

Abstract
Hydrogen sulphide (H2S) exerts a protective effect in hepatic ischemia-reperfusion (I/R) injury. However, the exact mechanism of H2S action remains largely unknown. This study was designed to investigate the role of the PtdIns3K-AKT1 pathways and autophagy in the protective effect of H2S against hepatic I/R injury. Primary cultured mouse hepatocytes and livers with or without NaHS (a donor of H2S) preconditioning were exposed to anoxia/reoxygenation (A/R) and I/R, respectively. In certain groups, they were also pretreated with LY294002 (AKT1-specific inhibitor), 3-methyladenine (3MA, autophagy inhibitor) or rapamycin (autophagy enhancer), alone or simultaneously. Cell viability, expression of P-AKT1, T-AKT1, LC3 and BECN1 were examined. The severity of liver injury was measured by the levels of serum aminotransferase and inflammatory cytokine, apoptosis and histological examination. GFP-LC3 redistribution and transmission electron microscopy were used to test the activity of autophagy. H2S preconditioning activated PtdIns3K-AKT1 signaling in hepatocytes. LY294002 could abolish the AKT1 activation and attenuate the protective effect of H2S on hepatocytes A/R and hepatic I/R injuries. H2S suppressed hepatic autophagy in vitro and in vivo. Further reducing autophagy by 3MA also diminished the protective effect of H2S, while rapamycin could reverse the autophagy inhibitory effect and enhance the protective effect of H2S against hepatocytes A/R and hepatic I/R injuries, consequently. Taken together, H2S protects against hepatocytic A/R and hepatic I/R injuries, at least in part, through AKT1 activation but not autophagy. An autophagy agonist could be applied to potentiate this hepatoprotective effect by reversing the autophagy inhibition of H2S.
Keywords: hydrogen sulphide, liver, ischemia-reperfusion injury, autophagy, mouse
Introduction
Hepatic ischemia-reperfusion (I/R) injury is an important clinical problem, and usually occurs in liver transplantation, trauma, shock and elective liver resection when inflow occlusion or total vascular exclusion is used to minimize bleeding. The pathophysiology of hepatic I/R injury includes direct cellular damage resulting from the ischemic insult and delayed dysfunction and damage caused by inflammatory pathway activation. Histopathological changes such as cellular swelling, vacuolization, endothelial cell disruption, neutrophil infiltration and cellular apoptosis and necrosis were also found in hepatic I/R injury.
Hydrogen sulphide (H2S) was known as a “toxic gas.” However, it is now a novel gaseous messenger.1 It possesses important physiological and pathophysiological functions, and exerts many effects on the pathogenesis of various diseases such as hypertension, shock or myocardial ischemia reperfusion injury.2-5 Our previous study shows that H2S displays a protective role in a rat model of hepatic I/R injury through anti-apoptosis and anti-inflammatory activities.6 However, the exact mechanism of H2S-attenuated hepatic I/R injury remains largely unknown.
The PtdIns3K-AKT1 pathway controls a variety of cellular processes, including cell survival and proliferation, and modulation of this pathway may be a potential strategy in clinical settings of ischemic liver injury to decrease organ damage.7 Recently, Hu et al. have reported that activation of the PtdIns3K-AKT1 pathway is involved in the protective role of H2S preconditioning in a mouse model of cardiac ischemia-reperfusion injury.8 We hypothesized that the anti-inflammatory and anti-apoptosis activities of H2S in hepatic I/R injury may be also mediated by activation of the PtdIns3K-AKT1 pathway.
The role of autophagy in ischemic cellular damage has recently begun to be investigated with the preponderance of work coming in the realm of liver I/R models. Cardinal et al. suggested that there is a protective role of cisplatin in ischemic liver injury caused through induction of autophagy.9 Kim et al. also reported that during anoxia/reoxygenation (A/R), CAPN2/calpain 2-mediated degradation of ATG7 and BECN1 impairs mitochondrial autophagy, and this subsequently leads to MPT-dependent hepatocyte death after A/R.10 In this study, we further elucidate the role of autophagy during the treatment of H2S in hepatic I/R injury.
Therefore, this study is designed to assess the role of AKT1 and autophagy in the protective effect of H2S against hepatic I/R injury. We show that preconditioning of NaHS (a donor of H2S) can activate the PtdIns3K-AKT1 pathways and reduce the A/R or I/R-induced injury both in vitro and in vivo. In addition, we also found that H2S treatment can degrade the level of autophagy in hepatocytes after A/R and I/R injuries. Furthermore, rapamycin could reverse the autophagy inhibitory effect and consequently enhance the protective effect of H2S against A/R and I/R injuries.
Results
In vitro and in vivo hepatotoxicity of H2S
Primary cultured mouse hepatocytes were treated with escalating concentrations of NaHS for 24 h, and then a cell proliferation and cytotoxicity assay (CCK-8) was performed to assess the cell viability. NaHS concentrations of less than 50 µm were not associated with decreased cell viability, and treatments with relatively high concentrations of NaHS (100 µM) showed cytotoxicity (p < 0.05) (Fig. 1A).
