

Abstract
AID/APOBEC family cytosine deaminases, known to function in diverse cellular processes from antibody diversification to mRNA editing, have also been implicated in DNA demethylation, an important process for transcriptional activation. While oxidation-dependent pathways for demethylation have been described, pathways involving deamination of either 5-methylcytosine (mC) or 5-hydroxymethylcytosine (hmC) have emerged as alternatives. Here, we have addressed the biochemical plausibility of deamination-coupled demethylation. We found that purified AID/APOBECs have substantially reduced activity on mC relative to cytosine, their canonical substrate, and no detectable deamination of hmC. This finding was explained by the reactivity of a series of modified substrates, where steric bulk was increasingly detrimental to deamination. Further, upon AID/APOBEC overexpression, the deamination product of hmC was undetectable in genomic DNA, while oxidation intermediates remained detectable. Our results indicate that the steric requirements for cytosine deamination are one intrinsic barrier to the proposed function of deaminases in DNA demethylation.
AID/APOBEC enzymes are well characterized for their ability to deaminate cytosine to uracil. In various cellular settings, this modification alters and expands the genome’s coding potential1, 2. In the immunoglobulin locus of the maturing B cell, activation-induced deaminase (AID) deaminates cytosine to trigger pathways that facilitate antibody affinity maturation or isotype switching. Similarly, APOBEC3 enzymes extensively hypermutate reverse-transcribed DNA as a powerful innate defense mechanism to antagonize retroviruses and retroelements. Finally, APOBEC1 deaminates cytosine to introduce a stop codon in the mRNA of an apolipoprotein, generating a protein variant that differentially influences lipid metabolism. In each of these canonical functions, the purposeful mutations achieved by AID/APOBEC enzymes are generated by the deamination of a canonical cytosine nucleobase in RNA or DNA (Fig. 1). However, the role of modified cytosine bases in epigenetic regulation of gene expression raises the pressing question as to whether these enzymes can also act on non-canonical 5-substituted cytosines1, 3, 4.
Figure 1. Proposed non-canonical role for AID/APOBEC enzymes acting on modified cytosine substrates in DNA.
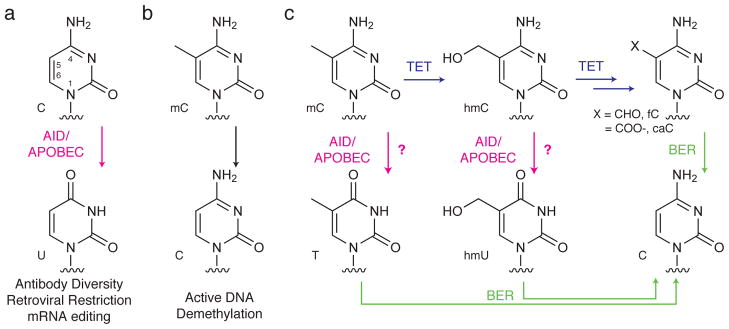
(a) Deamination of cytosine plays known physiological roles in adaptive immunity (AID), innate immunity against retroviruses (APOBEC3 enzymes), and mRNA editing (APOBEC1). These canonical roles involve deamination of cytosine to generate uracil. (b) By contrast, the proposed function of AID/APOBEC family members on modified cytosine residues remains poorly understood despite their implication in potential pathways for active DNA demethylation. (c) Proposed pathways for DNA demethylation. Deamination of 5-methylcytosine (mC) or 5-hydroxymethylcytosine (hmC), the product of TET-mediated oxidation, could generate thymidine or 5-hydroxymethyluracil (hmU), respectively. Base excision repair (BER) could subsequently excise the deaminated bases and replace them with unmodified cytosine. An alternative deamination-independent pathway involves iterative oxidation, generating 5-formylcytosine (fC) or 5-carboxylcytosine (caC). BER-mediated excision of the oxidized cytosine would result in reversion to unmodified cytosine.
The best understood cytosine modification is 5-methylcytosine (mC), which exerts transcriptionally repressive effects that are integral to such processes as genomic imprinting, X-chromosome inactivation, cellular differentiation and response to signaling stimuli5. This epigenetic modification is achieved through the action of DNA methyltransferase enzymes in mammalian cells, which introduce and maintain the methylation mark on the C5-position of cytosine bases within CpG motifs. An added layer of chemical complexity has emerged with the recent observation that mC is oxidized to 5-hydroxymethylcytosine (hmC) by the TET family of Fe(II)/α-ketoglutarate dependent oxygenase enzymes6, 7, though the consequences of hmC on gene expression are only starting to be elucidated8–10.
