

Abstract
The major cellular components of tumor microenvironment, referred to as the cancer stroma, are composed of cancer-associated fibroblasts that support tumor epithelial growth, invasion and therapeutic resistance. Thus when we speak of developing therapies that address tumor heterogeneity it is not only a matter of different mutations within the tumor epithelia. While individual mutations in the stromal compartment are controversial, the heterogeneity in fibroblastic population in a single tumor is not up for debate. Cooperative interaction among heterotypic fibroblasts and tumor cells contribute to cancer progression. Therefore to tackle solid tumors, we need to understand its complex microenvironment. Here we review some seminal developments in the field of tumor microenvironment, mainly focusing on cancer-associated fibroblast.
Keywords: carcinoma-associated fibroblast, fibroblast, microenvironment, paracrine signaling, stroma
Stromal Contribution to Cancer
A bilateral collaborative effort of normal epithelial cells and components of the stromal compartment (Fig. 1) maintain the integrity of a normal physiological system. A continuous cross talk between the stroma and epithelia dictates tissue differentiation.1,2 In the event of pathological conditions of wounding events, including cancer, the stroma takes on the role for repair and cross-compartmental paracrine signaling alters epithelial proliferation and differentiation.3,4 This review will address some seminal research done on tumor-associated stroma, mainly focused on cancer-associated fibroblasts (CAF) signaling.
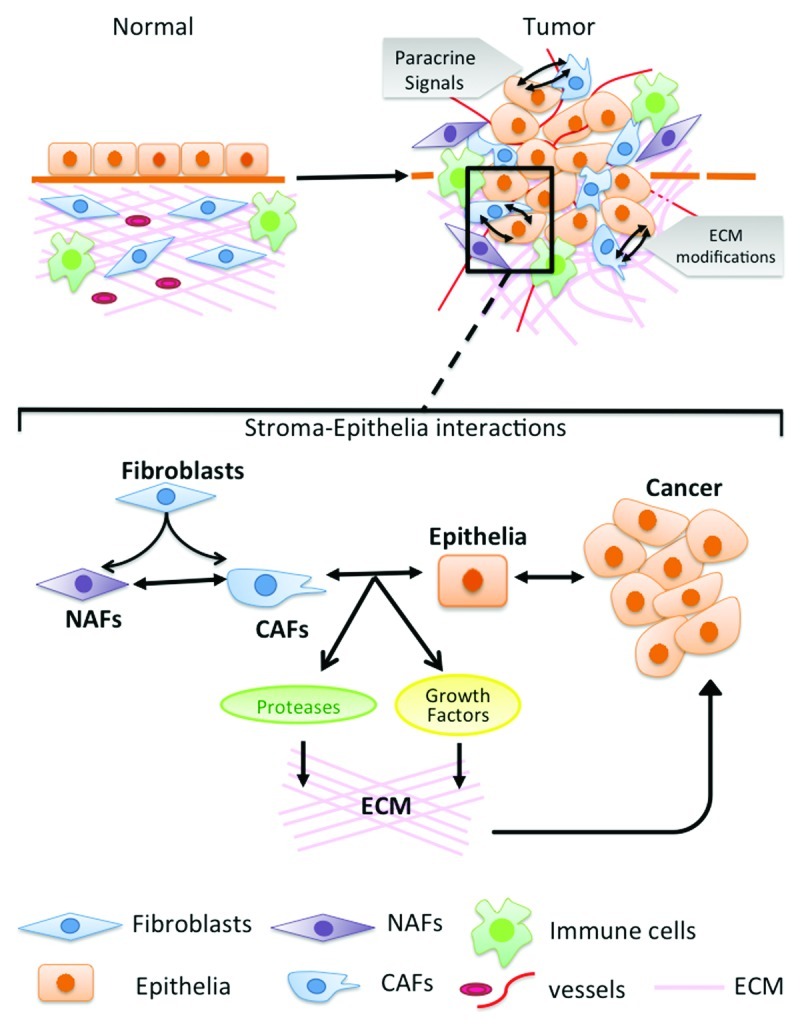
Figure 1. Stroma-epithelia interactions. Normal microenvironment contains fibroblasts, immune cells, blood vessels and extracellular matrix associated with the epithelial cells. During cancer progression the microenvironment is altered in multiple ways including increased number of fibroblasts, blood vessels, immune cells and ECM components. There are modifications of normal fibroblasts (NAFs) to cancer fibroblasts (CAFs) and remodeling of ECM by degradation and increased stiffness. The cross talk between normal fibroblasts, cancer fibroblasts and the epithelial cells leads to cancer progression.