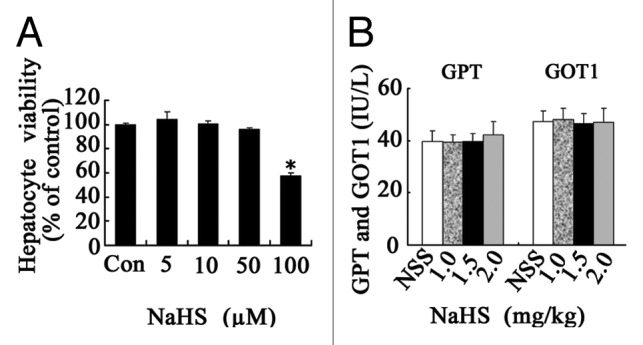
Figure 1. The hepatotoxity of H2S administration in vitro and in vivo. (A) Primary mouse hepatocytes were treated with escalating concentrations of NaHS for 24 h, and the cytotoxicity was assessed. (B) C57BL/6 male mice were given an ip injection of NSS or escalating doses of NaHS. Liver function was assessed 72 h later. Data are expressed as mean ± SD of 6 animals per group. *Significant difference in hepatocyte viability from control, p < 0.05.
C57BL/6 mice were given an IP injection of either NSS or NaHS (0, 1.0, 1.5 or 2.0 mg/kg). Liver functions were assessed 72 h after injection by measuring the levels of serum GPT [glutamic-pyruvate transaminase/alanine aminotransferase (ALT)] and GOT1 [glutamic-oxaloacetic transaminase 1, soluble/aspartate aminotransferase (AST)]. NaHS doses of up to 2 mg/kg did not raise the serum levels of GPT and GOT1 (Fig. 1B). Thus, we applied NaHS in the concentrations of 5 µM in vitro and 1.5 mg/kg in vivo for the remaining experiments.
H2S activates PtdIns3K-AKT1 signaling in hepatocytes
To determine the role of PtdIns3K-AKT1 signaling in the hepatoprotective effect of H2S, western blots for total-AKT1 (T-AKT1) and phospho-AKT1 (P-AKT1) were performed. As is shown in Figure 2A, A/R caused moderate activation of AKT1, and P-AKT1 levels increased significantly (p < 0.05) in the NaHS group compared with control, whereas pretreatment with NaHS further increased the activation of AKT1 in the A/R group (p < 0.05), and T-AKT1 remains unchanged. To extend the in vitro findings to the in vivo situation, we further investigated the expression levels of P-AKT1 in liver homogenates, which demonstrated that either NaHS or I/R could upregulate the levels of P-AKT1 in liver tissues in comparison with the sham control (p < 0.05), Administration of NaHS further increased the upregulation of P-AKT1 caused by I/R (p < 0.05) (Fig. 2B).
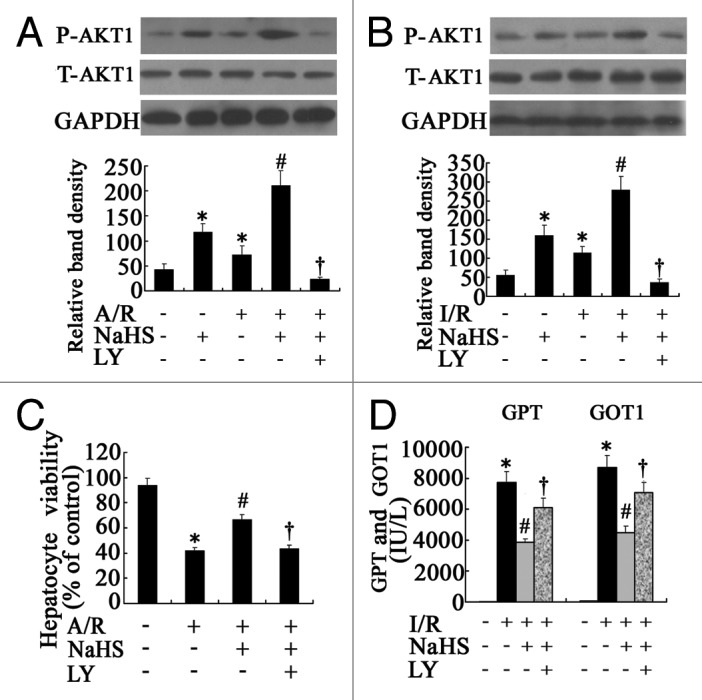
Figure 2. The effect of AKT1 in the protective effect of H2S on hepatocyte A/R and I/R injuries. (A) Representative western blots and quantitative evaluation of P-AKT1 expression of hepatocytes that were subjected to control, NaHS, A/R, NaHS+A/R and NaHS+A/R+LY at 3 h after reoxygenation; GAPDH was run as an internal standard (n = 3). (B) Representative western blots and quantitative evaluation of P-AKT1 expression in liver tissues from the mice subjected to sham-operated, NaHS, NSS+I/R, NaHS+I/R and NaHS+I/R+LY at 6 h after I/R. GAPDH was run as an internal standard (n = 3). (C) Cell viabilities of the hepatocytes that were subjected to control, A/R, NaHS+A/R and NaHS+A/R+LY were determined at 24 h after reoxygenation. The results are expressed as the mean ± SD (n = 3). #p < 0.05 compared with control, *p < 0.05 between two groups. LY, LY294002. (D) Liver function was assessed in the mice subjected to sham-operated, NSS+I/R, NaHS+I/R and NaHS+I/R+LY at 6 h after I/R; the results are expressed as the mean ± SD of 6 animals per group. *Significant difference from control, p < 0.05; #Significant difference from A/R or I/R group, p < 0.05; †Significant difference from NaHS+A/R or NaHS+I/R group, p < 0.05. LY, LY294002.