To achieve DNA demethylation – an important process for reversing cytosine methylation and restoring transcription – cytosine deamination, oxidation, and base excision repair (BER) have been invoked in a variety of possible combinations (Fig. 1)1, 11, 12. In this regard, two types of deamination-dependent mechanisms have been postulated. In one scenario, deamination of mC by an AID/APOBEC enzyme generates a T:G mismatch leading to subsequent repair by the BER enzyme thymidine DNA glycosylase (TDG)13. In the other scenario, deamination of hmC generates 5-hydroxymethyluracil (hmU), which could also be reverted to cytosine by BER14. Recent studies have also demonstrated the feasibility of a deamination-independent pathway for DNA demethylation involving oxidation of mC by TET enzymes. The product of this oxidation, hmC, undergoes iterative oxidation to yield both 5-formylcytosine (fC) and 5-carboxylcytosine (caC)15, 16. These higher oxidation products are detectable in the genome of embryonic stem cells and are good substrates for excision by TDG, which could ultimately regenerate unmodified cytosine15–19. Notably, deficiency in TDG, a potential common mediator in the various proposed pathways for DNA demethylation, is associated with developmental methylation defects and embryonic lethality20, 21.
The plausibility of deamination-dependent demethylation has been difficult to establish because of the poorly characterized activities of AID/APOBEC enzymes on C5-modified cytosines and a lack of knowledge about the functional redundancy between AID/APOBEC family members22. Although prior studies suggest that AID can deaminate mC at reduced levels relative to cytosine13, 23, other work proposes that the enzyme lacks any mC deaminase activity24. Additional ambiguity arises because the activities of other APOBEC enzymes on mC have not been directly investigated, and the biochemical activities of all AID/APOBECs against hmC remain entirely unknown.
In general, the presence of numerous AID/APOBEC family members presents a challenge to sorting out their potential roles in demethylation. A potential role for AID in demethylation of pluripotency promoters arises from heterokaryon-based systems for the generation of stem cells25, and the suggestion that AID deficiency perturbs the DNA methylome of primordial germ cells26. However, these observations are confounded by the finding that AID deficiency is viable 27, suggesting that other deaminases might serve functionally redundant roles in demethylation. In support of this proposal, APOBEC2 enzymes are postulated to play a role in zebrafish DNA demethylation28 and APOBEC1 is implicated in neuronal DNA demethylation14. Thus, biochemical characterization of the similarities and differences between these deaminases would address the functional redundancy of these enzymes in DNA demethylation.
The previous implications that deaminases might be involved in DNA demethylation1, 3, 4 make it important to examine their activity on 5-substituted cytosine bases in DNA. Here, we have elucidated the substrate preferences of AID/APOBEC family members and examined the plausibility of deamination-dependent versus oxidation-dependent pathways for cytosine demethylation. We found that all AID/APOBEC family members preferentially deaminate unmodified cytosine and strongly discriminate against 5-substituted cytosine substrates with increasing steric bulk. We also observed these preferences in cells, where the deamination products of the bulky base hmC are not detected when deaminases are overexpressed. Further, intermediates in the iterative oxidation pathway were readily found in genomic DNA, and their formation was not diminished by the overexpression of deaminases. We argue that, relative to the oxidation-mediated pathway, the potential for function of deaminases in DNA demethylation is limited.
Results
AID/APOBECs preferentially deaminate unmodified cytosine
We wished to profile the reactivity of representative AID/APOBEC family members with modified cytosine nucleobases. We chose to investigate the mouse enzyme family, which possesses only a single gene for each family member (Supplementary Results, Supplementary Fig. 1a), rather than the human family, where extensive gene duplication and specialization at the A3 locus have generated at least seven A3A-A3H variants. Mouse APOBEC1 (mA1), APOBEC2 (mA2), APOBEC3 (mA3) and AID (mAID) were generated as N-terminal maltose binding protein fusion constructs. Although mAID was inactive under these conditions (data not shown), we had previously expressed and characterized active human AID (hAID) by co-expression of the enzyme with the chaperone Trigger Factor in E. coli29. Using this expression system, the expanded cohort of AID/APOBEC enzymes were all soluble and were partially purified over amylose resin (Supplementary Methods, Supplementary Fig. 1b).
We designed DNA oligonucleotides containing a single cytosine residue with several criteria in mind. First, since each AID/APOBEC family member prefers to deaminate cytosine in a different trinucleotide sequence context29–31, we selected a universal sequence that would be acted upon by multiple family members (S30-TGC). Next, a guanine was introduced directly downstream of the cytosine to create a CpG motif, an important consideration given that epigenetic modifications via methylation, hydroxymethylation and demethylation are highly linked to CpG sites and islands in the mammalian genome. All oligonucleotides were synthesized using standard phosphoramidite chemistry (Supplementary Methods).