Stephen Paget pioneered the concept of tumor microenvironment in 1889 in the form of the seed and soil hypothesis. Paget analyzed more than 700 postmortem data from women that died of breast cancer and found the distribution of metastasis to organs to be nonrandom.5 He suggested that the site of metastasis preferred by the cancer cell (seed) is based on the microenvironment (soil) of that organ. Hart and Fiddler re-introduced the seed/soil hypothesis in the 1980s through studies done on mice.6 They grafted lung, ovary and kidney tissues under the skin or muscle of mice. B16 melanoma cells were then injected to these mice. They observed that tumor growth developed preferentially in the grafted lung and ovary tissues but not on the kidney grafts. The study suggested that the specificity of the organ for metastasis was due to the microenvironment (soil) of the organ that was preferred by the cancer cell (seed) for colonization. The preference of the location for metastasis by cancer cell makes the location a special “niche” for the tumor cells. Later on studies have shown that before the colonization of the cancer cells this niche undergoes a series of preparation for it to become the future site of metastasis, the phenomenon referred to as premetastatic niche.7 Therefore the early observations support current findings of microenvironmental changes characterized at the primary cancer site, as well as directly indicated the importance of the pre-metastatic niche. Another early concept, that a benign stromal tissue from embryonic mammary mesenchyme has the ability to revert the cancer phenotype of tumor cells by inducing it to a differentiated phenotype, was revealed through studies by Decosse and coworkers.8 Along the same lines, a seminal paper showed that the microenvironment of mouse blastocyst suppressed tumorigencity.9 Dolberg and Bissell demonstrated that components of the extracellular matrix alleviated the transforming ability of Rous sarcoma virus when injected to chick embryos.10 More recently, the Bissell lab showed that the microenvironment induction of a benign phenotype of cancer does not involve reversion of chromosomal alterations pre-existing in the epithelia.11 Together, these studies support that the stroma can be a driver of epithelial phenotype. CAF can induce a cancerous phenotype (discussed later in detail), just as stroma from benign tissue can restore a benign phenotype in neoplastic epithelia. Therefore the stroma could be a potential therapeutic target of carcinogenesis. Yet, therapeutic targeting of the microenvironment will only be possible as we learn the mechanisms involved in stromal changes that dictate epithelial fate.
Stromal Fibroblastic Changes Can Contribute to Cancer
Genetic changes in epithelial tumor cells remain an important driver of cancer; however, cells in the tumor stroma including fibroblasts, immune cells and endothelial cells are active partners in tumor growth and metastasis. The “reactive” nature of cancer associated stroma differ in structure and morphology compared with stroma associated with benign tissues, and provide a promoting environment for cancer progression in contrast to the suppressive role of the benign counterpart.12 Fibroblasts are the predominant cells in stroma, especially in case of breast, prostate and pancreatic carcinoma.13 CAFs are responsible for production of paracrine growth factors, proteolytic enzymes and ECM components.13 Early on, studies examining the stromal effects on tumor progression were done by Leland Chung’s group using grafts containing cancer epithelial cells with normal or modified fibroblast.14 The observation that the combination of cancer epithelia with fibroblasts developed larger tumors than cancer epithelia alone constituted the first study to suggest stromal fibroblastic cells can promote tumorigenesis. On the surface this may seem contrary to the statement that benign fibroblasts suppress tumorigenesis. However, there is clearly a crosstalk between the two compartments whereby there seems to be a grade of the cancer epithelia that cannot be reverted to a benign phenotype. The stromal fibroblasts also respond to the neoplastic epithelia by expressing elevated levels of collagen, growth factors and desmoplastic factors. The fibroblastic stroma of the mammary gland and smooth muscle stromal of the prostate differentiate to a similar myofibroblastic differentiation. This stromal differentiation response is akin to the development of the protypical CAF phenotype. Yet, just as there are grades of neoplastic differentiation of the epithelia, all CAF are not created equal. We have shown that the specific CAF phenotype of the loss of TGFβ receptor type II receptor expression, when recapitulated in transgenic mice cannot only promote tumor expansion of neoplastic epithelia, but also initiate neoplastic progression from benign epithelia.15 The epithelia and stromal fibroblastic cells are said to co-evolve through a mechanism that being revealed through current active research.