H2S protects hepatocytes against A/R and I/R injuries
Primary cultured mouse hepatocytes were treated with or without NaHS (5 µM) 1 h before A/R. As shown in Figure 2C, after 4 h of anoxia and 24 h of reoxygenation, the hepatocyte viability decreased from 94.13% ± 5.29% to 41.67% ± 2.52% (p < 0.05). However, H2S preconditioning significantly reduced the decrease in cell viability caused by A/R, from 41.67% ± 2.52% to 66.54% ± 3.42% (p < 0.05), and thus appeared to be protective.
To further confirm the hepatoprotective effect of H2S, we performed an in vivo assay using a murine hepatic I/R injury model. As is shown in Figure 2D, I/R significantly increased the levels of GOT1 and GPT 6 h after the operation in comparison with sham operated mice (p < 0.05). Administration of NaHS significantly reduced the GOT1 and GPT levels (Fig. 2D) in comparison with untreated hepatic I/R mice (p < 0.05). We further examined the histopathological changes in liver tissues. The sham operation did not show any effects on liver histology, as the H&E stained liver sections exhibited normal morphology (Fig. 3A). Histological alteration of the liver from NSS-treated I/R mice was characterized as inflammatory cell infiltration, hemorrhagic change and focal necrosis (Fig. 3A). In contrast, pretreatment of NaHS attenuated such pathological changes (Fig. 3A).

Figure 3. The histopathological changes and inflammatory cytokines production in hepatic I/R. (A) Representative photographs (40×) of H&E-stained liver sections were taken from mice subjected to sham-operated, NSS+I/R, NaHS+I/R and NaHS+I/R+LY conditions, 6 h after reperfusion (upper), and representative photographs (200 × ) of liver sections stained by TUNEL were taken from the mice in sham-operated, NSS+I/R, NaHS+I/R and NaHS+I/R+LY conditions (lower). (B) Histopathological scoring of hepatic injury was performed. (C) TUNEL-positive cells were counted as described in Materials and Methods. (D) Systemic IL6 and (E) TNF levels in sera from the blood samples of mice were assessed by ELISA. Data are expressed as mean ± SD of 6 animals per group. *Significant difference from control, p < 0.05; #Significant difference from A/R or I/R group, p < 0.05; †Significant difference from NaHS+I/R group, p < 0.05. LY, LY294002. Bar: 100 µm.
The injury scores of mouse livers in the I/R group were significantly greater than those in the sham group (p < 0.05), and NaHS significantly (p < 0.05) decreased the histological scores, in comparison with NSS-treated I/R mice (p < 0.05) (Fig. 3B). We also analyzed both apoptosis and necrosis in the collected tissues by TUNEL assay. The results of representative images and cell counting are shown in Figure 3A and C. While no TUNEL-positive cells were detected in the sham control, hepatic I/R induced a percentage of 84.23% ± 8.32%, which was decreased to 39.34% ± 5.65% by NaHS (p < 0.05) (Fig. 3C). The serum levels of inflammatory cytokines, including TNF and IL6, were measured at the same time. As shown in Figure 3D and E, hepatic I/R significantly increased the two parameters in comparison with the sham control (p < 0.05). Administration of NaHS attenuated these increases as the levels of the two parameters were significantly lower than those in untreated hepatic I/R mice (p < 0.05) (Fig. 3D and E).
AKT1 inhibition attenuates the hepatoprotective effect of H2S
To assess the relevance of the PtdIns3K-AKT1 pathway activation in the protective effect of H2S, both the cultured primary hepatocytes and mice were treated with the PtdIns3K inhibitor LY294002. As is shown in Figures 2 and 3, LY294002 administration significantly (p < 0.05) reduced the increase of AKT1 phosphorylation (Fig. 2A and B) as well as the hepatoprotective effect of H2S (Fig. 2C and D; Fig. 3) both in vitro and in vivo (p < 0.05), whereas T-AKT1 remains unchanged. The LY294002 was also used alone to set a relative control, which inhibited the PtdIns3K-AKT1 pathway without significant cytotoxicity (data not shown).
H2S suppressed hepatic autophagy in vitro and in vivo
Autophagy is a highly conserved cellular process, which can recycle long-lived and/or damaged proteins and organelles while cells are suffering from cellular stress.11 We wondered whether autophagy had been affected by NaHS treatment in vitro and in vivo. The levels of LC3, a protein that is lipidated upon activation of autophagy,12 and BECN1, a component of the PtdIns3K complex that is required for autophagy,13 were examined by western blotting analysis. As expected, both BECN1 and lipidated LC3 (LC3-II) levels were significantly decreased in NaHS-treated hepatocytes when compared with control (p < 0.05) (Fig. 4A). A/R caused moderate activation of autophagy (p < 0.05), while pretreatment of NaHS decreased the activation of autophagy in the A/R group (p < 0.05). We further detected autophagy by analyzing the formation of fluorescent puncta or autophagosomes in GFP-LC3-transfected hepatocytes. Some autophagosomes were detected, as characterized by punctate, green-fluorescing structures.14 As shown in Figure 4B, most control hepatocytes had an even and diffuse GFP-LC3 staining with occasional puncta. Similarly, there were almost no GFP-LC3 puncta in NaHS-treated hepatocytes. On A/R conditions, some cells displayed numerous unevenly distributed, cup- or ring-shaped green dots of various sizes, whereas NaHS markedly decreased the number of autophagosomes in A/R cells. The results indicated that A/R increased GFP-LC3-positive autophagosomes number from the basal level of 7.33 ± 1.53 to 15.33 ± 2.51 (p < 0.05), which was decreased to 4.33 ± 1.53 (p < 0.05) by NaHS pretreatment.

