

Abstract
Drug and radiation resistance represent a challenge for most anticancer therapies. Diverse experimental approaches have provided evidence that the tumor-associated microenvironment constitutes both a protective shell that impedes drug or radiation access and a permissive or promotive microenvironment that encourages a nurturing cancer (i.e., cancer stem cell) niche where tumor cells overcome treatment- and cancer-induced stresses. Better understanding of the effects of the tumor microenvironment on cancer cells before, during and immediately after chemo- or radiotherapy is imperative to design new therapies aimed at targeting this tumor-protective niche. This review summarizes some of the known mesenchymal stromal effects that account for drug resistance, the main signal transduction pathways associated with this resistance and the therapeutic efforts directed to increase the success of current therapies. Special emphasis is given to environment-mediated drug resistance in general and to cell adhesion-mediated drug resistance in particular.
Keywords: extracellular matrix, matrix-induced drug resistance, mesenchymal stroma, stromal-induced drug resistance, tumor microenvironment, tumor-associated or carcinoma-associated fibroblasts, tumor-stromal interactions
Introduction
Cancer cells use diverse strategies to decrease their sensitivity to drug therapy including alteration in drug-induced apoptosis, reduction of proliferation rates, expression of new drug-efflux pumps and failure to initiate DNA repair responses.1 These strategies largely rely on the ability of tumor cells to acquire a series of genetic changes that confer a survival advantage. Nevertheless, this genetic resistance takes a relatively long time to develop, whereas other “less permanent” or durable types of resistance mechanisms come into play earlier in treatment with a given drug. Following a selection of mutations, tumor cells become permanently resistant to the specific drug and to additional drug families because the selective pressure leads to new gene expression patterns that differ substantially from the expression patterns of the drug sensitive parental tumor cells.2
Ideally, cancer treatment would eliminate all malignant cells in order to avoid relapse and the increased aggressiveness that is often associated with tumor recurrence. However, even after “complete responses” evidenced by absence of macroscopic lesions, a small but significant number of cancer cells often survive chemotherapy.3 These surviving cells constitute minimal residual disease and represent a valuable diagnostic test to predict therapeutic outcomes, especially the probability of relapse. For instance, in acute lymphoblastic leukemia, the levels of these cells constitute the most important prognostic factor.4 Despite the clinical correlation between levels of minimal residual disease and probability of relapse, the mechanism whereby these cells escape the damaging effects of chemotherapeutic agents remains unclear.5
The presence of resistant subpopulations within the tumor mass and their subsequent selection and enrichment after treatment highlight the role of the genetic variability of these resistant cells. It has been proposed that cancer stem cells can divide asymmetrically producing the heterogeneous array of cells that compose a tumor while maintaining a resistant population of stem-like cells through self-renewal. In other words, it is possible that cancer stem cells may be the source of this heterogeneity, and thus the stem cells constitute the so-called “resistant population.”6 An alternative model suggests that cancer cells can undergo an epithelial-mesenchymal transition leading to the acquisition of stem cell properties.7 In this context, epithelial to mesenchymal transition may comprise epigenetic and/or a genetic change resulting in altered gene expressions. The difference between these two events is that epigenetic changes arise by alterations in RNA or protein expression independently of changes in the DNA sequence, while a genetic event originates from an alteration in the DNA sequence.8 For instance, epigenetic changes caused by extrinsic (i.e., microenvironmental), persistent or temporary cues may induce DNA methylation changes or alteration in some chromatin binding proteins leading to modifications in gene expression patterns that facilitate tumorigenesis and/or to drug resistance.9 In this case, gene expression changes, due to either microenvironmental regulation (i.e., epigenetic) and/or to mutations (i.e., genetic), convey alterations in gene expression that confer selective survival advantages.10,11 These statements are consistent with the traditional hypothesis of acquired resistance; however, this classical view solely considers resistance that is genetic.12,13 In contrast, new approaches to the study of drug resistance also consider microenvironmental influenced epigenetic changes.
The above-mentioned mechanisms require a complex interplay between environmental signals, gene mutations and selection pressures. Some models propose that a stepwise acquisition and accumulation of mutations take a long time,14-16 suggesting that cells destined to develop genetic alterations first require a sustained protection from the toxic effects of the drug through a non-genetic mechanism such as an altered tumor microenvironment prior to the acquisition of a resistant phenotype. Alternatively, a small population of cells within the drug naïve tumor could already possess specific genetic or epigenetic pre-dispositions, which will be selected for during the drug treatment. Hence, the drug-resistant population will in time present a different gene expression pattern from the one seen in the original drug naïve tumor. Under this time required scenario, it seems possible that the resistant cell population is protected by the tumor microenvironment (or selected for by the environmental pressures during treatment) providing a nurturing niche in which the cells could undergo mutagenesis or epigenetic changes. In both cases, the microenvironment is expected to play a role in assisting these cells to survive until the tumor becomes effectively drug resistant. In summary, newly genetic mutated, epigenetically modified or existent pre-disposed cells could all persist and regenerate resulting in what is known as an “acquired resistance” to therapy.2,17
It is important to note that de novo resistance could also include a microenvironmental protection mechanism or physical barrier that would limit the distribution (or the penetration) of an anticancer agent to tumor cell populations shielding cells from potential damage imparted by the drug.18,19 Note that this aspect of protection was reviewed elsewhere and therefore does not constitute a major discussion point.18,19 Nonetheless, several factors present in the tumor microenvironment induce gene transcription or activate post-translational modifications that reduce toxic drug effects. As this is considered a possibly transient and reversible type of resistance, therapeutic efforts to target these shielding environmental factors constitute an attractive approach to better access the tumor and to attempt to eliminate the cells that constitute the above-mentioned minimal residual disease. Consequently, it has been proposed that blockage of environment-mediated drug resistance (EM-DR) will lead to elimination of relapse or at least extend the time to disease relapse.20
The tumor-associated microenvironment that epithelial cancer cells often encounter during tumorigenesis (i.e., renal21 and ovarian22 cancers) and/or invasion (i.e., breast and other cancers23), consists of a rich assortment of cells (i.e, endothelial cells, plasma cells, macrophages, adipocytes and fibroblasts), extracellular matrix (ECM) fibers and stromal-derived soluble factors that contribute to the tumor-microenvironment interactions.24 De novo EM-DR falls into two broad categories: soluble factor-mediated resistance and cell adhesion-mediated resistance or CAM-DR. These two forms of resistance interact in a cooperative way. For instance, soluble factors may induce the expression of cellular adhesion molecules initiating a positive feedback loop and amplifying the resistance. A combination of both mechanisms may contribute to minimal residual disease making it difficult to separate one process from the other.25 In this review, we discuss aspects of CAM-DR in terms of its significant biological effects and list some promising therapeutic interventions aimed at counteracting these effects. Nevertheless, since soluble factors play an important role in facilitating, amplifying and strengthening CAM-DR, we will refer to selected factors whenever they significantly enhance this type of EM-DR.
Stages of EM-DR
A strong example of development of EM-DR is observed within the bone marrow microenvironment. Hematologic malignancies are established in the bone marrow while many solid (i.e., non-hematologic) tumors metastasize to the bone marrow. This compartment is rich in interleukins, especially IL-6 (see Table 1 for a list of drugs targeting this and many other EM-DR factors), and fibronectin both known to contribute to the acquisition of drug resistance.26 According to models that include tumors of primary bone origin or tumors metastatic to bone, bone marrow-like drug resistance develops in three discrete stages (see provided figure for representative example): homing, de novo resistance (including soluble factor mediated resistance and CAM-DR) and acquired resistance.27
Table 1. Examples of targets that could decrease tumor microenvironment-mediated drug resistance.
| Target | Therapeutic agent | Model and references |
|---|---|---|
| IL-6 |
Monoclonal antibody (siltuximab) |
Ovarian cancer xenografts and clinical trial149 |
| CXCR4 |
Plerixaflor (AMD3100) |
Myeloid acute leukemia cells140 |
| Glioblastoma and medulloblastoma xenografts142 | ||
| Breast cancer cells and xenografts146 | ||
| AMD 3465 |
Glioblastoma and medulloblastoma cell lines and xenografts143 |
|
| CTCE-9908 Inhibitory peptide |
Transgenic mouse model of breast cancer144 |
|
| siRNA |
Immunocompetent mouse model and cells for papillary epithelial ovarian cancer141 |
|
| Integrins |
Integrins (i.e., fibronectin synergy domain) antagonist penta-peptide (ATN-161) |
Clinical trial (solid tumors)120 |
| Anti β1-integrin monoclonal antibody (AIIB2) |
Breast cancer xenografts124 |
|
| General β1-integrin and natural HMG-CoA reductase inhibitor (Simvastatin) |
Head and neck squamous cell carcinoma cell lines151 |
|
| αVβ3-inregrin: etaracizumab |
Clinical trials (melanoma and advanced solid tumors)121,122 |
|
| α4β1- and α4β7: shRNA |
Multiple myeloma cells127 |
|
| Monoclonal anti α4-integrin antibody (Natalizumab) |
Multiple myeloma cells and mouse model128 |
|
| Cholesterol biosynthesis-dependent CAM-DR |
Statins, inhibitors of HMG-CoA reductase often through a Rho pathway inhibition manner (i.e., cerivastatin, simvastatin, lovastatin and fluvastatin) |
Breast cancer cells and xenografts152 Multiple myeloma cells153 Murine melanoma cells and xenografts156 |
| Hedgehog |
IPI-926 (oral smoothened inhibitor) |
Murine pancreatic cancer models69 |
| SPARC/ostenectin | SPARC analog N-terminal peptide |
Human colon, breast and pancreatic cancer cells and xenografts139 |
| Nanoparticles conjugated to albumin that specifically binds to stromal SPARC (i.e., nab-paclitaxel) | Breast cancer models and clinical trials131,132,137,138 Prostate cancer clinical trial133 Pancreatic cancer clinical trial134 |
Note that additional (not listed) reagents may be available. The relevant targets, therapeutic agents and models used are provided.11
Stage I
Homing, the first stage of EM-DR, normally refers to the adhesion of tumor cells to bone marrow hosted cells and/or to its ECM.27 Nevertheless, this step may not be necessary in solid tumors, while an alternative microenvironment, such as the lung, may play the same homing role during the metastatic establishment of tumors.28 Both normal and malignant hematopoietic cells and many epithelial solid tumor cells express a G-protein coupled receptor, CXCR4. CXCR4 is believed to be upregulated in response to pro-inflammatory factors often seen during tumor development and progression such as tumor necrosis α and IL-6.29 CXCR4 binds to ligands such as stroma-derived factor 1 (SDF-1) also known as CXCL12. This factor is believed to attract cells to the homing tissue (i.e., bone marrow or lung), retain them within this niche and stimulate their survival.27,30,31 In contrast to hematopoietic cells, solid tumor cells with greatest CXCR4 expression are those likely to metastasize favoring tumor cell migration, invasion and metastasis. These responses are believed to be triggered by the secondary metastatic tissue-derived SDF-1/CXCL12 known to be expressed by a predetermined secondary niche at the resident stromal location.28,30,32,33 Hence, the CXCR4-SDF-1/CXCL12 axis may constitute a general mechanism of tumor-stroma interaction that attracts neoplastic cells to the stroma, which can subsequently foster development of EM-DR.