AID/APOBEC family members were assayed against the cytosine-containing substrate S30-TGC using a discontinuous, uracil DNA glycosylase(UDG)-coupled assay (Fig. 2a). At the end of the deamination period, the reaction product was hybridized to a complementary strand, yielding duplexed DNA containing a U:G mismatch in the deaminated product. Treatment with UDG generated abasic sites in the deaminated oligonucleotides, while leaving unreacted substrates intact. Cleavage of the abasic sites under alkaline conditions allowed for specific detection of product after separation on a denaturing gel. Under these conditions the mA1, mA3 and hAID variants all showed deaminase activity against S30-TGC (Fig. 2b and Supplementary Fig. 2). As anticipated from prior studies mA2 showed no detectable cytosine deaminase activity32.
Figure 2. AID/APOBEC enzymes preferentially deaminate unmodified cytosine.

(a) Fluorophore(FAM)-labeled oligonucleotides (S30) synthesized with a single internal modified cytosine (red) embedded in a CpG motif were incubated with AID/APOBEC family members. After incubation, oligonucleotides were duplexed with a complementary strand generating U:G, T:G, or hmU:G mismatches with the deaminated substrates (blue).Treatment with UDG (reactive with U:G), TDG (reactive with T:G) or SMUG (reactive with hmU:G), respectively, followed by base-mediated cleavage fragments the deaminated products to a 15-mer (P15). (b) The reaction products resulting from incubation of S30-TGX substrates with mA1, mA2, mA3 or hAID are shown separated on a denaturing gel. The substrate and product controls, without incubation with AID/APOBEC enzymes, are shown on the left. Gels are shown without cropping in Supplementary Figure 2. (c) The fraction of deaminated substrates are plotted as a function of increasing concentrations of enzyme for mA1 and hAID-ΔC assayed against substrates containing a cytosine (magenta), mC (blue) or hmC (green). hAID-ΔC was incubated with the S30-TGX series of substrates, while mA1 was incubated with the S30-ATX substrates. Error bars represent standard deviation from the mean of at least three independent replicates. For relative comparison of substrates, the slope of each plot in the region where product formation is linear with enzyme (dashed line) is listed. The standard error for the measurement of enzyme dependent product formation is ±20% of the reported value for all measurements. The values given for hmC substrates are the lower limits of detection with these substrates.
To next examine the deamination activities on physiologically relevant 5-modified cytosines, we synthesized S30-TGmC and S30-TGhmC. Upon deamination, these target bases would convert to T and hmU, respectively. Alternate DNA glycosylases were used to detect these species: TDG, which excises thymidine from T:G mismatches in CpG contexts, and human SMUG1 (SMUG), which excises hmU mispaired to G (Fig. 2a). We established that these glycosylase-coupled assays accurately detect low levels of deamination products (as little as 0.5% product under the condition of our deamination reaction) (Supplementary Fig. 3). Using the TDG-coupled assay, deamination of S30-TGmC was detectable with all active AID/APOBEC enzymes, though product formation was decreased relative to S30-TGC (Fig. 2b). By contrast, no detectable deamination activity was evident against S30-TGhmC, despite robust deamination of S30-TGC under identical conditions with mA1, mA3 and hAID. mA2 was also inactive against both S30-TGmC and S30-TGhmC in vitro.
To assure that the discrimination against 5-substituted cytosine bases was not simply limited to a single sequence context, or perhaps the single enzyme concentration used in the above studies, we performed additional investigations. First, we tested a series of substrates containing C, mC and hmC in an ATX trinucleotide context, which is a preferred sequence for mA1. As expected, reaction of S30-ATC with mA1 led to robust deamination, while S30-ATmC was compromised and deamination of S30-AThmC was undetectable (Fig, 2c). With varying concentrations of mA1, deamination was linearly dependent on the amount of enzyme used. mA1 was estimated to have ~10-fold discrimination against mC relative to unmodified C, and >300-fold discrimination against hmC based on our detection limits. We conducted similar enzyme-dependent analysis of deamination of S30-TGX substrates with a C-terminal truncation variant of hAID (hAID-ΔC), which is associated with hyperactive deamination (~3-fold) without impacting sequence-dependent targeting29. We reasoned that low-level deamination might be easier to detect with this hyperactive variant. As with mA1, a similar discrimination against mC (~16-fold) and significant discrimination against hmC (>150-fold) was found (Fig. 2c). A quantitatively similar pattern can be observed with full-length hAID (Supplementary Fig. 4). Finally, mA3 also demonstrated clear discrimination against the naturally modified cytosine nucleobases (Supplementary Fig. 4). We conclude that discrimination against 5-substituted cytosines is an intrinsic property evident in the entire AID/APOBEC enzyme family regardless of the source organism or canonical function.