Figure 4. The activity of autophagy during the treatment of H2S in hepatic I/R in vitro and in vivo. (A) Representative western blots and quantitative evaluation of the expression of LC3-II and BECN1 in hepatocytes that were subjected to control, NaHS, A/R, NaHS+A/R, Rap+A/R, NaHS+A/R+Rap, 3MA+A/R and NaHS+A/R+3MA at 3 h after reoxygenation, with GAPDH as protein loading control (n = 3). (B) The average number of autophagosomes/cell ± SD counted from confocal microscopy images of primary mouse hepatocytes expressing GFP-LC3 in (A). Bar: 5 µm. (C) Representative western blots and quantitative evaluation of the expression of LC3-II and BECN1 in liver tissues from mice that were subjected to Sham, NaHS, I/R, NaHS+I/R, Rap+I/R, NaHS+I/R+Rap, 3MA+I/R and NaHS+I/R+3MA at 6 h after I/R. GAPDH was run as an internal standard (n = 3). (D) Representative electron micrographs showing autophagic vacuoles in liver sections from mice in (C) and the quantification of the number of autophagic vacuoles per 100 µm cytoplasm. Data are expressed as mean ± SD of 6 animals per group. Bar: 2 µm. *Significant difference from control, p < 0.05; #Significant difference from A/R or I/R group, p < 0.05; †Significant difference from single treatment of either agent, p < 0.05; ‡Significant difference from NaHS+A/R or NaHS+I/R group, p < 0.05. Rap, rapamycin; 3MA, 3-methyladenine.
To further assess autophagy activation in a murine hepatic I/R model, the expression of LC3 and BECN1 in liver tissues had been detected by western blotting. As expected in Figure 4C, NaHS significantly decreased the levels of LC3-II and BECN1 in comparison with sham-operated mice (p < 0.05), while hepatic I/R could cause an increase (p < 0.05). Administration of NaHS significantly reduced LC3-II and BECN1 levels 6 h after I/R (p < 0.05). To confirm the results of western blotting analysis, the autophagosomes and related autophagic vacuoles were detected by electron microscopy (Fig. 4D), the typical autophagosomes being characterized by double- or multiple-membrane structures containing cytoplasm or undigested organelles such as mitochondria, while the autolysosomes were identified as single- membrane structures with remnants of cytoplasmic components. The autophagic vacuoles were evaluated by morphometric methods.15 The amount of autophagic vacuoles per unit cytoplasmic area of 100 µm was evaluated. Compared with the basal level of 3.02 ± 0.81 in the sham control, fewer autophagic vacuoles (1.41 ± 0.42) (p < 0.05) were seen in NaHS-treated mice liver tissues. However, autophagic vacuoles were drastically increased following hepatic I/R, to 7.12 ± 0.82 (p < 0.05), which were decreased to 2.67 ± 0.91 (p < 0.05) by NaHS preconditioning. Thus, autophagy is suppressed by H2S in vitro and in vivo.
Autophagy activation enhanced the hepatoprotective effect of H2S
Because impaired autophagy contributes to mitochondrial dysfunction in redox-stressed hepatocytes both in vitro and in vivo,10 we wondered whether the protective effect of NaHS could be enhanced if autophagy was activated by rapamycin, an autophagy inducer. As expected, rapamycin administration increased both BECN1 and LC3-II levels, which were decreased by NaHS pretreatment in A/R cells and hepatic I/R mice (Fig. 4A and C). The GFP-LC3-positive autophagosome number was increased from 15.33 ± 2.51 to 37.66 ± 3.05 (p < 0.05) and 4.33 ± 1.53 to 27.33 ± 2.52 (p < 0.05) by rapamycin in A/R and NaHS-treated A/R cells, respectively (Fig. 4B). Rapamycin significantly increased the number of autophagic vacuoles from 7.12 ± 0.82 to 32.48 ± 3.74 (p < 0.05) and 2.67 ± 0.91 to 27.48 ± 2.63 (p < 0.05) in liver tissues of normal saline solution (NSS)-treated and NaHS-treated hepatic I/R mice, respectively (Fig. 4D).
As is shown in Figure 5A, after 4 h of anoxia and 24 h of reoxygenation, the cell viability was further increased from 40.13% ± 2.14% to 52.16% ± 3.33% (p < 0.05) and 65.34% ± 3.39% to 78.76% ± 4.11% (p < 0.05) by rapamycin in the A/R and NaHS-treated A/R cells, respectively. The severity of liver injury was also measured.

Figure 5. Evaluation of the role of autophagy in the protective effect of H2S on hepatic I/R in vitro and in vivo. (A) Cell viabilities of the hepatocytes that treated as described in Figure 4A were determined at 24 h after reoxygenation. Data are expressed as mean ± SD (n = 3). (B) The levels of GPT and GOT1 were measured in sera from the mice treated as described in Figure 4C. (C) Histopathological scoring of hepatic injury was performed. (D) Representative photographs (100×) of H&E-stained liver sections were taken from mice in (B) at 6 h after I/R(upper) and Liver sections stained by TUNEL were illustrated(lower). Histopathological scoring of hepatic injury was performed. (E) TUNEL-positive cells were counted as described in Materials and Methods. (F) Systemic IL6 and (G) TNF levels in sera from the blood samples of mice in (B) were assessed by ELISA. Data are expressed as mean ± SD of 6 animals per group. *Significant difference from control, p < 0.05; #Significant difference from A/R or I/R group, p < 0.05; †Significant difference from single treatment of either agent, p < 0.05; ‡Significant difference from NaHS+A/R or NaHS+I/R group, p < 0.05. Bar: 100 µm. Rap, rapamycin; 3MA, 3-methyladenine.