Increased CXCR4 expression in clinical specimens seems to be predictive of poor prognosis in pancreatic cancer.34 In experimental systems, stromal-produced SDF-1/CXCL12, together with additional factors such as IL-8, not only attract pancreatic cancer cells to the experimental stroma (i.e., fibroblasts) but also increase tumor cell survival favoring migration and invasion.35 CXCR4 positive cells show increased activation of Akt/PKB and ERK pathways in response to SDF-1/CXCL12 thus leading to Bad phosphorylation, which translates into resistance to gemcitabine (the current preferred drug approved for the treatment of advanced pancreatic cancer) induced apoptosis. In this context, the interaction between CXCR4 and SDF-1/CXCR12 may constitute clinical relevance since it elicits resistance to gemcitabine. The study in question did not directly show that gemcitabine induces pancreatic cancer CXCR4 or stromal SDF-1/CXCL12. Nevertheless, the study shows that a small-molecule antagonist to CXCR4 desensitizes cells to gemcitabine by an effect on apoptosis.36
Interestingly, the CXCR4 and SDF-1/CXCL12 axis has other biologic effects that are relevant to the development of drug resistance, which do not necessarily seem to be related to homing. For example, glioblastoma cells (which usually remain in the brain and almost never metastasize) express both CXCR4 and CXCR7, which result in Erk 1/2 phosphorylation that also mediate anti-apoptotic responses.37 In fact, stromal cells harvested from lymph nodes promote drug resistance in human colon cancer cells through a CXCR4-SDF-1/CXCL12 dependent mechanism.38 Moreover, epithelial CXCR4 and stromal SDF-1/CXCL12 have been shown to regulate various normal and pathologic processes such as development, organogenesis, tissue regeneration and tumorigenesis.31
Stage II
In the second stage of EM-DR, tumor cells also engage the microenvironment. In turn, the engaged microenvironment secretes well-defined soluble factors and provides a specific adhesive milieu for the cancer cells to establish growth (see Fig. 1). In this stage, soluble factors and adhesive substrates can lead to drug resistance, while multiple interactions between the two amplify the cells’ responses leading to a rapid yet reversible resistance to therapies.27 For example, stromal cells in the bone marrow are known to secrete IL-6, which stimulates myeloma cells to produce vascular endothelial growth factor known to activate endothelial (stromal) cells resulting in angiogenesis and stroma nourishment.39 In addition, IL-6 stimulates secretion of additional factors such as fibroblast growth factor, which is known for its stromal mitogenic effects thus promoting stromal fibroblast proliferation and activation. This reciprocal stimulation generates an amplifying loop in which stromal and tumor cells together acquire increased ability to proliferate and survive.39-41

Figure 1. Stages during the development of environment-dependent drug resistance. (A) Homing or attraction. This first step requires specific cell-cell or cell-extracellular matrix interactions. Soluble stromal factors, such as SDF-1 and IL-6, and receptor-mediated adhesion contribute to attract tumor cells to the stromal niche where the tumor will be established. This first step is necessary in many hematopoietic malignancies,27 as well as in the establishment of secondary (i.e., metastatic28) tumors. Although homing is often seen in primary bone marrow tumors as well as in secondary tumor establishments, this step may not be necessary during primary solid tumor development. (B) De novo resistance. In this second stage (first stage for primary tumors that are not established in the bone marrow), the main stress from the treatment is applied to the, until then, drug naïve tumor. This step is characterized by a series of cell responses and the modification of the composition of the ECM creating a positive feedback loop that amplifies the pro-survival and anti-apoptotic signals. (C) Acquired resistance. This stage is commonly regarded as being environmental-independent, yet the microenvironment still plays an important role; for example, it can act as a barrier that physically or biochemically prevents the effective access of drugs to the tumor cells (see ref. 18 for review). Note the presence of a small amount of cancer resistance cells at early stages of development of drug resistance. This small cell population in what sometimes is regarded as the first stage (i.e., environment-dependent) and represents the possibility of a predisposed (i.e., cancer stem) resistant cell or, alternatively, one that has undergone a drug resistant mutation, will be selected during the stress period rendering a genetically different tumor signature compared with the drug naïve tumor population.
CAM-DR involves the adhesion of integrins and other receptors with components of the ECM: various types of collagen such as collagen I and III, splice variants of fibronectin such as ED-A, as well as other proteins such as vitronectin, tenascin-C, SPARC and osteopontin. Drug resistance is associated with anti-apoptotic and anti-proliferative cues. Both strategies render cells insensitive to chemotherapeutics that stimulate apoptosis or that target pathways that are preferentially active in rapidly dividing cells. Although several integrins have been implicated in this process, a special emphasis has been placed on α4β1-integrin, especially in bone marrow metastases. This receptor seems to play a crucial role in the acquisition of de novo resistance,42-45 supported by evidence that adhesion via β1-integrins decreases drug-induced DNA damage, apoptosis and/or cell cycle arrest in small cell lung,46 breast47 and hematopoietic cancers.48 Inhibition of apoptosis and increased cell proliferation can also be triggered by the binding of tumoral Notch-1,49 with Jagged, which is often expressed as a membrane bound ligand by bone marrow stromal cells.50 Interestingly, inhibition of the Notch axis has been proposed as a possible approach to induce pancreatic cancer apoptosis, while inhibition of Notch interplaying factors such as Hedgehog have been suggested for enhancing drug delivery to this highly desmoplastic tumor.51 Moreover, Hedgehog transcriptional target ABCG2 is well known for its stroma-dependent drug tolerance in lymphoma.52 The expression of these types of stromal factors and their role in protecting cancer cells against toxic therapies support the notion that the mesenchymal tumor microenvironment promotes development of CAM-DR. Interestingly in a classic experiment, Teicher et al. produced resistant mammary carcinoma cell lines by treating cells with four anti-neoplastic alkylating agents and passing cells through four groups of animals where cells were orthotopically injected. Treated tumor cells were isolated and sequentially transferred to fresh animals. The resultant tumors that developed in vivo acquired resistance to the drugs.16 Strikingly, the resultant cells lost their resistance after several passages of culture in vitro indicating that the mechanisms responsible for the resistance were not only reversible but strictly dependent on in vivo conditions.16
Stage III
During EM-DR, the tumor microenvironment protects cells from harsh therapeutic conditions providing anti-apoptotic signals that help cells resist drug-mediated DNA damage and apoptosis.43,53-55 In addition, it has been suggested that the mesenchymal stromal anti-proliferative cues can prevent cells from being targeted by growth inhibitory therapies.56 Hence, it is possible that stressful conditions promote the acquisition of transient reversible resistance in tumor cells that enables them to gain time and set in motion a complex process of DNA mutation (and/or epigenetic changes) that will ultimately lead to a state of what is believed to be an irreversible resistance. This third stage of EM-DR is known as acquired resistance (see Fig. 1), and, as decades of experience demonstrate at this stage of drug resistance development, cells become particularly difficult to treat.20,57
Biological Effects of CAM-DR
Four decades ago, Durand and Sutherland demonstrated that, compared with single cells in suspension, CHO cells grown as 3D spheroids in which cells establish close contact with one another were more resistant to the toxic effects of radiation.58 While the authors used spheroids exclusively composed of epithelial cells without a stromal component, this seminal work illustrated the importance of cell-cell adhesions in drug resistance. Building on these observations, we recently utilized a discrete cell culture model in which we were able to differentiate between responses of cancer epithelial cells to drug exposure determined by cell-cell vs. cell-ECM interactions (Cukierman and colleagues, unpublished results). These types of tissue engineered in vitro experimental models should assist in better understanding mechanisms responsible for some aspects of CAM-DR and facilitate development of pre-clinical screens for agents that can modulate CAM-DR.
In contrast to classical two-dimensional cell culture systems, animal models provide the complex stromal network (including altered ECMs) and permit the study of stromal influences.23 In addition, various three-dimensional culture systems also recapitulate some of the in vivo biological properties observed in the tumor stroma.59-61 Culture of a myeloma cell line on fibronectin provided the first indication that resistance to chemotherapeutics may develop through cell adhesion molecules.62 Damiano et al. has long established that CAM-DR can arise from the interaction between α4β1-integrin expressed by the tumor epithelial cells and the ECM protein fibronectin expressed by stromal cells.62 This adhesion elicits post-translational modifications such as phosphorylations resulting in activation of survival signals such as activation of IκB kinase and others tending to inhibit or counteract drug-induced apoptosis.63 In addition, β1-integrin was shown to play an Akt/PKB-independent role in the observed mesenchymal matrix protective effects seen under cytotoxic drug treatments in a plethora of cancer cells cultured onto fibroblast-derived 3D ECMs in vitro.64 A recent study suggested that tumor ECM can impart resistance to drugs due to β1-integrin, talin and FAK-dependent nuclear activation of NFκB.65 Another study suggested that a combination of specific splice variant forms of fibronectin together with α5β1-integrin play a pivotal role in resistance to radiation in breast cancer and that inhibiting this specific fibronectin-integrin axis can be instrumental in ensuring a better response to radiation treatment.66 In addition, α5β1-integrin has been implicated in chemoresistance in pancreatic cancer.67 In addition, CAM-DR can result in reduced availability of pro-proliferative signal molecules and mitogenic activity favoring a quiescent state that protects cells from current cell cycle-dependent therapies.68 Moreover, inhibition of epithelial-stromal signals (i.e., by inhibiting the Hedgehog pathway) has been shown to induce increases in chemotherapy delivery using a murine model of pancreatic cancer.69 It is well established that tumor-associated fibroblasts (TAFs, also known as carcinoma-associated fibroblasts or CAFs) produce a tumor-altered ECM that differs from that associated with normal, nonmalignant ECMs.23,59,60,70-73 In fact, it has been suggested that TAFs are responsible for resistance to epidermal growth factor inhibition in lung cancers with epidermal growth factor receptor-activating mutations74 and TAFs have been blamed for supporting tumor growth and drug resistance in some cancers (i.e., melanoma75).