Deamination decreases with increasing steric bulk at C5
The molecular basis for recognition of cytosine by AID/APOBEC enzymes is a matter of speculation given the lack of structural information on these enzymes complexed with nucleic acid substrates. To probe the molecular impact of substitution at the 5-position of cytosine, we synthesized additional substrates with unnatural 5-substituents of varied steric and electronic character (Fig. 3a) using a sequence context appropriate for hAID (S30-TGX) or mA1 (S30-ATX). One potential determinant of reactivity in this series is the electron withdrawing ability of the C5-substituents. Electronegative C5 groups could potentially enhance deamination by making C4 of cytosine more electrophilic or by lowering the pKa of N3. Alternatively, hydrophobicity of the 5-position substituent could influence selectivity33. Finally, the size of the substituent at the 5-position could dominate the rate effect.
Figure 3. DNA deamination decreases as a function of increasing steric bulk at the 5-position of cytosine.

(a) Schematic of the deamination assay with DNA oligonucleotides containing unnatural modifications at C5. (b) At left, the denaturing gel for hAID-ΔC with S30-TGX substrate series is shown. At right, the denaturing gel with mAPOBEC1 assayed against S30-ATX substrates. Samples were run in order of increasing steric bulk of the 5-substituent. Gels are shown without cropping in Supplementary Figure 7 (c) Shown are the electrostatic potential maps of each of the modified cytosine bases as determined using the SPARTAN program (6–31G* basis set). The electrostatic potential is colored from maximal negative (red) to positive (blue). The volume (*) is determined based on linking the 5-position substituent to a single hydrogen atom and calculating the total volume. The hydrophobic substituent constant (**) is derived from partitioning studies of substituted benzenes between octanol and water, where negative values for hydroxyl and hydroxymethyl substituents represent less hydrophobicity 33. Reported are the values for enzyme dependent product formation (nM product/μM enzyme) for each substrate examined with each active deaminases (†). The data for C, mC and hmC deamination are consolidated from Fig. 2c and Supplementary Fig. 4; data for unnaturally modified substrates are consolidated from Supplementary Fig. 8. The relative activity for each modified substrate when compared to unmodified cytosine for each deaminase is reported parenthetically, allowing for comparisons across a row with each enzyme.
In order to study deamination of these unnatural cytosines, we first determined the specificity of UDG, TDG and SMUG against DNA substrates containing either the modified cytosine or the corresponding 5-substituted uracil. Based on the determined substrate preferences, we selected UDG to assay the 5-fluoro substrates and SMUG for the 5-hydroxy, 5-bromo and 5-iodo substrates (Supplementary Fig. 5). We also constructed standard curves to allow for accurate quantification of deamination of each unnatural 5-modified substrate using our glycosylase-coupled assay (Supplementary Fig. 6).
Each 5-substituted S30-TGX substrate was incubated with hAID-ΔC, then duplexed and treated with the appropriate glycosylase to assay for deamination. For hAID-ΔC, any substitution resulted in decreased efficiency of deamination relative to unmodified cytosine (Fig. 3b and Supplementary Fig, 7). To allow for relative comparison of substrates, we next calculated the product formation under conditions where deamination was linearly proportional to enzyme concentration. Across these series of substrates, representing a >150-fold difference in reactivity, the size of the substituent at the 5-position appeared to be an important determinant of deamination (Fig. 3c and Supplementary Fig. 8). The smallest unnatural substituent, S30-TG(5F)C, was deaminated most readily, although it remained half as reactive as unmodified cytosine. Bulkier halogen substituents were relatively poor substrates compared to the smaller mC. In addition to the influence of sterics, the poor hydrophobic character of hmC may play an additional role in the reactivity decrease seen between (5I)C, which has detectable deamination, and hmC, which has no detectable deamination.
To assess the generality of reactivity determinants in the deaminase family, we additionally profiled mA1 against a series of unnatural substrates in its preferred S30-ATX context (Fig. 3). Again, (5F)C was a good substrate for deamination, though approximately only one quarter as reactive as cytosine, while bulkier substituents were increasingly poor substrates (Fig. 3c). While smaller size remained a prerequisite for efficient deamination with mA1, hydrophobicity also appears to play an important role. This is most strikingly notable with (5OH)C, which undergoes negligible deamination, while the larger mC is readily deaminated. Our extensive data show that for two distinct, active APOBEC family enzymes, efficient deamination of cytosine has steric requirements at the 5-position that contribute to the lack of any detectable enzymatic activity on hmC.