As is shown in Figure 5, both NaHS and rapamycin used alone significantly reduced the increase of GOT1 and GPT levels caused by hepatic I/R (Fig. 5B) in mice (p < 0.05). The combined NaHS+rapamycin treatment I/R group demonstrated an improved hepatoprotective effect, as compared with the monotherapy group (p < 0.05). The injury scores of mouse livers in the NSS-treated and NaHS-treated hepatic I/R mice were decreased from 13.67 ± 2.52 to 9.67 ± 2.08 (p < 0.05) and 6.33 ± 1.53 to 3.33 ± 0.58 (p < 0.05) by rapamycin administration, respectively (Fig. 5C). The pathological changes of mouse livers in NaHS-treated mice were further attenuated by rapamycin (Fig. 5D). For the TUNEL assay, rapamycin administration decreased the percentage of TUNEL-positive cells in liver tissue sections, from 78.67% ± 6.11% to 56.33% ± 7.09% (p < 0.05) and 43.67% ± 5.51% to 27.33% ± 4.04% (p < 0.05) in NSS-treated and NaHS-treated I/R mice, respectively (Fig. 5D and E). Serum obtained from rapamycin + NaHS-treated mice contained significantly less TNF and IL6 at the protein level than that obtained from monotherapy mice (p < 0.05) (Fig. 5F and G). To further evaluate the role of autophagy in the hepatoprotective effect of H2S, we applied 3-methyladenine (3MA), an autophagy inhibitor, to further decrease the activity of autophagy during the treatment with H2S in hepatic I/R.16 As is shown in Figures 4 and 5, 3MA administration significantly reduced the activity of autophagy (p < 0.05) as well as the hepatoprotective effect of H2S (p < 0.05). It is worth noticing that 3MA alone significantly aggravated the severity of hepatocyte A/R and hepatic I/R injuries. The rapamycin and 3MA were also used alone under normoxia or in a sham group to set the relative control, which showed activation or inhibition of autophagy without significant cytotoxicity (data not shown).
Discussion
Recently, several research groups, including ours, have reported that H2S displays anti-inflammatory and cytoprotective activities in various I/R injuries.4,17 However, the underlying mechanism remains largely unknown.
PtdIns3K-AKT1 is one of the most potent cell survival signaling pathways and has been shown to be involved in the ischemic tolerance seen in heart and in neurons.18-21 It has been shown that the PtdIns3K-AKT1-RPS6KB2/p70S6 kinase pathway promotes cell survival against oxidative stress-induced apoptosis in H9c2 cells.18 Yano et al. suggest that AKT1 activation is induced by a sublethal ischemic insult in gerbil hippocampus and contributes to neuroprotective ischemic tolerance in CA1 pyramidal neurons.19 Takatani et al. reported that taurine prevents ischemia-induced apoptosis in cardiomyocytes through an AKT1-mediated CASP9 inactivation.20 Jonassen and coworkers reported that insulin administration at reperfusion reduces myocardial infarction, and that this is mediated via AKT1- and RPS6KB2-dependent signaling pathways.21
A recent study reported that H2S preconditioning produces cardioprotective effects against ischemia in rat cardiac myocytes by activating the PtdIns3K-AKT1 pathway.8 In addition, studies have also demonstrated a protective role of AKT1 activation against ischemic injury in the liver.22,23 Izuishi et al. reported that the PtdIns3K-AKT1 pathway plays an essential role in the protective effects of ischemic preconditioning in hepatic I/R injury, and modulation of this pathway may be a potential strategy in clinical settings of ischemic liver injury to decrease organ damage.24 Therefore, the PtdIns3K signaling cascade may contribute to the recruitment of multiple endogenous protective pathways to reduce tissue damage and inflammatory cytokines production after ischemia and reperfusion.25,26 However, whether the activation of AKT1 is the key mechanism of the protection conferred by H2S preconditioning in liver I/R injury remains unclear. Therefore, the aim of our study was to determine if the PtdIns3K-AKT1 pathway mediates the protective effects of H2S preconditioning in the liver. In the current study, we found that (1) H2S preconditioning resulted in increased AKT1 activation in hepatocytes in vitro and in vivo (Fig. 2); (2) the protective effects of H2S preconditioning were abolished with AKT1 inhibition in vitro and in vivo (Figs. 2 and 3). These findings reveal that the hepatoprotective effect of H2S against hepatic I/R injury depends, at least in part, on the activation of the PtdIns3K-AKT1 pathway.