Initial profiling of stromal compartments of various tissues indicated tissue-specific differences.16 However, the specific differences in gene expression in cancer-associated fibroblasts (CAF) and normal tissue associated fibroblasts (NAF) are much more complicated due to the definition of CAF. Loose definitions of CAF, include cells that surround cancer epithelia. However, stricter definitions of CAF involve those fibroblastic cells that have the ability to promote tumorigenesis.13 This latter definition was a result of careful studies done in in vivo tissue recombination systems and in vitro co-culture systems by Gerald Cunha’s group where NAF had little effect on the growth of a non-tumorigenic prostatic cell line. But the tissue recombination of the specific primary CAF cells caused explosive tumorigenic transformation and growth. Authors reported that a genetically initiated but non-tumorigenic human prostate cancer cells could undergo a malignant transformation due to its exposure to CAFs. Thus fibroblasts from tumors had acquired the ability to promote cancer progression.17,18 The Bhowmick lab demonstrated that human prostate cancer associated fibroblastic cells lose expression of the transforming growth factor β receptor type II (Tgfbr2) gene expression in close to 70% of the cases irrespective of Gleason grade.15 The clinical observation supported a specific fibroblast signaling pathways that could initiate cancer, as demonstrated by a conditional knockout of Tgfbr2 in stromal fibroblastic cells, in transgenic mice (Tgfbr2fspKO).19 Inactivation of Tgfbr2 in a population of stromal fibroblasts resulted in prostatic neoplasia and invasive squamous cell carcinoma in the forestomach with 100% penetrance. The ablation TGFβ fibroblastic responsiveness led to increased HGF expression, which in turn induced c-Met activation in adjacent epithelia.19 Other paracrine factors overexpressed in the fibroblasts due to conditional ablation of Tgfbr2 include macrophage stimulating 1 (Mst1), multiple Wnt ligands and TGFα. The Hayward group demonstrated the upregulation of cyclin D1 in prostatic fibroblasts could promote tumorigenesis.20 Not surprisingly all cells that circumscribe a tumor do not have the ability to promote tumor growth. However, microarray analysis of prostatic CAF, by the stricter definition, suggested the differential expression of genes often attributed to changes observed in cancer epithelia.20 The endogenous mechanism for gene regulation in stromal cells is less clear.
The phenotypic heterogeneity of the stromal fibroblasts has a biologic role on tumor initiation and progression. Paracrine factors regulate cell morphology, survival and death of cancer cells.13 Several studies have shown the role of factors secreted by stroma in tumor growth like, in human breast tumors, CAFs secrete elevated levels of stromal derived factor (SDF1, CXCL12) that binds to CXCR4 receptor on the breast cancer cells and promote growth and invasion.21 Tissue recombination experiments of fibroblasts with tumor cells in mouse xenograft models, demonstrate an increase in tumor size by addition of stromal cells.15,17,20,22,23 Although less studied, the role of stromal heterogeneity is acknowledged since it is found that human prostate cancer has heterogeneous stroma in terms of loss of Tgfbr2 in the stromal compartment.15 To dissect the role of stromal heterogeneity in disease progression, different stromal subpopulations were mixed with epithelial cells. Along these lines, Franco et al. demonstrated that the expression of dominant-negative Tgfbr2 in 50% of NAF stromal cell population caused malignant transformation of BPH1 cells (nontumorigenic human prostate epithelial cells) associated with further myofibroblastic differentiation of the stroma, similar to that found in CAF.24 The mixture of NAF cells with downregulated TGF-β responsiveness were found to upregulate SDF1 and members of the fibroblast growth factor and bone morphogenic families.24 In a very different approach to investigate the role of stromal heterogeneity in cancer, Kiskowski et al. used a combination of experimental data and mathematical modeling.25 Here the prostatic fibroblasts from Tgfbr2fspKO mice were grown in culture to generate a 100% Tgfbr2 knockout fibroblastic population (Tgfbr2-KO).25 These Tgfbr2-KO fibroblasts were mixed