Deaminases do not alter global levels of epigenetic bases
Among the multiple potential pathways for DNA demethylation is the possibility of collaboration between oxidation and deamination to generate hmU from mC14. To complement our in vitro findings, we addressed whether hmC deamination could be detected in genomic DNA. Accordingly, we overexpressed one isoform of the TET oxidase family, TET2, in HEK 293T cells, the result of which generates a high prevalence of genomic hmC in a genome otherwise devoid of detectable oxidized cytosine bases15. TDG and the individual AID/APOBEC family members were co-transfected into HEK 293T cells along with TET2 to determine impact on modified nucleoside levels. Similar levels of expression for each enzyme were confirmed by Western blot (Supplementary Fig. 9). Using methodology we previously employed to detect the products of iterative oxidation by TET15, the genomic DNA was isolated and digested to generate a genomic nucleoside pool. For highly sensitive detection of the modified bases, the obtained nucleosides were subjected to LC-MS/MS in multiple reaction monitoring mode and quantified by comparison to known standards, including genomic cytosine to allow for approximation of the genomic prevalence of modified bases. This methodology enables detection of 10 fmol of hmU from 0.75 μg of genomic DNA, which converts to a detection limit of approximately 2–3 hmU bases in 100,000 dC bases (Supplementary Fig. 10 and 11).
As has been previously observed, TET2 overexpression in isolation leads to hmC levels that are about 1/6 that of genomic mC. The products of iterative oxidation, fC and caC, can also be detected at about 1/100 the level of mC15, 16. We posited that if AID/APOBEC enzymes could deaminate hmC, three changes should be observed: hmU should be detected in genomic DNA, hmC levels should decrease, and fC/caC levels should also decrease due to the deamination of their precursor hmC. However, none of these three predicted changes were observed when TET2 was overexpressed along with either mA1 or hAID (Fig. 4a). In cells overexpressing TET2 and hAID or mA1, we conclude that the genome contains less than 2–3 hmU bases per 105 dC bases. To further examine whether other family members might influence the genomic levels of modified bases, we screened all mouse AID/APOBEC enzymes with similar results (Supplementary Fig. 11). To determine whether our inability to detect hmU could be due to its rapid enzymatic excision from the genome, we measured the hmU glycosylase activity from 293T nuclear extracts. We observed negligible hmU glycosylase activity under conditions where robust nuclear uracil glycosylase activity was observed (Supplementary Fig. 12). Together these data suggest that if deamination of hmC occurs, it falls below the detection limits of this sensitive methodology.
Figure 4. AID/APOBEC enzymes do not perturb levels of mC oxidation intermediates in genomic DNA.

(a) Genomic DNA was extracted from HEK 293T cells co-expressing TET2 along with an empty vector control, TDG, mA1 or hAID, digested to single nucleosides, and analyzed via mass spectrometry for the presence of C, mC, hmC, fC, caC, and hmU. The left axis depicts the absolute fmol of nucleoside on a logarithmic scale, while the right axis represents the approximate conversion from absolute fmol to the genomic prevalence of modified bases for every 105 dC bases. The dashed line demonstrates the lowest examined amount of fC, caC, and hmU standards. Error bars indicate the standard deviation from the mean for two to three biological replicates. Asterisks: p ≤ 10−3 for fC and caC in samples with TDG in comparison to plasmid only control. (b) To confirm that overexpressed hAID and mA1 are catalytically active, nuclear extracts were tested for deaminase activity against unmodified cytosine. At left, TET2-hAID nuclear extracts and negative controls (no extract, nuclear extract from untransfected 293T cells, and TET2-hAID E58A) were incubated with S30-TGC substrate; At right, TET2-mA1 nuclear extracts and negative controls (no extract and nuclear extract from untransfected 293T cells) were incubated with S30-ATC substrate. The lanes demonstrating deamination of S30-TGC by TET2-hAID and S30-ATC substrate by TET2-mA1 extracts are highlighted (red). Nuclear extracts were also incubated with S30-TGU or S30-ATU to verify robust uracil excision activity. Gels are shown without cropping in Supplementary Fig. 13.
To confirm that the overexpressed deaminases were active despite the lack of detectable genomic hmU, we first examined nuclear lysates for deaminase activity using oligonucleotides containing unmodified cytosine. In line with our biochemical studies, deamination of cytosine was only detectable in nuclear lysates from cells overexpressing either hAID or mA1, but not a catalytic mutant of hAID (E58A) (Fig. 4b and Supplementary Fig. 13). As a second validation of deaminase activity in the cellular setting, we examined if overexpressed deaminases have an appreciable impact on genomic deoxyuridine (dU) levels. To overcome the rapid and efficient processing of genomic dU by multiple DNA repair pathways, we inhibited the major pathway involving UDG by overexpressing the small protein uracil DNA glycosylase inhibitor (UGI) along with hAID, hAID-E58A, mA1 or an empty plasmid control (Supplementary Fig. 14). The nuclear lysates associated with UGI overexpression were unable to excise uracil from duplexed oligonucleotides, confirming that UGI effectively inhibited the major pathway for uracil excision. Notably, this observation also independently suggested low levels of TDG and SMUG activity in 293T cells, as low levels of uracil excision were seen despite the fact that these enzymes are not inhibited by UGI34. In these cells, we next quantified the level of genomic dU using our highly sensitive LC-MS/MS methodology and demonstrated a consistent increase in genomic dU in hAID samples relative to hAID-E58A or the empty plasmid control. Unlike hAID, it is unknown if mA1 can act upon genomic cytosine. In our analysis, mA1 overexpression also yielded increases in genomic dU levels compared to an empty vector control. Thus, by two measures, in lysates and analysis of genomic DNA, the deaminases were active on unmodified cytosine under conditions where no deamination of hmC was detectable.