Strikingly, the resistance observed in CAM-DR proved to be similar to the so-called acquired cell-derived drug resistance (de novo resistance of cells maintained in suspension). Using a human myeloma cell line, Hazlehurst et al. showed a significant increase in resistance to the alkylating agent melphalan when cells were cultured on fibronectin (i.e., CAM-DR) and compared these levels with levels of resistance imparted upon cells grown in suspension (i.e., acquired to melphalan cell-derived resistance). The levels of resistance on these two cultures were comparable. Only 69 genes were differentially expressed in CAM-DR (e.g., cultured on fibronectin) with respect to the cells grown in suspension. Among these CAM-DR 69 genes, the resistance mechanisms most frequently observed were related to decreased melphalan-induced mitochondrial depolarization and impaired caspase activation. These results highlight not only the magnitude of CAM-DR but also its reversible nature, since both the polarization state of the mitochondrial membrane and the activation of an inactive protein can be reverted using appropriate agents.15 Furthermore, the limited number of genes activated during CAM-DR suggests that therapies directed against relevant cell-matrix interactions could successfully target cancer cells administered alone or together with other anticancer agents.
Cancer cells may alter the composition of the ECM to modulate cell-matrix interactions accelerating the acquisition of CAM-DR. For example, collagen VI, a microfibrillar collagen associated with proliferation,76 is highly expressed in advanced metastatic ovarian cancer77 and cisplatin-resistant ovarian cancer cell lines. On the other hand, an ovarian cancer cell line, A2780, proved to increase its resistance to cisplatin when grown onto collagen VI. In this context, ovarian cancer cell lines favoring the acquisition of resistance synthesize ECM components that provide proliferative and adhesive advantages in the presence of cisplatin. The expression of collagen VI increases the adhesion to decorin, a component of the ECM, providing a specific binding to stromal components facilitating the adhesion of ovarian cancer cells to distant sites and favoring metastasis. Yet again, collagen VI decreases the expression of Bax, a pro-apoptotic protein, increasing cell survival. This paracrine mechanism initiated by alteration of the ECM by the tumor cells establishes a self- amplifying loop between the stroma and the tumor cells.78 Interestingly, it is important to highlight that different stromal cells and stromal ECMs can induce alternative cancer cell behaviors/phenotypes, which in turn are believed to be important in the manner that cancer cells respond to drug treatments.79 The notion that the degree of change in the stromal components of a tumor may be predictive of tumor behavior and also of response to treatment, leads to the concept of “stromal staging.”23,59,64,80-82 To this end, work has emerged in various types of cancers such as pancreatic82 and renal59 cancers demonstrating the potential clinical utility of stromal staging (see Fig. 2).
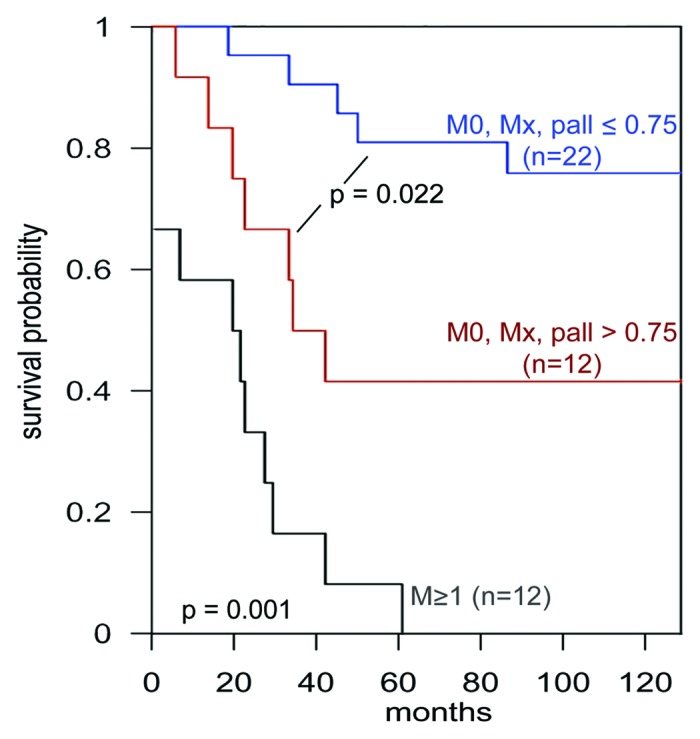
Figure 2. Activated stroma comprises a risk factor in non metastatic renal cell carcinoma. Multivariate CART-based sorting of a cohort where non metastatic patients (M0, Mx) were sorted by their stromal palladin expression levels representing non-activated (stromal palladin ≤ 0.75) vs. activated (stromal palladin > 0.75) stromal levels and were compared with metastatic patients (M ≥ 1) in a Kaplan-Meier curve showing time-scale (in months) at the x-axis and corresponding survival probability (e.g., survival fraction) at y-axis. The corresponding p values are provided. This figure was adapted from reference 59.
Signaling Pathways Associated with CAM-DR
As stated before, the interaction between the tumor cells and the stroma initiates post-translation mechanisms that confer a survival advantage. Hence, CAM-DR does not only function as a mere attachment of the tumor cell to the stroma but also as a powerful stimulus that triggers several signal transduction pathways leading to decreased sensitivity to apoptosis and, in some cases, inhibition of cell proliferation. Inhibition of cell proliferation (i.e., dormancy) may render cells resistant to anti-cancer therapies that target the cell cycle.3,83 Moreover, epithelial cancer cell signaling is likely to differ when engaged with the microenvironment.84 Therefore, the implication will be that amplification loops are not only initiated by the tumor microenvironment, but unique signals are educed within cancer cells under stimuli from both the soluble and physical microenvironment.84
Integrins and their downstream signaling pathways have often been implicated in cancer growth and invasion. Moreover, direct integrin inhibition or blockade of integrin-dependent pathways have been shown to assist in vitro to overcome drug resistance or to better drug treatments.47,64,85 In myeloma68 and in colon cancer86 cell lines, adhesion of α4β1- or α5β1-integrins to fibronectin results in increased levels of expression of p27Kip1, which inhibits the activity of the cyclin A and cyclin E associated kinases. Inactivation of these kinases decreases the activity of cyclins and leads to arrest in G1 phase. Interestingly, the levels of p27Kip1 return to normal (i.e., low) basal levels within two hours after disruption of a specific fibronectin-integrin interaction.68,86 This rapid response to cell-ECM adhesion relies on post-translational processes rather than on transcriptional activation of p27Kip1. In fact, adhesion of these integrins to fibronectin activates the ubiquitin ligase complex APC-Cdh1 increasing the ubiquitinilation and subsequent degradation of Skp-2, a negative modulator of p27Kip1.87,88 Decreased levels of Skp-2 results in increased p27Kip1 stability leading to its accumulation in the nucleus89 where it drives growth arrest in myeloma and lymphoma cell lines.90 A similar mechanism has been proposed for α2β1-integrin mediated cell growth arrest in a metastatic melanoma.91 Adhesion of melanoma cells to polymerized fibrillar collagen I results in cell growth arrest.91 As suggested above, growth arrest represents a useful strategy to decrease sensitivity to drugs that target the cell cycle. However, in solid tumors and in the context of drugs that promote apoptosis, β1-integrin engagement exerts the opposite effect: it overrides cell cycle arrest, decreases caspase-3 activation and prevents cells from undergoing apoptosis.92,93 Again, in this context, the ECM plays a crucial role since it provides the ligand/substrate for engaging integrin activities.
Increased and/or altered production of ECM proteins such as fibronectin, collagen IV, tenascin C and others predict poor prognosis in small cell lung carcinoma, a malignancy characterized by a good initial response to therapy but a high incidence of relapse indicative of CAM-DR.94 In fact in this malignancy, increased proliferation and survival play an important role in the aggressiveness of stromal rich tumors.94 Moreover, small cell lung carcinoma expresses different combinations of β1-integrins known for their abilities to bind and to respond to changes in collagen, laminin and fibronectin. This type of lung carcinoma expresses increased and differential spliced forms of fibronectin potentiating the effects of signal transduction pathways and, hence, of tumorigenic cellular responses.94 Others have shown that the normal response to etoposide and ionizing radiation increases levels of negative regulators of the G2/M checkpoint including p21Cip1 and p27Kip1, which leads to decreased stability of cyclins A, B and E and to reduced phosphorylation of CDK2 with the net effect of cell cycle arrest.46 By contrast, increased adhesion of β1-integrins to the tumor ECM triggers the activation of the non-receptor and integrin-dependent tyrosine kinase FAK, which in turn activates PI3 Kinase, Akt/PKB and GSK3β. These events trigger an increase in cyclin and a decrease in both p21 and p27 stability thus resulting in bypass of the G2/M checkpoint and eluding the pro-apoptotic effects seen following treatment with the combination of etoposide and ionizing radiation.46
Other signal transduction pathways driven through PI3 Kinase lead to resistance to apoptosis. Adhesion of β1-integrins activates the PI3 Kinase recruiting Akt/PKB to the membrane and leading to Akt-mediated pro-survival signals such as inactivation of the pro-apoptotic proteins Bad, Bim and Noxa,95,96 or to matrix-induced resistance to paclitaxel treatment.47 Some of the above-mentioned pro-apoptotic proteins were shown to be ubiquitinilated for degradation (BcL-2, Bim) while others were phosphorylated.
