;82:157–163. doi: 10.1016/s0301-4622(99)00115-5. [DOI] [PubMed] [Google Scholar]
- Shapiro L, et al. Structural basis of cell-cell adhesion by cadherins. Nature. 1995;374:327–337. doi: 10.1038/374327a0. [DOI] [PubMed] [Google Scholar]
- Shikata Y, et al. S1P induces FA remodeling in human pulmonary endothelial cells: role of Rac, GIT1, FAK, and paxillin. J Appl Physiol. 2003;94:1193–1203. doi: 10.1152/japplphysiol.00690.2002. [DOI] [PubMed] [Google Scholar]
- Sieg DJ, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- Singleton PA, et al. Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J. 2005;19:1646–1656. doi: 10.1096/fj.05-3928com. [DOI] [PubMed] [Google Scholar]
- Sovova V, et al. Transactivation of E-cadherin is not involved in the activity of EGF receptor in colorectal carcinoma cells. Int J Oncol. 2004;25:1459–1464. [PubMed] [Google Scholar]
- Stewart A, et al. The focal adhesion kinase amino-terminal domain localises to nuclei and intercellular junctions in HEK 293 and MDCK cells independently of tyrosine 397 and the carboxy-terminal domain. Biochem Biophys Res Commun. 2002;299:62–73. doi: 10.1016/s0006-291x(02)02547-0. [DOI] [PubMed] [Google Scholar]
- Stockinger A, et al. E-cadherin regulates cell growth by modulating proliferation-dependent beta-catenin transcriptional activity. J Cell Biol. 2001;154:1185–1196. doi: 10.1083/jcb.200104036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, et al. Enhanced interaction between focal adhesion and adherens junction proteins: involvement in sphingosine 1-phosphate-induced endothelial barrier enhancement. Microvasc Res. 2009;77:304–313. doi: 10.1016/j.mvr.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunita Bhattacharya SQ, Bhattacharya Jahar. Endothelial-matrix interactions in the lung. In: Patterson CE, editor. Perspectives on Lung Endothelial Barrier Function. Vol. 35. Elsevier B.V; 2005. pp. 237–250. [Google Scholar]
- Takahashi K, Suzuki K. Density-dependent inhibition of growth involves prevention of EGF receptor activation by E-cadherin-mediated cell-cell adhesion. Exp Cell Res. 1996;226:214–222. doi: 10.1006/excr.1996.0221. [DOI] [PubMed] [Google Scholar]
- Takemoto H, et al. Localization of IQGAP1 is inversely correlated with intercellular adhesion mediated by e-cadherin in gastric cancers. Int J Cancer. 2001;91:783–788. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1121>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Tamura K, et al. Structure-function analysis of cell adhesion by neural (N-) cadherin. Neuron. 1998;20:1153–1163. doi: 10.1016/s0896-6273(00)80496-1. [DOI] [PubMed] [Google Scholar]
- Taylor JM, et al. Selective expression of an endogenous inhibitor of FAK regulates proliferation and migration of vascular smooth muscle cells. Mol Cell Biol. 2001;21:1565–1572. doi: 10.1128/MCB.21.5.1565-1572.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CE. Paxillin and focal adhesion signalling. Nat Cell Biol. 2000;2:E231–E236. doi: 10.1038/35046659. [DOI] [PubMed] [Google Scholar]
- Turner CE, Brown MC. Cell motility: ARNOand ARF6 at the cutting edge. Curr Biol. 2001;11:R875–R877. doi: 10.1016/s0960-9822(01)00521-8. [DOI] [PubMed] [Google Scholar]
- Uehata M, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- van Nimwegen MJ, van de Water B. Focal adhesion kinase: a potential target in cancer therapy. Biochem Pharmacol. 2007;73:597–609. doi: 10.1016/j.bcp.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Volk T, Kox WJ. Endothelium function in sepsis. Inflamm Res. 2000;49:185–198. doi: 10.1007/s000110050579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder SJ. Structural insights into actin-binding, branching and bundling proteins. Curr Opin Cell Biol. 2003;15:14–22. doi: 10.1016/s0955-0674(02)00002-9. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard B, et al. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci. 2001;114:1343–1355. doi: 10.1242/jcs.114.7.1343. [DOI] [PubMed] [Google Scholar]
- Wu X, et al. Focal adhesion kinase regulation of N-WASP subcellular localization and function. J Biol Chem. 2004;279:9565–9576. doi: 10.1074/jbc.M310739200. [DOI] [PubMed] [Google Scholar]
- Xiao K, et al. p120-Catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol Biol Cell. 2005;16:5141–5151. doi: 10.1091/mbc.E05-05-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, Nelson WJ. Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell-cell adhesion. J Cell Biol. 2007;178:517–527. doi: 10.1083/jcb.200701058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, et al. Deconstructing the cadherin-catenin-actin complex. Cell. 2005;123:889–901. doi: 10.1016/j.cell.2005.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H, et al. Roles played by a subset of integrin signaling molecules in cadherin-based cell-cell adhesion. J Cell Biol. 2004;166:283–295. doi: 10.1083/jcb.200312013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap AS, Kovacs EM. Direct cadherin-activated cell signaling: a view from the plasma membrane. J Cell Biol. 2003;160:11–16. doi: 10.1083/jcb.200208156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachary I. VEGF signalling: integration and multi-tasking in endothelial cell biology. Biochem Soc Trans. 2003;31:1171–1177. doi: 10.1042/bst0311171. [DOI] [PubMed] [Google Scholar]
- Zantek ND, et al. E-cadherin regulates the function of the EphA2 receptor tyrosine kinase. Cell Growth Differ. 1999;10:629–638. [PubMed] [Google Scholar]
- Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- Zigmond S. Formin' adherens junctions. Nat Cell Biol. 2004;6:12–14. doi: 10.1038/ncb0104-12. [DOI] [PubMed] [Google Scholar]