By contrast to our results with overexpression of AID/APOBEC family members, we reasoned that if iterative oxidation coupled to BER is a feasible pathway for demethylation, levels of modified cytosine bases should be readily perturbed by co-expression of TET and TDG. Indeed, we found that TDG overexpression led to dramatic reductions in the highly oxidized fC and caC species. Interestingly, no overall change was observed in the levels of mC and hmC. While this observation could be explained by the sparsity of highly oxidized species relative to mC and hmC, it also suggests that TDG-mediated depletion of fC and caC does not promote further oxidation and consumption of genomic hmC. Potential explanations include the possibility that some genomic hmC is sheltered from further oxidation or plays roles independent of demethylation10. While the dynamics of iterative oxidation will require further intensive study, from our data we conclude that the global prevalence of modified cytosine nucleobases in the genome can be altered by overexpression of the players in the deamination-independent pathway, but not by those in the deamination-dependent pathway.
Discussion
The well-established roles of DNA deaminase enzymes in modulating the genome all involve deamination of unmodified cytosine: in antibody diversity (AID), retroviral restriction (A3) and RNA editing (A1). Here, we have considered the possibility that deaminases could also have a role in DNA demethylation via deamination of mC or hmC by assessing the ability of AID/APOBEC enzymes to deaminate modified forms of cytosine. Our observation that steric bulk of the 5-position substituent decreased the efficiency of deamination presents a mechanistic rationale for discrimination against mC and hmC. Although these enzymes have diverged to assume distinctive cellular functions, the consistent pattern of substrate selectivity across the AID/APOBEC family indicates a conserved active site architecture that is primarily tuned for the recognition and deamination of unmodified cytosine.
Both steric and electronic effects of pyrimidine 5-substitutions are known to impact various enzymes that act on DNA. For the BER enzyme TDG, electron withdrawing effects from the C5-substituent promote nucleobase hydrolysis by weakening the N-glycosidic bond35. This finding fits nicely with its epigenetic role in removing fC and caC from DNA, because these bases have 5-substituents that activate cleavage of the glycosidic bond17. By contrast, steric exclusion of 5-substituents is important for the BER glycosylase UDG which uses a tyrosine side chain to selectively exclude thymidine and excise uracil with high proficiency36. It will be intriguing to see if structural studies on the AID/APOBEC enzymes in complex with DNA will demonstrate a similar molecular mechanism for discrimination against bulky 5-position substituents on cytosine.
We can now reconcile the known characteristics of AID/APOBEC enzymes with their proposed function in DNA demethylation by examining the multiple levels of targeting involved in deamination. At the level of the nucleobase, our results suggest that deaminase enzymes have all evolved an active site that is designed to deaminate unmodified cytosine preferentially. However, deamination of mC to T can occur, albeit at ~10-fold reduced rate relative to cytosine deamination. Thus, strictly from the perspective of biochemical feasibility, deamination of mC may constitute a viable pathway for demethylation in some situations, though other constraints are important to consider (see below). By contrast, we demonstrated that deamination of hmC was not detectable in vitro nor in cells when relevant enzymes were overexpressed. Our results contrast with those of a prior study that used immunoblotting of DNA, a method of uncertain specificity, to report detection of genomic hmU37. However, our findings are in good agreement with studies on embryonic stem cells, where hmU is not detectable in genomic DNA when probed with a sensitive and specific mass spectrometry methodology18, 38, and with other groups who have not detected deamination of hmC by AID in vitro (personal communication, Svend Petersen-Mahrt). Together, our biochemical and cellular data provide a strong argument against the proposed collaboration between oxidation, deamination and BER as a pathway for DNA demethylation in mammalian cells.