Cells can also evade apoptosis by decreasing the proteolytic-dependent activation of caspase 8, a post-translational modulator of caspase 3. For example, in multiple myeloma cells grown in suspension, the death receptor CD-95 can be found to be associated with the adaptor protein FADD. In turn, this complex attracts and tethers the procaspase 8 to the plasma membrane where it is activated by a limited and specific proteolysis. In turn, activated caspase 8 cleaves and therefore activates caspase 3 which promotes apoptosis. When cells bind to fibronectin, cFLIPL is released from the endo-membrane system to the cytoplasm competing with procaspase 8 for FADD binding. In this case, reduced binding of procaspase 8 to the FADD results in decreased activation of this pro-apoptotic enzyme and, thus, to ECM-dependent increased survival.97
Therapeutic Interventions
Once cells acquire resistance to a drug, relapse of the disease seems inevitable thus making it imperative to change treatment. Unfortunately, each time an individual develops a specific resistance to a given drug (or drug family) and relapse is observed, the relapse-free time decreases and the possibility of patient benefit diminishes.20,98 In order to prevent or delay the acquisition of drug resistance, several therapeutic approaches have been proposed.99 These include: (1) interference with direct tumor-stromal interactions,51 (2) impairment of cell-ECM interactions,100-102 (3) hampering of both expression and activity of paracrine factors secreted by stromal cells,103,104 (4) inhibition of stromal nuclear receptor super-family molecules (i.e., ligand-activated transcription factors known to regulate lipid metabolism and other processes105) or (5) general blockade of tumorigenic signaling pathways.48,68,90,106-108
Integrin inhibitors have been in clinical use for two decades, for example, in prevention of αIIβ3-dependent platelet aggregation in patients with acute coronary thrombosis.109 Other anti-integrin antibodies with efficacy in the treatment of psoriasis, Crohn disease and multiple sclerosis target the α4-integrins. Unfortunately, these agents were associated with progressive multifocal encelopathy and were withdrawn from the market.110 However, one of these antibodies, natalizumab, was eventually reintroduced for clinical use because of its effectiveness in decreasing the rate of relapse in subjects with multiple sclerosis. Despite this initial low level of success, new understanding of the function of integrins,111-114 and the development of less toxic drugs points to a brighter future for these therapies. Hence, integrin inhibitors are even being considered as tumor-stromal interaction inhibitors to attempt targeting dormant cancer cells.115
Integrins play a crucial role in tumor development, angiogenesis and survival, as well as cell-cell and cell-ECM interactions.116-119 As such, several inhibitors of integrin function have been developed to stop tumor progression, and some of these are being tested in the clinic.120 Etaracizumab, a monoclonal antibody specific for αVβ3-integrin, is under clinical development for the treatment of advanced metastatic melanoma121 and for additional solid metastatic tumors.122 Downstream effectors of integrin activation such as FAK also constitute novel therapeutic targets.123 The mechanism of action of these inhibitors includes inhibition of angiogenesis, migration and survival of the epithelial cells. However, the growing awareness that this interaction (i.e., that regulate integrin-ligand communications or affect integrin functional conformational changes) leads ultimately to the acquisition of CAM-DR makes it essential to consider the biology of cell-ECM interactions for the development of new drugs.
Cilentigide, a cyclic peptide that blocks the pro-angiogenic actions of αVβ3-integrin, proved to be non-toxic stimulating investigators to search for new therapeutic approaches that target the interaction between integrins and their ligands. Inhibition of the β1-integrin activity by means of the use of the AIIB2 monoclonal antibody resulted in increased sensitivity to ionizing radiation in breast cancer cell lines.124 Furthermore, abrogation of β1-integrin signaling increased sensitivity to trastuzumab, pertuzumab and lapatinib in 3D but not in 2D environments despite the similar levels of β1-integrin expression in these environments125,126 suggesting an important role for in vivo-like microenvironmental settings in pre-clinical drug testing.61 As further evidence of microenvironmental influence, cells grown in 2D conditions showed changes in proliferation and apoptosis through Akt/PKB and MAPK pathway signaling in response to HER2-targeting agent exposure. Strikingly, cells shifted to a predominantly pro-proliferative behavior in 3D environments pointing to their ability to adapt to different microenvironments.126 These results validate integrins from the β1 family as valuable targets for future therapeutic interventions. Multiple myelomas are known to express α4-integrins such as α4β1 and α4β7. Therefore, knockdown expression of α4-integrins in multiple myeloma cells constitutes an effective way to sensitize these cells to bortezomib, a proteome inhibitor drug often used in multiple myeloma patients.127 To this end, natalizumab, a novel small adhesion molecule inhibitor that interferes with α4β1- and α4β7-integrins, is believed to prevent multiple myeloma cell interactions with both stromal cells and stromal ECM as well as indirectly interfere with VEGF secretion and insulin-like growth factor induced signaling in the bone marrow where it increases sensitivity to bortezomib and dexamethasone.128 These observations point to the potential clinical use of natalizumab for patients with relapsing multiple myeloma and other malignancies.129,130
Alternative strategies today utilize specific ECM proteins to target drug treatments. For example, after a series of clinical trials, the use of paclitaxel delivered through nanoparticles conjugated to albumin “nab-paclitaxel” has been approved for treatment of breast cancer.131 Albumin binds efficiently to osteonectin/SPARC, which is commonly upregulated in the activated (or desmoplastic) stroma of many cancers such as breast,132 prostate133 and pancreatic134 cancers where its upregulation is also associated with bad prognostics.135,136 In a metastatic breast cancer model, nab-paclitaxel showed synergy when used in combination with bevacizumab, a monoclonal antibody known for blocking angiogenesis.137 To this end, the combination of adjuvant dose-dense doxorubicin plus cyclophosphamide followed by dose-dense nab-paclitaxel was recently found to be safe for use in women with early-stage breast cancer.138 Interestingly, it has also been suggested that SPARC may be a stromal tumor suppressor protein which conveys resistance to therapies. In fact, a recent work demonstrated that a peptide analogous to SPARC has tumor-regressing and chemo-sensitizing activities in vitro and in pre-clinical animal models.139
Soluble factors involved in de novo acquired drug resistance such as SDF1/CXCL12 represent alternative targets for cancer therapy. In lymphoblastic leukemia cells, a CXCR4-SDF1/CXCL12 antagonist decreased the adhesion of cancer cells to their tumor microenvironment resulting in decreased survival and increased differentiation.140 In another preclinical model, treatment of intraperitoneally xenotransplanted ovarian cancer cells with a selective CXCR4 antagonist resulted in decreased tumor proliferation, increased sensitivity to drug-induced apoptosis and enhanced activity of cytotoxic activity of T-lymphocytes.141 This strategy has been implemented in a series of preclinical tumor models and cells such as in oral squamous cell carcinoma,36 glioblastoma142,143 and breast cancer144-146 models with similar positive results. IL-6 is another stromal soluble factor that is typically upregulated during inflammatory diseases and stromal implicated cancers (i.e., desmoplastic or containing activated stromal cancers).147 A small molecule inhibitor of the pleotropic serine/threonine kinase CK2 known for its regulation of IL-6 expression in inflammatory breast cancer was currently assessed in a small clinical trial where it was shown to effectively reduce IL-6 in plasma.148 Moreover, in a platinum-resistant ovarian cancer phase II clinical trial, it was recently demonstrated that use of the anti-IL-6 antibody siltuximab is effective in downregulating IL-6 induced stromal factors such as CCL2, SDF1/CXCL12 and VEGF.149
An alternative approach considers downstream pathways activated in CAM-DR. For example, genes involved in cholesterol biosynthesis are upregulated in CAM-DR and in acquired drug resistance.15 Simvastatin, one of the 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors, was shown to decrease tumor cell proliferation and induce cell cycle arrest at G1/S phase.150 Simvastatin resulted in decreased expression of β1-integrin, impaired FAK phosphorylation and detachment of tumor cells from the ECM leading to cell death in several head and neck squamous cell carcinoma cell lines.151 Furthermore, inhibition of cholesterol biosynthesis resulted in decreased activity of the small GTPases Rho, Ras and RAP 1152,153 resulting in weaker focal adhesions while decreasing the affinity of integrins for their ligands.154-155
Inhibition of Wnt, Notch and Hedgehog pathways is considered a promising therapeutic approach because each is implicated in tumor-stromal interaction and may contribute to the protection of cancer stem cells by the microenvironment.157 Some investigators are considering Notch blockage as a CAM-DR inhibition strategy51 while Hedgehog pathway inhibitors have already gone through massive translational research considerations.158 Also, the feasibility and pharmacokinetics of the Hedgehog pathway inhibitor GDC-0449 were demonstrated in a basal-cell carcinoma phase I trial159 while a single medulloblastoma case study showed a temporary regression of the disease.160 Another novel approach involves the use of low, less toxic concentrations of naturally occurring stromal cytokines such as gamma interferon in combination with blockade of NFκB, a stromal-induced epithelial cell survival factor. This combination has been found to induce cancer necroptosis in vitro.161
Conclusions
Drug resistance represents a challenge to any therapy. Unfortunately, once a cell acquires a drug resistance mutation and the change becomes irreversible, readjusted doses are no longer sufficient and alternative therapeutic approaches are needed. Alternatively, and often by means of epigenetic changes, tumor cells can eventually overcome the toxic effects of chemotherapeutics through evading apoptosis and other treatment-derived toxic effects. To this end, the stroma in general and the tumor-associated ECM in particular provide a permissive environment that stimulates pro-survival pathways and an effective barrier against many of these chemotherapeutics. Therefore, the tumor microenvironment is considered a nurturing setting that protects tumor cells from drug exposure and reduces cytotoxicity. In conclusion, inhibition of tumor-stromal interactions could be a useful approach for preventing drug resistance and improving cancer treatment.
Acknowledgments
We would like to thank Drs A. Klein-Szanto, A. O’Reilly for discussions, G. Hudes for consultation and discussions, as well as E. Ragan for proofreading. This review was supported by NIH/NCI CA113451 and by The Keystone Program in Personalized Kidney Cancer Therapy at Fox Chase Cancer Center.