Recently, many studies have shown that autophagy is upregulated during hepatic IR injury.10 Autophagy is an essential cellular process that mediates continuous recycling of intracellular components. As a response to stress conditions such as I/R, autophagy can digest cytoplasmic materials to generate essential metabolic substrates and energy to keep the cells alive.27-31 There are profound interactions between autophagy and apoptosis: apoptosis is inhibited when autophagy is activated, whereas inhibition of autophagy can promote cell death and the activity of caspase proteins.32,33 Recent evidence supports the view that enhancing autophagy may be a novel approach to improve hepatocyte viability and function after I/R injury, and such a hepatoprotective role of autophagy may be associated with its anti-apoptotic and anti-inflammatory activity.9 In the present study, we found that: (1)Autophagy was inhibited by H2S preconditioning in hepatocytes in vitro and in vivo (Fig. 4); (2) rapamycin alone afforded protection during hepatocyte A/R and hepatic I/R, and it could reverse the autophagy inhibition of H2S. The combined NaHS+rapamycin treatment group contributes an improved hepatoprotective effect, which is significantly different from the monotherapy group; (3) Further inhibition of autophagy by 3MA reduced the protective effect of H2S during hepatocyte A/R and hepatic I/R injuries (Figs. 4 and 5). These results suggest that autophagy is also a key mediator in the hepatoprotective effect of H2S.
Interestingly, a recent paper has shown that in three different in vitro models of insulin-induced necrotic cell death, activation of PtdIns3K-AKT1 signaling can promote necrotic cell death via suppression of autophagy.34 One of the key regulators of autophagy is the mechanistic target of rapamycin, MTOR kinase, which is the major inhibitory signal that shuts off autophagy in the presence of growth factors and abundant nutrients.35 The class I PtdIns3K-AKT1 signaling molecules link receptor tyrosine kinases to MTOR activation.36 Thereby, the activation of the PtdIns3K-AKT1 pathway may work as a double-edged sword; it plays a protective role against hepatic I/R, but on the other hand, it also inhibits autophagy, which is another important protective mechanism against hepatic I/R. This may be applied to interpret our findings that although autophagy was inhibited by H2S, H2S still showed a protective role against I/R. So, reversing the autophagy inhibitory effect of H2S by rapamycin could enhance its protective effect against hepatocyte A/R and hepatic I/R injury.
In summary, our findings demonstrate that H2S protects against hepatocyte A/R and hepatic I/R injuries, at least in part, through AKT1 activation but not autophagy. The autophagy agonist rapamycin could be applied to potentiate this hepatoprotective effect by reversing the autophagy inhibition of H2S. Therefore, enhancing autophagy may be a promising strategy to improve the hepatoprotective ability of H2S in hepatic I/R injury.
Materials and Methods
Chemicals and antibodies
The following reagents were purchased from Sigma-Aldrich: NaHS (161527), LY294002 (L9908), rapamycin (R0395), 3-MA (M9281) and sodium pentobarbital (P3761). All culture media were purchased from HyClone China Ltd: DMEM (HyClone, SH30022. 01B), fetal bovine serum (GIBCO, 16000). The following antibodies were purchased from Cell Signaling Technology: P-AKT1 (Ser473, 9271), T-AKT1 (Ser473, 9272), BECN1 (3738), LC3 (2775) and GAPDH (2118). Both LY294002 and rapamycin were dissolved in DMSO to make a stock solution of 50 mM.
Animals
Male C57BL/6 (18–20 g) mice were supplied by the Animal Research Center at the First Clinical Medical School of Harbin Medical University (Harbin, China). All surgical procedures and care administered to the animals were approved by the institutional ethic committee, and this study also complied with the criteria in Guide for the Care and Use of Laboratory Animals.37
Hepatocyte isolation
Mouse hepatocytes were isolated by a modified in situ collagenase perfusion technique as described.9 Hepatocyte purity and viability typically exceeded 99% and 95%, respectively.
A/R and Cell viability assay
To simulate tissue I/R, hepatocytes were treated as described previously.10 Some hepatocytes were pretreated with NaHS (5, 10, 50, 100 µM) alone, or together with LY294002 (25 µM) or rapamycin (1 µM) or 3MA (10 mM), respectively, before A/R. Twenty-four h after A/R, the cell viability was measured with Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, CK04–13) according to the instructions of the manufacturer. The experiments were repeated three times.
Analysis of autophagy by GFP-LC3 redistribution
To monitor the formation of GFP-LC3 puncta, primary hepatocytes were transiently transfected with 1.0 µg GFP-LC3 plasmid and then treated as described above. After treatment, the number of autophagosomes/cell was recorded for quantification as described previously.38
Hepatic I/R model
The procedures of hepatic I/R have been described previously.9 Briefly, the mice were anesthetized by intraperitoneal injection of sodium pentobarbital (60 mg/kg), and a midline laparotomy was performed. Then, the left lateral and median lobes of the liver were clamped at its base using an atraumatic clip. Throughout anesthesia, body temperature was monitored by a rectal probe and maintained at 37°C by a heating lamp. After 90 min of ischemia, the clip was removed, initiating hepatic reperfusion.
In vivo design
The mice in hepatic I/R group and NaHS+I/R group were given an intraperitoneal injection(ip) of NSS and NaHS (1.5 mg/kg) 1 h before the onset of liver I/R, respectively; a NaHS+I/R+LY294002 group in which mice were given a tail vein injection of LY294002 (1.5 mg/kg) 0.5 h before the administration of NaHS and subsequent liver I/R; an I/R+NaHS+rapamycin group in which rapamycin (1 mg/kg, ip) were given 0.5 h before the administration of NaHS and subsequent liver I/R; an I/R+NaHS+3MA group in which 3MA (30 mg/kg, ip) was given 0.5 h before the administration of NaHS and subsequent liver I/R. Sham operations (control) involved administration of anesthesia, laparotomy and exposure of the portal triad without hepatic ischemia. Mice were sacrificed at 6 h after reperfusion for serum and liver samples.