Beyond the target cytosine, the local sequence context provides an additional potential barrier to efficient deamination. Each AID/APOBEC family member acts at preferred trinucleotide hotspots29–31, 39 (Supplementary Fig. 1a), yet methylated CpG motifs can be found in all common sequence contexts, even those that may be disfavored by the individual deaminases. Further, since all known AID/APOBEC enzymes are specific for single-stranded DNA23, 40, it remains unclear how methylated CpG motifs in genomic DNA might be sufficiently targeted by AID/APOBECs. Although transcription or replication could generate single-stranded DNA, active demethylation of CpG islands does not always require replication or transcription41, 42. Further, only AID and human APOBEC3A have been shown to deaminate mammalian host genomic DNA43, 44, and these mutations appear to be localized to expressed genes. Finally, expression of the various deaminase family members is restricted to particular cell lineages, suggesting that a single deaminase is unlikely to play a universal role in demethylation.
Integrating across these layers of targeting, from the nucleobase to the cellular level, enables us to assess the plausibility of the early steps in the various proposed DNA demethylation pathways. First, our findings show that deamination of mC remains a plausible demethylation pathway based on enzymatic function, although other known limitations in targeting would seem to present several barriers to its efficient function. It is possible that deamination of mC operates in non-physiological systems, such as heterokaryon-based reprogramming25. In other physiological settings, it will be important to examine whether these enzymes may play other niche roles21. Although our findings exclude hmC deamination as a detectable enzymatic activity, a requirement for AID/APOBEC enzymes in conversion of hmC to C in neurons has been suggested37. Notably, in that study, AID/APOBEC enzymes were not shown to be directly capable of hmC deamination, and a requirement for catalytically active AID in conversion of hmC to C was also not established. If catalysis were to be required, it is feasible that AID/APOBEC-mediated deamination of C or mC indirectly stimulates TET or DNA damage response pathways to promote excision of genomic hmC. Our biochemical and cellular data make it unlikely that deamination of hmC by AID/APOBEC enzymes is involved in DNA demethylation. Finally, our data suggest that under the very same conditions where the product of hmC deamination (hmU) is below detectable limits, the proposed intermediates in the iterative-oxidation pathway, fC and caC, are readily detected. As we have addressed for the AID/APOBEC enzymes, this work raises the question of targeting by the TET enzymes. From their relative preferences for oxidation of mC, hmC and fC, to their regulated expression in the appropriate cellular settings, much remains to be understood. These questions form an important area of focus for further explorations on the mechanism of DNA demethylation.
Methods
AID/APOBEC Protein Expression and Purification
Expression vectors contained the mA1, mA2, and mA3 genes37 downstream of an N-terminal maltose-binding protein (MBP) in pET41 (Novagen), as previously described for hAID29, and were generously provided by Junjie Guo and Hongjun Song (Johns Hopkins University). AID/APOBEC constructs were expressed and purified as described in Supplementary Methods.
Deamination Assays
Synthesis of oligonucleotides is detailed in Supplementary Methods. Deamination reactions were carried out in Buffer DA (20 mM Tris-Cl, pH 8.0, 1 mM dithiothreitol, 5 mM EDTA) with 0.1 μg/μL RNaseA, using 200 nM oligonucleotide substrate. Unless otherwise detailed, incubations were performed for 12 hours with 2 μM final enzyme concentration. Reactions with mA1 against AT-hotspot oligonucleotides were incubated for 15 min given higher overall catalytic activity. Reactions were incubated at 30 °C and terminated by 20 minutes at 95 °C. Subsequently, excess complementary oligonucleotide was added (150 nM reaction oligonucleotide to 250 nM complement) and annealed by slow cooling from 95 °C. Duplexed DNA (final 100 nM) was incubated with the appropriate glycosylase using either 0.25 units/μL UDG (New England Biolabs) or 140 nM hSMUG in 1 × Buffer DA with 0.1 mg/mL BSA and incubated at 37 °C for 45 minutes. For TDG reactions, 1.6 μM TDG was used in 1 × Buffer DA supplemented with 0.1 mg/mL BSA, and reactions were incubated at 16 °C for 12 hours. The selection of the glycosylase used was based upon comprehensive survey of glycosylases (Supplementary Figure 5), selecting a glycosylase with robust activity against a given uracil analog, with no detectable activity on the associated cytosine analog. C and (5F)C substrates were assayed for deamination with UDG; mC substrates were assayed with TDG; and (5OH)C, (5Br)C, (5I)C and hmC were assayed with SMUG. The duration of the incubation times were selected to maximize the excision of the deaminated bases. DNA glycosylase reactions were quenched in 50% formamide (v/v) and 150 mM NaOH and heated to 95 °C for 20 minutes to cleave the abasic sites generated by glycosylases.