Glossary
Abbreviations:
- CAM-DR
cell adhesion-mediated resistance
- ECM
extracellular matrix
- EM-DR
environment-mediated drug resistance
- nab-paclitaxel
paclitaxel delivered through nanoparticles conjugated to albumin
- TAFs
tumor-associated fibroblasts (also known as carcinoma-associated fibroblasts or CAFs)
- SDF-1
stroma-derived factor 1
Footnotes
Previously published online: www.landesbioscience.com/journals/celladhesion/article/20210
References
- 1.Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–27. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 2.Baguley BC. Multiple drug resistance mechanisms in cancer. Mol Biotechnol. 2010;46:308–16. doi: 10.1007/s12033-010-9321-2. [DOI] [PubMed] [Google Scholar]
- 3.Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target? Nat Rev Cancer. 2010;10:871–7. doi: 10.1038/nrc2933. [DOI] [PubMed] [Google Scholar]
- 4.Borowitz MJ, Devidas M, Hunger SP, Bowman WP, Carroll AJ, Carroll WL, et al. Children’s Oncology Group Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children’s Oncology Group study. Blood. 2008;111:5477–85. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834–46. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Royer C, Lu X. Epithelial cell polarity: a major gatekeeper against cancer? Cell Death Differ. 2011;18:1470–7. doi: 10.1038/cdd.2011.60. [quest] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slatkin M. Epigenetic inheritance and the missing heritability problem. Genetics. 2009;182:845–50. doi: 10.1534/genetics.109.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang AD, Fan F, Camp ER, van Buren G, Liu W, Somcio R, et al. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res. 2006;12:4147–53. doi: 10.1158/1078-0432.CCR-06-0038. [DOI] [PubMed] [Google Scholar]
- 11.Basu D, Nguyen TT, Montone KT, Zhang G, Wang LP, Diehl JA, et al. Evidence for mesenchymal-like sub-populations within squamous cell carcinomas possessing chemoresistance and phenotypic plasticity. Oncogene. 2010;29:4170–82. doi: 10.1038/onc.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coldman AJ, Goldie JH. A stochastic model for the origin and treatment of tumors containing drug-resistant cells. Bull Math Biol. 1986;48:279–92. doi: 10.1007/BF02459682. [DOI] [PubMed] [Google Scholar]
- 13.Goldie JH, Coldman AJ. The genetic origin of drug resistance in neoplasms: implications for systemic therapy. Cancer Res. 1984;44:3643–53. [PubMed] [Google Scholar]
- 14.Bellamy WT, Dalton WS, Gleason MC, Grogan TM, Trent JM. Development and characterization of a melphalan-resistant human multiple myeloma cell line. Cancer Res. 1991;51:995–1002. [PubMed] [Google Scholar]
- 15.Hazlehurst LA, Enkemann SA, Beam CA, Argilagos RF, Painter J, Shain KH, et al. Genotypic and phenotypic comparisons of de novo and acquired melphalan resistance in an isogenic multiple myeloma cell line model. Cancer Res. 2003;63:7900–6. [PubMed] [Google Scholar]
- 16.Teicher BA, Herman TS, Holden SA, Wang YY, Pfeffer MR, Crawford JW, et al. Tumor resistance to alkylating agents conferred by mechanisms operative only in vivo. Science. 1990;247:1457–61. doi: 10.1126/science.2108497. [DOI] [PubMed] [Google Scholar]
- 17.Borovski T, De Sousa E Melo F, Vermeulen L, Medema JP. Cancer stem cell niche: the place to be. Cancer Res. 2011;71:634–9. doi: 10.1158/0008-5472.CAN-10-3220. [DOI] [PubMed] [Google Scholar]
- 18.Cukierman E, Khan DR. The benefits and challenges associated with the use of drug delivery systems in cancer therapy. Biochem Pharmacol. 2010;80:762–70. doi: 10.1016/j.bcp.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tannock IF, Lee CM, Tunggal JK, Cowan DS, Egorin MJ. Limited penetration of anticancer drugs through tumor tissue: a potential cause of resistance of solid tumors to chemotherapy. Clin Cancer Res. 2002;8:878–84. [PubMed] [Google Scholar]
- 20.Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. 2009;9:665–74. doi: 10.1038/nrc2714. [DOI] [PubMed] [Google Scholar]
- 21.Lohi J, Leivo I, Oivula J, Lehto VP, Virtanen I. Extracellular matrix in renal cell carcinomas. Histol Histopathol. 1998;13:785–96. doi: 10.14670/HH-13.785. [DOI] [PubMed] [Google Scholar]
- 22.Capo-Chichi CD, Smith ER, Yang DH, Roland IH, Vanderveer L, Cohen C, et al. Dynamic alterations of the extracellular environment of ovarian surface epithelial cells in premalignant transformation, tumorigenicity, and metastasis. Cancer. 2002;95:1802–15. doi: 10.1002/cncr.10870. [DOI] [PubMed] [Google Scholar]
- 23.Goetz JG, Minguet S, Navarro-Lérida I, Lazcano JJ, Samaniego R, Calvo E, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011;146:148–63. doi: 10.1016/j.cell.2011.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beacham DA, Cukierman E. Stromagenesis: the changing face of fibroblastic microenvironments during tumor progression. Semin Cancer Biol. 2005;15:329–41. doi: 10.1016/j.semcancer.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Hazlehurst LA, Landowski TH, Dalton WS. Role of the tumor microenvironment in mediating de novo resistance to drugs and physiological mediators of cell death. Oncogene. 2003;22:7396–402. doi: 10.1038/sj.onc.1206943. [DOI] [PubMed] [Google Scholar]
- 26.Nefedova Y, Landowski TH, Dalton WS. Bone marrow stromal-derived soluble factors and direct cell contact contribute to de novo drug resistance of myeloma cells by distinct mechanisms. Leukemia. 2003;17:1175–82. doi: 10.1038/sj.leu.2402924. [DOI] [PubMed] [Google Scholar]
- 27.Meads MB, Hazlehurst LA, Dalton WS. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008;14:2519–26. doi: 10.1158/1078-0432.CCR-07-2223. [DOI] [PubMed] [Google Scholar]
- 28.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–48. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 29.Zhao C, Lu X, Bu X, Zhang N, Wang W. Involvement of tumor necrosis factor-alpha in the upregulation of CXCR4 expression in gastric cancer induced by Helicobacter pylori. BMC Cancer. 2010;10:419. doi: 10.1186/1471-2407-10-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203:2201–13. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ratajczak MZ, Zuba-Surma E, Kucia M, Reca R, Wojakowski W, Ratajczak J. The pleiotropic effects of the SDF-1-CXCR4 axis in organogenesis, regeneration and tumorigenesis. Leukemia. 2006;20:1915–24. doi: 10.1038/sj.leu.2404357. [DOI] [PubMed] [Google Scholar]
- 32.Juarez J, Dela Pena A, Baraz R, Hewson J, Khoo M, Cisterne A, et al. CXCR4 antagonists mobilize childhood acute lymphoblastic leukemia cells into the peripheral blood and inhibit engraftment. Leukemia. 2007;21:1249–57. doi: 10.1038/sj.leu.2404684. [DOI] [PubMed] [Google Scholar]
- 33.Spiegel A, Kollet O, Peled A, Abel L, Nagler A, Bielorai B, et al. Unique SDF-1-induced activation of human precursor-B ALL cells as a result of altered CXCR4 expression and signaling. Blood. 2004;103:2900–7. doi: 10.1182/blood-2003-06-1891. [DOI] [PubMed] [Google Scholar]
- 34.Maréchal R, Demetter P, Nagy N, Berton A, Decaestecker C, Polus M, et al. High expression of CXCR4 may predict poor survival in resected pancreatic adenocarcinoma. Br J Cancer. 2009;100:1444–51. doi: 10.1038/sj.bjc.6605020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuo Y, Ochi N, Sawai H, Yasuda A, Takahashi H, Funahashi H, et al. CXCL8/IL-8 and CXCL12/SDF-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int J Cancer. 2009;124:853–61. doi: 10.1002/ijc.24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh S, Srivastava SK, Bhardwaj A, Owen LB, Singh AP. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: a novel target for therapy. Br J Cancer. 2010;103:1671–9. doi: 10.1038/sj.bjc.6605968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hattermann K, Held-Feindt J, Lucius R, Müerköster SS, Penfold ME, Schall TJ, et al. The chemokine receptor CXCR7 is highly expressed in human glioma cells and mediates antiapoptotic effects. Cancer Res. 2010;70:3299–308. doi: 10.1158/0008-5472.CAN-09-3642. [DOI] [PubMed] [Google Scholar]
- 38.Margolin DA, Silinsky J, Grimes C, Spencer N, Aycock M, Green H, et al. Lymph node stromal cells enhance drug-resistant colon cancer cell tumor formation through SDF-1α/CXCR4 paracrine signaling. Neoplasia. 2011;13:874–86. doi: 10.1593/neo.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dankbar B, Padró T, Leo R, Feldmann B, Kropff M, Mesters RM, et al. Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood. 2000;95:2630–6. [PubMed] [Google Scholar]
- 40.Bisping G, Leo R, Wenning D, Dankbar B, Padró T, Kropff M, et al. Paracrine interactions of basic fibroblast growth factor and interleukin-6 in multiple myeloma. Blood. 2003;101:2775–83. doi: 10.1182/blood-2002-09-2907. [DOI] [PubMed] [Google Scholar]
- 41.Gupta D, Treon SP, Shima Y, Hideshima T, Podar K, Tai YT, et al. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: therapeutic applications. Leukemia. 2001;15:1950–61. doi: 10.1038/sj.leu.2402295. [DOI] [PubMed] [Google Scholar]
- 42.de la Fuente MT, Casanova B, Moyano JV, Garcia-Gila M, Sanz L, Garcia-Marco J, et al. Engagement of alpha4beta1 integrin by fibronectin induces in vitro resistance of B chronic lymphocytic leukemia cells to fludarabine. J Leukoc Biol. 2002;71:495–502. [PubMed] [Google Scholar]