Measurement of parameters in sera
The levels of GPT and GOT1 in sera were measured with an autobiochemical analyzer (Toshiba, TBA-200FR), as described previously.39 The serum levels of TNF (TNFα) and IL6 (interleukin 6) were measured with enzyme-linked immunosorbent assay (ELISA) kits: TNF (R&D Systems, MTA00B) and IL6 (R&D Systems, M6000B) according to the manufacturer’s instructions.
Histological examination
Liver specimens were fixed in 10% buffered formalin, embedded in paraffin, stained with hematoxylin and eosin (H&E), and examined with a light microscope. The histopathological scoring analysis was performed blindly according to previously described methods.6
Western blotting analysis
Protein lysates of liver or primary hepatocytes were prepared, separated onto SDS-polyacrylamide gels and transferred to PVDF membrane as previously described.6 western blotting was performed using appropriate primary antibodies and horseradish peroxidase-conjugated suitable secondary antibodies, followed by detection with enhanced chemiluminescence (Pierce Chemical, 34080). GAPDH was used as protein loading control, and the levels of proteins were normalized with respect to GAPDH band density.
Transmission electron microscopy
Mice were treated as described above and after 6 h of reperfusion, laparotomy was performed under isoflurane anesthesia. The liver was flushed with 1 ml NSS, then perfused with 2 ml 2.5% glutaraldehyde in PBS. Livers were sectioned and photographed using a transmission electron microscope (JEOL, JEM 1210) at 80 or 60 kV onto electron microscope film (Kodak, ESTAR thick base) and printed onto photographic paper. For quantification, 20 to 30 fields of low magnification (×1000) were randomly selected from each liver, and digital images with scale bars were taken. Using Axio-Vision 4.0 software, the amount of autophagic vacuoles per unit cytoplasmic area of 100 µm was evaluated.
Terminal dUTP Nick-End Labeling (TUNEL) assay
The methodology has been described previously.6 The TUNEL (Roche, 11684795910) staining of sections was performed according to manufacturer’s instructions, and the TUNEL-positive cells were counted in 10 randomly selected ×400 high-power fields under microscopy and expressed as a percentage of the total hepatocytes.
Statistics analysis
Qualitative data including immunoblots and cell images are representatives of at least three experiments. Quantitative data were expressed as means ± SD. Statistical differences in multiple groups were determined by multiple comparisons with analysis of variance followed by Tukey’s post-tests. Statistical differences between two groups were determined by two-tailed unpaired Student’s t-test. p < 0.05 was considered significantly different.
Acknowledgments
We thank Dr. Noboru Mizushima (The Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan) for the LC3 cDNA. We also thank Dr. Shengkan Jin at UMDNJ-Robert Wood Johnson Medical School for technical help and suggestions. This work was supported in part by grants from the National Natural Scientific Foundation of China (81100305 to Y.M.; 30972938 to H.C.J.).
Glossary
Abbreviations:
- I/R
ischemia-reperfusion
- H2S
hydrogen sulphide
- A/R
anoxia/reoxygenation
- NaHS
sodium hydrosulfide
- GPT
glutamic-pyruvate transaminase/alanine aminotransferase
- GOT1
glutamic-oxaloacetic transaminase 1, soluble/aspartate aminotransferase
- TNF
tumor necrosis factor(-α)
- IL6
interleukin 6
- PtdIns3K
phosphatidylinositol 3-kinase
- LC3
microtubule-associated protein 1 light chain 3
- 3MA
3-methyladenine
- TUNEL
TdT-mediated dUTP nick-end labeling
- ELISA
enzyme-linked immunosorbent assay
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/19927
References
- 1.Guidotti TL. Hydrogen sulphide. Occup Med (Lond) 1996;46:367–71. doi: 10.1093/occmed/46.5.367. [DOI] [PubMed] [Google Scholar]
- 2.Yan H, Du J, Tang C. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem Biophys Res Commun. 2004;313:22–7. doi: 10.1016/j.bbrc.2003.11.081. [DOI] [PubMed] [Google Scholar]
- 3.Mok YY, Atan MS, Yoke Ping C, Zhong Jing W, Bhatia M, Moochhala S, et al. Role of hydrogen sulphide in haemorrhagic shock in the rat: protective effect of inhibitors of hydrogen sulphide biosynthesis. Br J Pharmacol. 2004;143:881–9. doi: 10.1038/sj.bjp.0706014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A. 2007;104:15560–5. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geng B, Chang L, Pan C, Qi Y, Zhao J, Pang Y, et al. Endogenous hydrogen sulfide regulation of myocardial injury induced by isoproterenol. Biochem Biophys Res Commun. 2004;318:756–63. doi: 10.1016/j.bbrc.2004.04.094. [DOI] [PubMed] [Google Scholar]
- 6.Kang K, Zhao M, Jiang H, Tan G, Pan S, Sun X. Role of hydrogen sulfide in hepatic ischemia-reperfusion-induced injury in rats. Liver Transpl. 2009;15:1306–14. doi: 10.1002/lt.21810. [DOI] [PubMed] [Google Scholar]
- 7.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–7. doi: 10.1016/S0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 8.Hu Y, Chen X, Pan TT, Neo KL, Lee SW, Khin ES, et al. Cardioprotection induced by hydrogen sulfide preconditioning involves activation of ERK and PI3K/Akt pathways. Pflugers Arch. 2008;455:607–16. doi: 10.1007/s00424-007-0321-4. [DOI] [PubMed] [Google Scholar]