in different ratios with Tgfbr2floxE2/floxE2 (behave like wild-type) and found that the mixture with 50% of each, Tgfbr2 responsive and nonresponsive stroma, when associated with wild-type mouse prostatic epithelia, resulted in neoplastic development. However, the recombination with 100% Tgfbr2-KO fibroblasts only supported a pre-neoplastic differentiation of the epithelia. Using a computational model the mechanism of tumor progression that involved two independent steps mediated by the two distinct stromal cell populations was elucidated. What emerged from this study, was that the TGFβ responsive and TGFβ nonresponsive fibroblastic populations were interacting through the IL-1β production to cooperatively upregulate SDF1 production. The data suggested that the second step in the neoplastic initiation, then cancer progression, paradigm was dependent on this SDF1 mediated epithelial differentiation. Thus, the long recognized stromal heterogeneity has a role in cancer progression.25
Tumor stromal co-evolution involves selective genetic changes. Conceptually, random somatic mutations could occasionally generate a stromal cell with a selective advantage such that it is able to clonally expand and replace those cells without the mutation (occurring in a cell autonomous fashion). One of the initial studies in this direction showed that genetic events like loss of heterozygosity (loss) could occur in breast tumor stroma. This study, using 11 breast samples of ductal carcinoma in situ with five cases of infiltrating ductal carcinoma (IDC), showed LOH at several loci exclusively in stroma along with cases with LOH common to both epithelia and stroma. Further, the LOH frequency in stroma of invasive cancer was higher as compared with DCIS.26 This study was supported by the observations by other groups in colon and breast cancer samples.27,28 Studies by Charis Eng and colleagues on a larger scale supported the earlier results of LOH in the stroma of breast cancer samples.29-31 Terry Van Dyke’s group observed independent Trp53 LOH in the fibroblastic cells, in a prostate epithelial tumorigenesis mouse model.32 PTEN loss in stromal fibroblast resulted in mammary epithelial tumor initiation and progression and malignant transformation.33 Michael Ostrowski and colleagues describe the loss in PTEN expression in breast CAF associated with poorer prognosis of the disease.33,34 Similarly, a transgenic conditional knockout of Tgfbr2 in breast cancer fibroblasts caused increased proliferation, angiogenesis, and invasion of adjacent cancer cells in vivo.35 The co-evolution hypothesis necessarily invokes a model in which a mutation in the stromal cell provides no direct selective advantage to itself. Whereas proponents of this model would argue that the selective advantage derives from the symbiotic carcinoma cells, requiring only mutant fibroblast to respond to the signals emanating from the carcinoma cells in order for that fibroblast clone to dominate the stromal population. This acquired trait to outgrow the neighboring fibroblasts and generate a dominant clonal population of CAFs would suggest the simultaneous generation of two symbiotic malignancies: (1) generation of carcinoma and (2) generation of sarcomas. If a mutation resulted in clonal expansion of the CAF, the incidence of sarcomas would be more prevalent. This would mean that the stroma would run into “crisis” before populating millimeters of the tissue.36 Cellular crisis could involve aberrant chromosomal content, like end-to-end fusion and unstable dicentric chromosome, associated with an apoptotic check-point. However, the idea of genetic-stromal co-evolution is not universally accepted based on the lack of identification of such alterations in CAFs in multiple studies.37-39 Kornelia Polyak and many others feel the controversy surrounding the presence of somatic mutations in fibroblastic cells may be the result of technical methods of analysis and not necessarily the result of true genetic changes in the fibroblasts.37-39 We found that the loss of TGFβ responsiveness in the prostatic fibroblasts to be associated with promoter methylation changes in growth factor regulation.25 Such epigenetic changes could have a wider effect on other pathways in CAF. Regardless of the mechanism, the expression profiles of CAF are clearly different from that of NAF.