- 43.Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9:1158–65. doi: 10.1038/nm909. [DOI] [PubMed] [Google Scholar]
- 44.Taylor ST, Hickman JA, Dive C. Epigenetic determinants of resistance to etoposide regulation of Bcl-X(L) and Bax by tumor microenvironmental factors. J Natl Cancer Inst. 2000;92:18–23. doi: 10.1093/jnci/92.1.18. [DOI] [PubMed] [Google Scholar]
- 45.Weekes CD, Kuszynski CA, Sharp JG. VLA-4 mediated adhesion to bone marrow stromal cells confers chemoresistance to adherent lymphoma cells. Leuk Lymphoma. 2001;40:631–45. doi: 10.3109/10428190109097661. [DOI] [PubMed] [Google Scholar]
- 46.Hodkinson PS, Elliott T, Wong WS, Rintoul RC, Mackinnon AC, Haslett C, et al. ECM overrides DNA damage-induced cell cycle arrest and apoptosis in small-cell lung cancer cells through beta1 integrin-dependent activation of PI3-kinase. Cell Death Differ. 2006;13:1776–88. doi: 10.1038/sj.cdd.4401849. [DOI] [PubMed] [Google Scholar]
- 47.Aoudjit F, Vuori K. Integrin signaling inhibits paclitaxel-induced apoptosis in breast cancer cells. Oncogene. 2001;20:4995–5004. doi: 10.1038/sj.onc.1204554. [DOI] [PubMed] [Google Scholar]
- 48.Hazlehurst LA, Argilagos RF, Dalton WS. Beta1 integrin mediated adhesion increases Bim protein degradation and contributes to drug resistance in leukaemia cells. Br J Haematol. 2007;136:269–75. doi: 10.1111/j.1365-2141.2006.06435.x. [DOI] [PubMed] [Google Scholar]
- 49.Li L, Milner LA, Deng Y, Iwata M, Banta A, Graf L, et al. The human homolog of rat Jagged1 expressed by marrow stroma inhibits differentiation of 32D cells through interaction with Notch1. Immunity. 1998;8:43–55. doi: 10.1016/S1074-7613(00)80457-4. [DOI] [PubMed] [Google Scholar]
- 50.Walker L, Lynch M, Silverman S, Fraser J, Boulter J, Weinmaster G, et al. The Notch/Jagged pathway inhibits proliferation of human hematopoietic progenitors in vitro. Stem Cells. 1999;17:162–71. doi: 10.1002/stem.170162. [DOI] [PubMed] [Google Scholar]
- 51.Ristorcelli E, Lombardo D. Targeting Notch signaling in pancreatic cancer. Expert Opin Ther Targets. 2010;14:541–52. doi: 10.1517/14728221003769895. [DOI] [PubMed] [Google Scholar]
- 52.Singh RR, Kunkalla K, Qu C, Schlette E, Neelapu SS, Samaniego F, et al. ABCG2 is a direct transcriptional target of hedgehog signaling and involved in stroma-induced drug tolerance in diffuse large B-cell lymphoma. Oncogene. 2011;30:4874–86. doi: 10.1038/onc.2011.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mraz M, Zent CS, Church AK, Jelinek DF, Wu X, Pospisilova S, et al. Bone marrow stromal cells protect lymphoma B-cells from rituximab-induced apoptosis and targeting integrin α-4-β-1 (VLA-4) with natalizumab can overcome this resistance. Br J Haematol. 2011;155:53–64. doi: 10.1111/j.1365-2141.2011.08794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- 55.Zucchetto A, Benedetti D, Tripodo C, Bomben R, Dal Bo M, Marconi D, et al. CD38/CD31, the CCL3 and CCL4 chemokines, and CD49d/vascular cell adhesion molecule-1 are interchained by sequential events sustaining chronic lymphocytic leukemia cell survival. Cancer Res. 2009;69:4001–9. doi: 10.1158/0008-5472.CAN-08-4173. [DOI] [PubMed] [Google Scholar]
- 56.Taylor ST, Hickman JA, Dive C. Survival signals within the tumour microenvironment suppress drug-induced apoptosis: lessons learned from B lymphomas. Endocr Relat Cancer. 1999;6:21–3. doi: 10.1677/erc.0.0060021. [DOI] [PubMed] [Google Scholar]
- 57.Mimeault M, Hauke R, Batra SK. Recent advances on the molecular mechanisms involved in the drug resistance of cancer cells and novel targeting therapies. Clin Pharmacol Ther. 2008;83:673–91. doi: 10.1038/sj.clpt.6100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Durand RE, Sutherland RM. Effects of intercellular contact on repair of radiation damage. Exp Cell Res. 1972;71:75–80. doi: 10.1016/0014-4827(72)90265-0. [DOI] [PubMed] [Google Scholar]
- 59.Gupta V, Bassi DE, Simons JD, Devarajan K, Al-Saleem T, Uzzo RG, et al. Elevated expression of stromal palladin predicts poor clinical outcome in renal cell carcinoma. PLoS One. 2011;6:e21494. doi: 10.1371/journal.pone.0021494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quiros RM, Valianou M, Kwon Y, Brown KM, Godwin AK, Cukierman E. Ovarian normal and tumor-associated fibroblasts retain in vivo stromal characteristics in a 3-D matrix-dependent manner. Gynecol Oncol. 2008;110:99–109. doi: 10.1016/j.ygyno.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yamada KM, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell. 2007;130:601–10. doi: 10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 62.Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93:1658–67. [PMC free article] [PubMed] [Google Scholar]
- 63.Lin Y, Bai L, Chen W, Xu S. The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin Ther Targets. 2010;14:45–55. doi: 10.1517/14728220903431069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Serebriiskii I, Castelló-Cros R, Lamb A, Golemis EA, Cukierman E. Fibroblast-derived 3D matrix differentially regulates the growth and drug-responsiveness of human cancer cells. Matrix Biol. 2008;27:573–85. doi: 10.1016/j.matbio.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eberle KE, Sansing HA, Szaniszlo P, Resto VA, Berrier AL. Carcinoma matrix controls resistance to cisplatin through talin regulation of NF-kB. PLoS One. 2011;6:e21496. doi: 10.1371/journal.pone.0021496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nam JM, Onodera Y, Bissell MJ, Park CC. Breast cancer cells in three-dimensional culture display an enhanced radioresponse after coordinate targeting of integrin alpha5beta1 and fibronectin. Cancer Res. 2010;70:5238–48. doi: 10.1158/0008-5472.CAN-09-2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sebens Müerköster S, Kötteritzsch J, Geismann C, Gast D, Kruse ML, Altevogt P, et al. alpha5-integrin is crucial for L1CAM-mediated chemoresistance in pancreatic adenocarcinoma. Int J Oncol. 2009;34:243–53. [PubMed] [Google Scholar]
- 68.Hazlehurst LA, Damiano JS, Buyuksal I, Pledger WJ, Dalton WS. Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR) Oncogene. 2000;19:4319–27. doi: 10.1038/sj.onc.1203782. [DOI] [PubMed] [Google Scholar]
- 69.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Castelló-Cros R, Cukierman E. Stromagenesis during tumorigenesis: characterization of tumor-associated fibroblasts and stroma-derived 3D matrices. Methods Mol Biol. 2009;522:275–305. doi: 10.1007/978-1-59745-413-1_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123:4195–200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Amatangelo MD, Bassi DE, Klein-Szanto AJ, Cukierman E. Stroma-derived three-dimensional matrices are necessary and sufficient to promote desmoplastic differentiation of normal fibroblasts. Am J Pathol. 2005;167:475–88. doi: 10.1016/S0002-9440(10)62991-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang W, Li Q, Yamada T, Matsumoto K, Matsumoto I, Oda M, et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2009;15:6630–8. doi: 10.1158/1078-0432.CCR-09-1001. [DOI] [PubMed] [Google Scholar]
- 75.Flach EH, Rebecca VW, Herlyn M, Smalley KS, Anderson AR. Fibroblasts contribute to melanoma tumor growth and drug resistance. Mol Pharm. 2011;8:2039–49. doi: 10.1021/mp200421k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Atkinson JC, Rühl M, Becker J, Ackermann R, Schuppan D. Collagen VI regulates normal and transformed mesenchymal cell proliferation in vitro. Exp Cell Res. 1996;228:283–91. doi: 10.1006/excr.1996.0328. [DOI] [PubMed] [Google Scholar]
- 77.Ismail RS, Baldwin RL, Fang J, Browning D, Karlan BY, Gasson JC, et al. Differential gene expression between normal and tumor-derived ovarian epithelial cells. Cancer Res. 2000;60:6744–9. [PubMed] [Google Scholar]
- 78.Sherman-Baust CA, Weeraratna AT, Rangel LB, Pizer ES, Cho KR, Schwartz DR, et al. Remodeling of the extracellular matrix through overexpression of collagen VI contributes to cisplatin resistance in ovarian cancer cells. Cancer Cell. 2003;3:377–86. doi: 10.1016/S1535-6108(03)00058-8. [DOI] [PubMed] [Google Scholar]
- 79.Lee HO, Mullins SR, Franco-Barraza J, Valianou M, Cukierman E, Cheng JD. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer. 2011;11:245. doi: 10.1186/1471-2407-11-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Castelló-Cros R, Khan DR, Simons J, Valianou M, Cukierman E. Staged stromal extracellular 3D matrices differentially regulate breast cancer cell responses through PI3K and beta1-integrins. BMC Cancer. 2009;9:94. doi: 10.1186/1471-2407-9-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kwon Y, Cukierman E, Godwin AK. Differential expressions of adhesive molecules and proteases define mechanisms of ovarian tumor cell matrix penetration/invasion. PLoS One. 2011;6:e18872. doi: 10.1371/journal.pone.0018872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Erkan M, Michalski CW, Rieder S, Reiser-Erkan C, Abiatari I, Kolb A, et al. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol. 2008;6:1155–61. doi: 10.1016/j.cgh.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 83.Klein CA. Framework models of tumor dormancy from patient-derived observations. Curr Opin Genet Dev. 2011;21:42–9. doi: 10.1016/j.gde.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 84.Shain KH, Yarde DN, Meads MB, Huang M, Jove R, Hazlehurst LA, et al. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: implications for microenvironment influence on tumor survival and proliferation. Cancer Res. 2009;69:1009–15. doi: 10.1158/0008-5472.CAN-08-2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sansing HA, Sarkeshik A, Yates JR, Patel V, Gutkind JS, Yamada KM, et al. Integrin αβ1, αvβ, α6β effectors p130Cas, Src and talin regulate carcinoma invasion and chemoresistance. Biochem Biophys Res Commun. 2011;406:171–6. doi: 10.1016/j.bbrc.2011.01.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gong J, Ko TC, Brattain MG. Disruption of fibronectin binding to the alpha 5 beta 1 integrin stimulates the expression of cyclin-dependent kinases and DNA synthesis through activation of extracellular signal-regulated kinase. J Biol Chem. 1998;273:1662–9. doi: 10.1074/jbc.273.3.1662. [DOI] [PubMed] [Google Scholar]