- 9.Cardinal J, Pan P, Dhupar R, Ross M, Nakao A, Lotze M, et al. Cisplatin prevents high mobility group box 1 release and is protective in a murine model of hepatic ischemia/reperfusion injury. Hepatology. 2009;50:565–74. doi: 10.1002/hep.23021. [DOI] [PubMed] [Google Scholar]
- 10.Kim JS, Nitta T, Mohuczy D, O’Malley KA, Moldawer LL, Dunn WA, Jr., et al. Impaired autophagy: A mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology. 2008;47:1725–36. doi: 10.1002/hep.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001;2:330–5. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395–8. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 15.Tasdemir E, Galluzzi L, Maiuri MC, Criollo A, Vitale I, Hangen E, et al. Methods for assessing autophagy and autophagic cell death. Methods Mol Biol. 2008;445:29–76. doi: 10.1007/978-1-59745-157-4_3. [DOI] [PubMed] [Google Scholar]
- 16.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–92. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jha S, Calvert JW, Duranski MR, Ramachandran A, Lefer DJ. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol. 2008;295:H801–6. doi: 10.1152/ajpheart.00377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong F, Kwon SJ, Jhun BS, Kim SS, Ha J, Kim SJ, et al. Insulin-like growth factor-1 protects H9c2 cardiac myoblasts from oxidative stress-induced apoptosis via phosphatidylinositol 3-kinase and extracellular signal-regulated kinase pathways. Life Sci. 2001;68:1095–105. doi: 10.1016/S0024-3205(00)01012-2. [DOI] [PubMed] [Google Scholar]
- 19.Yano S, Morioka M, Fukunaga K, Kawano T, Hara T, Kai Y, et al. Activation of Akt/protein kinase B contributes to induction of ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab. 2001;21:351–60. doi: 10.1097/00004647-200104000-00004. [DOI] [PubMed] [Google Scholar]
- 20.Takatani T, Takahashi K, Uozumi Y, Matsuda T, Ito T, Schaffer SW, et al. Taurine prevents the ischemia-induced apoptosis in cultured neonatal rat cardiomyocytes through Akt/caspase-9 pathway. Biochem Biophys Res Commun. 2004;316:484–9. doi: 10.1016/j.bbrc.2004.02.066. [DOI] [PubMed] [Google Scholar]
- 21.Jonassen AK, Sack MN, Mjøs OD, Yellon DM. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ Res. 2001;89:1191–8. doi: 10.1161/hh2401.101385. [DOI] [PubMed] [Google Scholar]
- 22.Müller C, Dünschede F, Koch E, Vollmar AM, Kiemer AK. Alpha-lipoic acid preconditioning reduces ischemia-reperfusion injury of the rat liver via the PI3-kinase/Akt pathway. Am J Physiol Gastrointest Liver Physiol. 2003;285:G769–78. doi: 10.1152/ajpgi.00009.2003. [DOI] [PubMed] [Google Scholar]
- 23.Carini R, Grazia De Cesaris M, Splendore R, Baldanzi G, Nitti MP, Alchera E, et al. Role of phosphatidylinositol 3-kinase in the development of hepatocyte preconditioning. Gastroenterology. 2004;127:914–23. doi: 10.1053/j.gastro.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 24.Izuishi K, Tsung A, Hossain MA, Fujiwara M, Wakabayashi H, Masaki T, et al. Ischemic preconditioning of the murine liver protects through the Akt kinase pathway. Hepatology. 2006;44:573–80. doi: 10.1002/hep.21298. [DOI] [PubMed] [Google Scholar]
- 25.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 26.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–5. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 27.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lieberthal W. Macroautophagy: a mechanism for mediating cell death or for promoting cell survival? Kidney Int. 2008;74:555–7. doi: 10.1038/ki.2008.325. [DOI] [PubMed] [Google Scholar]
- 30.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–73. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 31.Sadoshima J. The role of autophagy during ischemia/reperfusion. Autophagy. 2008;4:402–3. doi: 10.4161/auto.5924. [DOI] [PubMed] [Google Scholar]
- 32.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 33.González-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquère S, et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- 34.Jiang H, Meng F, Li W, Tong L, Qiao H, Sun X. Splenectomy ameliorates acute multiple organ damage induced by liver warm ischemia reperfusion in rats. Surgery. 2007;141:32–40. doi: 10.1016/j.surg.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 35.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–95. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guide for the Care and Use of Laboratory Animals. Bethesda, MD: National Institutes of Health; 1985. [Google Scholar]
- 38.Farkas T, Høyer-Hansen M, Jäättelä M. Identification of novel autophagy regulators by a luciferase-based assay for the kinetics of autophagic flux. Autophagy. 2009;5:1018–25. doi: 10.4161/auto.5.7.9443. [DOI] [PubMed] [Google Scholar]
- 39.Zhang F, Tong L, Qiao H, Dong X, Qiao G, Jiang H, et al. Taurine attenuates multiple organ injury induced by intestinal ischemia reperfusion in rats. J Surg Res. 2008;149:101–9. doi: 10.1016/j.jss.2007.12.781. [DOI] [PubMed] [Google Scholar]