Emerging Concepts in the Tumor Microenvironment
The mechanism for tumor metastatic tropism, tendency to metastasize to specific tissues, is yet to be clearly defined. Studies have identified some sets of genes that can predict metastasis to the lung, bone or brain in certain breast cancer cells.40-43 For example, in case of breast cancer cells with increased expression of genes like inhibitor of differentiation 1 (Id1) and MMP1 had higher propensity for lung metastasis.43 The same group isolated cells that metastasize to the brain from breast cancer patients and did gene expression analysis. They found that cyclooxygenase COX2, HBEGF and α2,6sialyltransferase (ST6GALNAC5) as mediator of tumor cell passage across the blood-brain barrier.41 Thus, the expression profile of the cancer cells can help guide cells to a specific organ or site. However, the other critical half of metastatic tropism is the microenvironment of the metastatic site. In an elegant study, David Lyden’s group demonstrated that essential cellular events leading to the formation of the pre-metastatic niche to be important.7 They showed that bone marrow derived hematopoietic progenitor cells preferentially localize to future sites of metastasis and precondition them for the arrival and growth of circulating tumor cells.7 These bone marrow derived cells express VEGFR1 and other hematopoietic markers like, CD34, CD11b, c-Kit and Sca-1. Further, the VEGFR1+ hematopoietic progenitor cells preferentially localize to areas of elevated fibronectin. Since VEGF antagonists have not proven to effectively prevent metastasis in patients, such as those with renal cancer, it is likely other mechanisms of niche development exist and must be pursued. Along the same lines, another group has demonstrated that factors like VEGF-A, TNFα and TGFβ from the primary tumor induce expression of chemo-attractants S100A8 and S100A9 that help recruit CD11b positive myeloid cells to pre-metastatic niche.44 These researchers further show that these chemo-attractants led to induction of serum amyloid 3 in the premetastatic lung that facilitated metastasis.45 More recently, lysyl oxidase (LOX) has been implicated in formation of pre-metastatic niche. In breast cancer, LOX co-localizes with fibronectin in the pre-metastatic niche (lung) where it helps in collagen IV crosslinking at the basement membrane that in turn helps in CD11b+ myeloid cell recruitment. The CD11b+ cells help in collagen IV cleavage by producing MMP-2, which led to enhanced invasion and recruitment of BMDCs, all contributing to attracting the metastasizing cell.46
Generally speaking, the concept of the niche, whether it be for cancer stem cells, pre-metastatic, or the niche in which the primary or secondary tumor resides is at the fore front of current cancer therapeutic and preventative medicine. As we get better at exploiting the weaknesses of the cancer epithelia, the development of therapeutic resistance will emerge from the adaptations of the microenvironment. The tumor microenvironment has been shown to be capable of inducing cancer, as in the Tgfbr2fspKO transgenic model, involving multiple paracrine factors such as HGF and Wnt ligands.15,19,22 The colon cancer microenvironment supports cancer stem cell expansion.47 These colon cancer stem cells had elevated Wnt activity, particularly located in close proximity to the stromal fibroblasts with HGF expression.47 These results support the potential for endogenous cancer initiation from altered stromal cells. If so, killing the cancer epithelia will not be enough in preventing a recurrence. Even, if stromal initiation of cancer does not hold true, it is clear that CAF provide the growth factors and possibly the matrix environment that is conducive to cancer initiation.
Conclusion and Future Perspective
Finally, the stromal fibroblasts are a vital component of tumor progression. Although a lot of research done in this tumor microenvironment field is compelling, translation of these discoveries to clinic will require further progress prior to implementation. We need to better understand the cross talk of fibroblast and epithelial compartments. These include co-culture studies with stromal components (fibroblast, ECM or immune cells) in the cancer epithelial cell studies and studies related to altered microenvironment done using matrigel or hydrogel based models. Studies in immune-competent in vivo models would be needed. Signaling pathways identified in the fibroblastic cells need to be targeted in a cell type specific manner. Many advanced preclinical studies currently use targeting methodologies to the cancer epithelia. However, development of targeting to the CAF may be worthwhile. A challenge initially would be to define the CAF population and distinguish it from that of wound healing and inflammatory events occurring normally. Focus for therapeutic targets should involve the metastatic niche in order to block colonization and growth at the secondary site. This area of targeting will possibly complement the current and future therapeutic approaches based on tumor cell targeting and alleviate side effects, drug resistance and recurrence.