- 87.Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, et al. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–5. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 88.Shirane M, Harumiya Y, Ishida N, Hirai A, Miyamoto C, Hatakeyama S, et al. Down-regulation of p27(Kip1) by two mechanisms, ubiquitin-mediated degradation and proteolytic processing. J Biol Chem. 1999;274:13886–93. doi: 10.1074/jbc.274.20.13886. [DOI] [PubMed] [Google Scholar]
- 89.Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000;19:2069–81. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lwin T, Hazlehurst LA, Dessureault S, Lai R, Bai W, Sotomayor E, et al. Cell adhesion induces p27Kip1-associated cell-cycle arrest through down-regulation of the SCFSkp2 ubiquitin ligase pathway in mantle-cell and other non-Hodgkin B-cell lymphomas. Blood. 2007;110:1631–8. doi: 10.1182/blood-2006-11-060350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Henriet P, Zhong ZD, Brooks PC, Weinberg KI, DeClerck YA. Contact with fibrillar collagen inhibits melanoma cell proliferation by up-regulating p27KIP1. Proc Natl Acad Sci U S A. 2000;97:10026–31. doi: 10.1073/pnas.170290997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Estrugo D, Fischer A, Hess F, Scherthan H, Belka C, Cordes N. Ligand bound beta1 integrins inhibit procaspase-8 for mediating cell adhesion-mediated drug and radiation resistance in human leukemia cells. PLoS One. 2007;2:e269. doi: 10.1371/journal.pone.0000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shakibaei M, John T, Seifarth C, Mobasheri A. Resveratrol inhibits IL-1 beta-induced stimulation of caspase-3 and cleavage of PARP in human articular chondrocytes in vitro. Ann N Y Acad Sci. 2007;1095:554–63. doi: 10.1196/annals.1397.060. [DOI] [PubMed] [Google Scholar]
- 94.Sethi T, Rintoul RC, Moore SM, MacKinnon AC, Salter D, Choo C, et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat Med. 1999;5:662–8. doi: 10.1038/9511. [DOI] [PubMed] [Google Scholar]
- 95.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381–95. doi: 10.1016/S0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 97.Shain KH, Landowski TH, Dalton WS. Adhesion-mediated intracellular redistribution of c-Fas-associated death domain-like IL-1-converting enzyme-like inhibitory protein-long confers resistance to CD95-induced apoptosis in hematopoietic cancer cell lines. J Immunol. 2002;168:2544–53. doi: 10.4049/jimmunol.168.5.2544. [DOI] [PubMed] [Google Scholar]
- 98.Gilbert LA, Hemann MT. Chemotherapeutic resistance: surviving stressful situations. Cancer Res. 2011;71:5062–6. doi: 10.1158/0008-5472.CAN-11-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7:513–20. doi: 10.1016/j.ccr.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 100.Stupp R, Ruegg C. Integrin inhibitors reaching the clinic. J Clin Oncol. 2007;25:1637–8. doi: 10.1200/JCO.2006.09.8376. [DOI] [PubMed] [Google Scholar]
- 101.Preusser M, de Ribaupierre S, Wöhrer A, Erridge SC, Hegi M, Weller M, et al. Current concepts and management of glioblastoma. Ann Neurol. 2011;70:9–21. doi: 10.1002/ana.22425. [DOI] [PubMed] [Google Scholar]
- 102.Cox D, Brennan M, Moran N. Integrins as therapeutic targets: lessons and opportunities. Nat Rev Drug Discov. 2010;9:804–20. doi: 10.1038/nrd3266. [DOI] [PubMed] [Google Scholar]
- 103.Lin B, Podar K, Gupta D, Tai YT, Li S, Weller E, et al. The vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584 inhibits growth and migration of multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2002;62:5019–26. [PubMed] [Google Scholar]
- 104.Bisping G, Wenning D, Kropff M, Gustavus D, Müller-Tidow C, Stelljes M, et al. Bortezomib, dexamethasone, and fibroblast growth factor receptor 3-specific tyrosine kinase inhibitor in t(4;14) myeloma. Clin Cancer Res. 2009;15:520–31. doi: 10.1158/1078-0432.CCR-08-1612. [DOI] [PubMed] [Google Scholar]
- 105.Sherman MH, Downes M, Evans RM. Nuclear receptors as modulators of the tumor microenvironment. Cancer Prev Res (Phila) 2012;5:3–10. doi: 10.1158/1940-6207.CAPR-11-0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fu Y, Fang Z, Liang Y, Zhu X, Prins P, Li Z, et al. Overexpression of integrin beta1 inhibits proliferation of hepatocellular carcinoma cell SMMC-7721 through preventing Skp2-dependent degradation of p27 via PI3K pathway. J Cell Biochem. 2007;102:704–18. doi: 10.1002/jcb.21323. [DOI] [PubMed] [Google Scholar]
- 107.Piperdi B, Ling YH, Liebes L, Muggia F, Perez-Soler R. Bortezomib: understanding the mechanism of action. Mol Cancer Ther. 2011;10:2029–30. doi: 10.1158/1535-7163.MCT-11-0745. [DOI] [PubMed] [Google Scholar]
- 108.San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, et al. VISTA Trial Investigators Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359:906–17. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 109.Bonaca MP, Steg PG, Feldman LJ, Canales JF, Ferguson JJ, Wallentin L, et al. Antithrombotics in acute coronary syndromes. J Am Coll Cardiol. 2009;54:969–84. doi: 10.1016/j.jacc.2009.03.083. [DOI] [PubMed] [Google Scholar]
- 110.Warnke C, Menge T, Hartung HP, Racke MK, Cravens PD, Bennett JL, et al. Natalizumab and progressive multifocal leukoencephalopathy: what are the causal factors and can it be avoided? Arch Neurol. 2010;67:923–30. doi: 10.1001/archneurol.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bagley RG, Hudson LG, Stack MS. Integrins and Cancer. In: Teicher BA, ed. The Tumor Microenvironment: Springer New York, 2010:509-29. [Google Scholar]
- 112.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yamada KM, Pankov R, Cukierman E. Dimensions and dynamics in integrin function. Braz J Med Biol Res. 2003;36:959–66. doi: 10.1590/S0100-879X2003000800001. [DOI] [PubMed] [Google Scholar]
- 114.Larsen M, Artym VV, Green JA, Yamada KM. The matrix reorganized: extracellular matrix remodeling and integrin signaling. Curr Opin Cell Biol. 2006;18:463–71. doi: 10.1016/j.ceb.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 115.Barkan D, Chambers AF. β1-integrin: a potential therapeutic target in the battle against cancer recurrence. Clin Cancer Res. 2011;17:7219–23. doi: 10.1158/1078-0432.CCR-11-0642. [DOI] [PubMed] [Google Scholar]
- 116.Moschos SJ, Drogowski LM, Reppert SL, Kirkwood JM. Integrins and cancer. Oncology (Williston Park) 2007;21(Suppl 3):13–20. [Williston Park] [PubMed] [Google Scholar]
- 117.Jin H, Varner J. Integrins: roles in cancer development and as treatment targets. Br J Cancer. 2004;90:561–5. doi: 10.1038/sj.bjc.6601576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8:604–17. doi: 10.1038/nrc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Katz BZ. Adhesion molecules--The lifelines of multiple myeloma cells. Semin Cancer Biol. 2010;20:186–95. doi: 10.1016/j.semcancer.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 120.Cianfrocca ME, Kimmel KA, Gallo J, Cardoso T, Brown MM, Hudes G, et al. Phase 1 trial of the antiangiogenic peptide ATN-161 (Ac-PHSCN-NH(2)), a beta integrin antagonist, in patients with solid tumours. Br J Cancer. 2006;94:1621–6. doi: 10.1038/sj.bjc.6603171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hersey P, Sosman J, O’Day S, Richards J, Bedikian A, Gonzalez R, et al. Etaracizumab Melanoma Study Group A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin alpha(v)beta(3), + or - dacarbazine in patients with stage IV metastatic melanoma. Cancer. 2010;116:1526–34. doi: 10.1002/cncr.24821. [DOI] [PubMed] [Google Scholar]
- 122.Delbaldo C, Raymond E, Vera K, Hammershaimb L, Kaucic K, Lozahic S, et al. Phase I and pharmacokinetic study of etaracizumab (Abegrin), a humanized monoclonal antibody against alphavbeta3 integrin receptor, in patients with advanced solid tumors. Invest New Drugs. 2008;26:35–43. doi: 10.1007/s10637-007-9077-0. [DOI] [PubMed] [Google Scholar]
- 123.Stokes JB, Adair SJ, Slack-Davis JK, Walters DM, Tilghman RW, Hershey ED, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther. 2011;10:2135–45. doi: 10.1158/1535-7163.MCT-11-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Park CC, Zhang HJ, Yao ES, Park CJ, Bissell MJ. Beta1 integrin inhibition dramatically enhances radiotherapy efficacy in human breast cancer xenografts. Cancer Res. 2008;68:4398–405. doi: 10.1158/0008-5472.CAN-07-6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang XH, Flores LM, Li Q, Zhou P, Xu F, Krop IE, et al. Disruption of laminin-integrin-CD151-focal adhesion kinase axis sensitizes breast cancer cells to ErbB2 antagonists. Cancer Res. 2010;70:2256–63. doi: 10.1158/0008-5472.CAN-09-4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Weigelt B, Lo AT, Park CC, Gray JW, Bissell MJ. HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res Treat. 2010;122:35–43. doi: 10.1007/s10549-009-0502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Noborio-Hatano K, Kikuchi J, Takatoku M, Shimizu R, Wada T, Ueda M, et al. Bortezomib overcomes cell-adhesion-mediated drug resistance through downregulation of VLA-4 expression in multiple myeloma. Oncogene. 2009;28:231–42. doi: 10.1038/onc.2008.385. [DOI] [PubMed] [Google Scholar]
- 128.Podar K, Zimmerhackl A, Fulciniti M, Tonon G, Hainz U, Tai YT, et al. The selective adhesion molecule inhibitor Natalizumab decreases multiple myeloma cell growth in the bone marrow microenvironment: therapeutic implications. Br J Haematol. 2011;155:438–48. doi: 10.1111/j.1365-2141.2011.08864.x. [DOI] [PubMed] [Google Scholar]
- 129.Ransohoff RM. Natalizumab for multiple sclerosis. N Engl J Med. 2007;356:2622–9. doi: 10.1056/NEJMct071462. [DOI] [PubMed] [Google Scholar]
- 130.Rutgeerts P, Vermeire S, Van Assche G. Biological therapies for inflammatory bowel diseases. Gastroenterology. 2009;136:1182–97. doi: 10.1053/j.gastro.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 131.Vishnu P, Roy V. nab-paclitaxel: a novel formulation of taxane for treatment of breast cancer. Womens Health (Lond Engl) 2010;6:495–506. doi: 10.2217/whe.10.42. [DOI] [PubMed] [Google Scholar]
- 132.Montero AJ, Adams B, Diaz-Montero CM, Glück S. Nab-paclitaxel in the treatment of metastatic breast cancer: a comprehensive review. Expert Rev Clin Pharmacol. 2011;4:329–34. doi: 10.1586/ecp.11.7. [DOI] [PubMed] [Google Scholar]
- 133.Shepard DR, Dreicer R, Garcia J, Elson P, Magi-Galluzzi C, Raghavan D, et al. Phase II trial of neoadjuvant nab-paclitaxel in high risk patients with prostate cancer undergoing radical prostatectomy. J Urol. 2009;181:1672–7, discussion 1677. doi: 10.1016/j.juro.2008.11.121. [DOI] [PubMed] [Google Scholar]
- 134.Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011;29:4548–54. doi: 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Infante JR, Matsubayashi H, Sato N, Tonascia J, Klein AP, Riall TA, et al. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J Clin Oncol. 2007;25:319–25. doi: 10.1200/JCO.2006.07.8824. [DOI] [PubMed] [Google Scholar]
- 136.Mantoni TS, Schendel RR, Rödel F, Niedobitek G, Al-Assar O, Masamune A, et al. Stromal SPARC expression and patient survival after chemoradiation for non-resectable pancreatic adenocarcinoma. Cancer Biol Ther. 2008;7:1806–15. doi: 10.4161/cbt.7.11.6846. [DOI] [PubMed] [Google Scholar]
- 137.Volk LD, Flister MJ, Chihade D, Desai N, Trieu V, Ran S. Synergy of nab-paclitaxel and bevacizumab in eradicating large orthotopic breast tumors and preexisting metastases. Neoplasia. 2011;13:327–38. doi: 10.1593/neo.101490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Robert N, Krekow L, Stokoe C, Clawson A, Iglesias J, O’Shaughnessy J. Adjuvant dose-dense doxorubicin plus cyclophosphamide followed by dose-dense nab-paclitaxel is safe in women with early-stage breast cancer: a pilot study. Breast Cancer Res Treat. 2011;125:115–20. doi: 10.1007/s10549-010-1187-2. [DOI] [PubMed] [Google Scholar]
- 139.Rahman M, Chan AP, Tai IT. A peptide of SPARC interferes with the interaction between caspase8 and Bcl2 to resensitize chemoresistant tumors and enhance their regression in vivo. PLoS One. 2011;6:e26390. doi: 10.1371/journal.pone.0026390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tavor S, Eisenbach M, Jacob-Hirsch J, Golan T, Petit I, Benzion K, et al. The CXCR4 antagonist AMD3100 impairs survival of human AML cells and induces their differentiation. Leukemia. 2008;22:2151–5158. doi: 10.1038/leu.2008.238. [DOI] [PubMed] [Google Scholar]
- 141.Righi E, Kashiwagi S, Yuan J, Santosuosso M, Leblanc P, Ingraham R, et al. CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovarian cancer. Cancer Res. 2011;71:5522–34. doi: 10.1158/0008-5472.CAN-10-3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rubin JB, Kung AL, Klein RS, Chan JA, Sun Y, Schmidt K, et al. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci U S A. 2003;100:13513–8. doi: 10.1073/pnas.2235846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yang L, Jackson E, Woerner BM, Perry A, Piwnica-Worms D, Rubin JB. Blocking CXCR4-mediated cyclic AMP suppression inhibits brain tumor growth in vivo. Cancer Res. 2007;67:651–8. doi: 10.1158/0008-5472.CAN-06-2762. [DOI] [PubMed] [Google Scholar]
- 144.Hassan S, Buchanan M, Jahan K, Aguilar-Mahecha A, Gaboury L, Muller WJ, et al. CXCR4 peptide antagonist inhibits primary breast tumor growth, metastasis and enhances the efficacy of anti-VEGF treatment or docetaxel in a transgenic mouse model. Int J Cancer. 2011;129:225–32. doi: 10.1002/ijc.25665. [DOI] [PubMed] [Google Scholar]
- 145.Wu X, Lee VC, Chevalier E, Hwang ST. Chemokine receptors as targets for cancer therapy. Curr Pharm Des. 2009;15:742–57. doi: 10.2174/138161209787582165. [DOI] [PubMed] [Google Scholar]
- 146.Greco SJ, Patel SA, Bryan M, Pliner LF, Banerjee D, Rameshwar P. AMD3100-mediated production of interleukin-1 from mesenchymal stem cells is key to chemosensitivity of breast cancer cells. Am J Cancer Res. 2011;1:701–15. [PMC free article] [PubMed] [Google Scholar]
- 147.Neurath MF, Finotto S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 2011;22:83–9. doi: 10.1016/j.cytogfr.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 148.Drygin D, Ho CB, Omori M, Bliesath J, Proffitt C, Rice R, et al. Protein kinase CK2 modulates IL-6 expression in inflammatory breast cancer. Biochem Biophys Res Commun. 2011;415:163–7. doi: 10.1016/j.bbrc.2011.10.046. [DOI] [PubMed] [Google Scholar]
- 149.Coward J, Kulbe H, Chakravarty P, Leader D, Vassileva V, Leinster DA, et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin Cancer Res. 2011;17:6083–96. doi: 10.1158/1078-0432.CCR-11-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Keyomarsi K, Sandoval L, Band V, Pardee AB. Synchronization of tumor and normal cells from G1 to multiple cell cycles by lovastatin. Cancer Res. 1991;51:3602–9. [PubMed] [Google Scholar]
- 151.Takeda I, Maruya S, Shirasaki T, Mizukami H, Takahata T, Myers JN, et al. Simvastatin inactivates beta1-integrin and extracellular signal-related kinase signaling and inhibits cell proliferation in head and neck squamous cell carcinoma cells. Cancer Sci. 2007;98:890–9. doi: 10.1111/j.1349-7006.2007.00471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Denoyelle C, Albanese P, Uzan G, Hong L, Vannier JP, Soria J, et al. Molecular mechanism of the anti-cancer activity of cerivastatin, an inhibitor of HMG-CoA reductase, on aggressive human breast cancer cells. Cell Signal. 2003;15:327–38. doi: 10.1016/S0898-6568(02)00124-9. [DOI] [PubMed] [Google Scholar]
- 153.Schmidmaier R, Baumann P, Simsek M, Dayyani F, Emmerich B, Meinhardt G. The HMG-CoA reductase inhibitor simvastatin overcomes cell adhesion-mediated drug resistance in multiple myeloma by geranylgeranylation of Rho protein and activation of Rho kinase. Blood. 2004;104:1825–32. doi: 10.1182/blood-2003-12-4218. [DOI] [PubMed] [Google Scholar]
- 154.Kinbara K, Goldfinger LE, Hansen M, Chou FL, Ginsberg MH. Ras GTPases: integrins’ friends or foes? Nat Rev Mol Cell Biol. 2003;4:767–76. doi: 10.1038/nrm1229. [DOI] [PubMed] [Google Scholar]
- 155.Azab AK, Azab F, Blotta S, Pitsillides CM, Thompson B, Runnels JM, et al. RhoA and Rac1 GTPases play major and differential roles in stromal cell-derived factor-1-induced cell adhesion and chemotaxis in multiple myeloma. Blood. 2009;114:619–29. doi: 10.1182/blood-2009-01-199281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kidera Y, Tsubaki M, Yamazoe Y, Shoji K, Nakamura H, Ogaki M, et al. Reduction of lung metastasis, cell invasion, and adhesion in mouse melanoma by statin-induced blockade of the Rho/Rho-associated coiled-coil-containing protein kinase pathway. J Exp Clin Cancer Res. 2010;29:127. doi: 10.1186/1756-9966-29-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8:97–106. doi: 10.1038/nrclinonc.2010.196. [DOI] [PubMed] [Google Scholar]
- 158.Bisht S, Brossart P, Maitra A, Feldmann G. Agents targeting the Hedgehog pathway for pancreatic cancer treatment. Curr Opin Investig Drugs. 2010;11:1387–98. [PubMed] [Google Scholar]
- 159.Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164–72. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 160.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–8. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Thapa RJ, Basagoudanavar SH, Nogusa S, Irrinki K, Mallilankaraman K, Slifker MJ, et al. NF-kappaB protects cells from gamma interferon-induced RIP1-dependent necroptosis. Mol Cell Biol. 2011;31:2934–46. doi: 10.1128/MCB.05445-11. [DOI] [PMC free article] [PubMed] [Google Scholar]