Footnotes
Previously published online: www.landesbioscience.com/journals/celladhesion/article/20419
References
- 1.Polyak K, Kalluri R. The role of the microenvironment in mammary gland development and cancer. Cold Spring Harb Perspect Biol. 2010;2:a003244. doi: 10.1101/cshperspect.a003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Despars G, Tan J, Periasamy P, O’Neill HC. The role of stroma in hematopoiesis and dendritic cell development. Curr Stem Cell Res Ther. 2007;2:23–9. doi: 10.2174/157488807779317017. [DOI] [PubMed] [Google Scholar]
- 3.Cichon MA, Degnim AC, Visscher DW, Radisky DC. Microenvironmental influences that drive progression from benign breast disease to invasive breast cancer. J Mammary Gland Biol Neoplasia. 2010;15:389–97. doi: 10.1007/s10911-010-9195-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–52. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;1:571–3. doi: 10.1016/S0140-6736(00)49915-0. [DOI] [PubMed] [Google Scholar]
- 6.Hart IR, Fidler IJ. Role of organ selectivity in the determination of metastatic patterns of B16 melanoma. Cancer Res. 1980;40:2281–7. [PubMed] [Google Scholar]
- 7.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–7. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeCosse JJ, Gossens CL, Kuzma JF, Unsworth BR. Breast cancer: induction of differentiation by embryonic tissue. Science. 1973;181:1057–8. doi: 10.1126/science.181.4104.1057. [DOI] [PubMed] [Google Scholar]
- 9.Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci U S A. 1975;72:3585–9. doi: 10.1073/pnas.72.9.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dolberg DS, Bissell MJ. Inability of Rous sarcoma virus to cause sarcomas in the avian embryo. Nature. 1984;309:552–6. doi: 10.1038/309552a0. [DOI] [PubMed] [Google Scholar]
- 11.Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–45. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rønnov-Jessen L, Petersen OW, Bissell MJ. Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol Rev. 1996;76:69–125. doi: 10.1152/physrev.1996.76.1.69. [DOI] [PubMed] [Google Scholar]
- 13.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 14.Gleave M, Hsieh JT, Gao CA, von Eschenbach AC, Chung LW. Acceleration of human prostate cancer growth in vivo by factors produced by prostate and bone fibroblasts. Cancer Res. 1991;51:3753–61. [PubMed] [Google Scholar]
- 15.Li X, Placencio V, Iturregui JM, Uwamariya C, Sharif-Afshar AR, Koyama T, et al. Prostate tumor progression is mediated by a paracrine TGF-beta/Wnt3a signaling axis. Oncogene. 2008;27:7118–30. doi: 10.1038/onc.2008.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. 2002;99:12877–82. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–11. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayward SW, Wang Y, Cao M, Hom YK, Zhang B, Grossfeld GD, et al. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res. 2001;61:8135–42. [PubMed] [Google Scholar]
- 19.Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–51. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 20.He Y, Franco OE, Jiang M, Williams K, Love HD, Coleman IM, et al. Tissue-specific consequences of cyclin D1 overexpression in prostate cancer progression. Cancer Res. 2007;67:8188–97. doi: 10.1158/0008-5472.CAN-07-0418. [DOI] [PubMed] [Google Scholar]
- 21.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–48. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 22.Placencio VR, Sharif-Afshar AR, Li X, Huang H, Uwamariya C, Neilson EG, et al. Stromal transforming growth factor-beta signaling mediates prostatic response to androgen ablation by paracrine Wnt activity. Cancer Res. 2008;68:4709–18. doi: 10.1158/0008-5472.CAN-07-6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayward SW, Haughney PC, Lopes ES, Danielpour D, Cunha GR. The rat prostatic epithelial cell line NRP-152 can differentiate in vivo in response to its stromal environment. Prostate. 1999;39:205–12. doi: 10.1002/(SICI)1097-0045(19990515)39:3<205::AID-PROS9>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 24.Franco OE, Jiang M, Strand DW, Peacock J, Fernandez S, Jackson RS, 2nd, et al. Altered TGF-β signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res. 2011;71:1272–81. doi: 10.1158/0008-5472.CAN-10-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiskowski MA, Jackson RS, 2nd, Banerjee J, Li X, Kang M, Iturregui JM, et al. Role for stromal heterogeneity in prostate tumorigenesis. Cancer Res. 2011;71:3459–70. doi: 10.1158/0008-5472.CAN-10-2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moinfar F, Man YG, Arnould L, Bratthauer GL, Ratschek M, Tavassoli FA. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res. 2000;60:2562–6. [PubMed] [Google Scholar]
- 27.Wernert N, Löcherbach C, Wellmann A, Behrens P, Hügel A. Presence of genetic alterations in microdissected stroma of human colon and breast cancers. J Mol Med (Berl) 2000;78:B30. [PubMed] [Google Scholar]
- 28.Wernert N, Löcherbach C, Wellmann A, Behrens P, Hügel A. Presence of genetic alterations in microdissected stroma of human colon and breast cancers. Anticancer Res. 2001;21(4A):2259–64. [PubMed] [Google Scholar]
- 29.Weber F, Shen L, Fukino K, Patocs A, Mutter GL, Caldes T, et al. Total-genome analysis of BRCA1/2-related invasive carcinomas of the breast identifies tumor stroma as potential landscaper for neoplastic initiation. Am J Hum Genet. 2006;78:961–72. doi: 10.1086/504090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukino K, Shen L, Matsumoto S, Morrison CD, Mutter GL, Eng C. Combined total genome loss of heterozygosity scan of breast cancer stroma and epithelium reveals multiplicity of stromal targets. Cancer Res. 2004;64:7231–6. doi: 10.1158/0008-5472.CAN-04-2866. [DOI] [PubMed] [Google Scholar]
- 31.Fukino K, Shen L, Patocs A, Mutter GL, Eng C. Genomic instability within tumor stroma and clinicopathological characteristics of sporadic primary invasive breast carcinoma. JAMA. 2007;297:2103–11. doi: 10.1001/jama.297.19.2103. [DOI] [PubMed] [Google Scholar]
- 32.Hill R, Song Y, Cardiff RD, Van Dyke T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell. 2005;123:1001–11. doi: 10.1016/j.cell.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 33.Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature. 2009;461:1084–91. doi: 10.1038/nature08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wallace JA, Li F, Leone G, Ostrowski MC. Pten in the breast tumor microenvironment: modeling tumor-stroma coevolution. Cancer Res. 2011;71:1203–7. doi: 10.1158/0008-5472.CAN-10-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng N, Bhowmick NA, Chytil A, Gorksa AE, Brown KA, Muraoka R, et al. Loss of TGF-beta type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-alpha-, MSP- and HGF-mediated signaling networks. Oncogene. 2005;24:5053–68. doi: 10.1038/sj.onc.1208685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DePinho RA, Polyak K. Cancer chromosomes in crisis. Nat Genet. 2004;36:932–4. doi: 10.1038/ng0904-932. [DOI] [PubMed] [Google Scholar]
- 37.Wolf M, Mousses S, Hautaniemi S, Karhu R, Huusko P, Allinen M, et al. High-resolution analysis of gene copy number alterations in human prostate cancer using CGH on cDNA microarrays: impact of copy number on gene expression. Neoplasia. 2004;6:240–7. doi: 10.1593/neo.03439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6:17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 39.Campbell IG, Qiu W, Polyak K, Haviv I. Breast-cancer stromal cells with TP53 mutations. N Engl J Med. 2008;358:1634–5, author reply 1636. doi: 10.1056/NEJMc086024. [DOI] [PubMed] [Google Scholar]
- 40.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–49. doi: 10.1016/S1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 41.Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–9. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Minn AJ, Gupta GP, Padua D, Bos P, Nguyen DX, Nuyten D, et al. Lung metastasis genes couple breast tumor size and metastatic spread. Proc Natl Acad Sci U S A. 2007;104:6740–5. doi: 10.1073/pnas.0701138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–24. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8:1369–75. doi: 10.1038/ncb1507. [DOI] [PubMed] [Google Scholar]
- 45.Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10:1349–55. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- 46.Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vermeulen L, De Sousa E Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–76. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]