Analysis of Deamination Assays
Reaction products were separated by denaturing PAGE (20% acrylamide/TBE/7M urea), run at 50°C and imaged on a Typhoon 9410 (Amersham Biosciences). The intensities of product and substrate bands were determined in QuantityOne (BioRad) after subtracting background intensities. To account for incomplete excision of deaminated bases, standard curves for quantifying the fraction of deaminated product were generated using S30-ATX substrates (X = C/mC/hmC) and S30-ATY products (Y = U/T/hmU), mixed in various ratios and treated with UDG (for C/U), SMUG (for hmC/hmU) or TDG (for mC/T). Identical conditions were used to generate standard curves with the unnaturally modified S30-TGX substrates and S30-TGY products, with UDG (for (5F)C/(5F)U) and SMUG (for (5OH)C/5(OH)U, (5Br)C/(5Br)U and (5I)C/(5I)U). The calculated fraction of cleaved product was determined by the intensity of the fluorescent product over the total product plus substrate. For all deamination assays the actual fraction of deaminated product was determined with reference to the standard curve generated for the corresponding glycosylase (Supplementary Fig. 3 and 6).
Transient Expression of Candidate Enzymes
TDG, mA1 and mA3 from pCMV-SPORT6 vectors (Open Biosystems) were each cloned into a FLAG-tagged pcDNA3 vector (pcDNA3β-FLAG). mA2 amplified from mouse embryonic stem cell cDNA was cloned into pcDNA3β-FLAG. The cloned TET2 construct, untagged human AID and the catalytic mutant E58A of hAID were previously described15, 39. A synthetic gene encoding UGI was cloned into pIRESneo3 (Clontech). HEK 293T cells were transfected 18 hours after plating using Fugene HD transfection reagents (Roche) and harvested 48 hours after transfection. Additional details may be found in Supplementary Methods.
Mass spectrometric experiments
2.5 μg genomic DNA, isolated using the DNeasy Kit (Qiagen), was heat-denatured, hydrolyzed with 90 U of Nuclease S1 (Sigma) in Buffer (0.5 mM ZnSO4, 14 mM sodium acetate, pH 5.2) at 37 °C for 1 hour, followed by the addition of 5 μL 10 × Buffer 2 (560 mM Tris-Cl, 30 mM NaCl, 10 mM MgCl2, pH 8.3), 0.5 μg of phosphodiesterase I (Worthington) and 2 U of Calf Intestinal Alkaline Phosphatase (New England Biolabs) for an additional 1 hour (final volume 50 μL). Digested DNA was then filtered with Nanosep3K (Pall Corporation) and 15 μL of filtered samples were subjected to LC-MS/MS analysis as described previously15 with an additional transition for hmU (m/z 259.0 to 125.0) and for dU (m/z 229.0 to 113.0). The absolute amount of nucleoside was quantified by comparison to known standards.
Deamination and Base Excision Activity of Nuclear Lysates
Nuclear lysates were prepared from HEK 293T cells as previously described45. For analysis of deaminase activity, lysates (final protein concentration 0.3 μg/μL) were incubated with single-stranded oligonucleotide substrates (2 μM) in 1 × Buffer DA and 0.1 mg/μL BSA, for 30 minutes. Reactions were quenched by addition of phenol:chloroform:isoamyl alcohol (25:24:1). Formamide and NaOH were added to DNA extracted from the aqueous phase (50% v/v, 150 mM final concentrations). Samples were processed and analyzed as described for deamination assays. For analysis of base excision activity against uracil or hmU in nuclear lysates, lysates (final concentration 0.2–0.3 μg/μL as detailed) were pre-incubated at 37 °C for 20 minutes before addition of 1 μM duplex DNA. Reactions were quenched and analyzed as described above.
Statistical Analysis
All data shown represent the mean value for a given number of replicates, as specified in each figure legend. Error bars represent standard deviation from the mean. Student’s T-test determined statistical significance between groups.
Supplementary Material
Acknowledgments
We are grateful to Marisa Bartolomei for helpful discussions, Kiran Gajula for technical assistance, and the UNC Biomarker Mass Spectrometry Facility for guidance. We are also grateful for Alex Drohat (University of Maryland), Amy Guminski (Johns Hopkins University) and Hongjun Song (Johns Hopkins University) for providing reagents. This work was supported in part by the Rita Allen Foundation (to R.M.K), the W. W. Smith Charitable Trust (to R.M.K.), and NIH grants K08-AI089242 (to R.M.K), GM056834 (to J.T.S.), and U01DK089565 (to Y.Z.). Y.Z. is an investigator of the HHMI.
Footnotes
Author Contributions
R.M.K., J.T.S. and Y.Z. conceived the project. R.M.K., J.T.S., Y.Y., C.S.N. and Y.Z. designed the experiments. C.S.N., H.J., Y.Y., L.S., H.L.G. performed the experiments. C.S.N., H.J., L.S., analysed the data. C.S.N., H.J., L.S., H.L.G., J.T.S., Y.Z., and R.M.K interpreted the data. C.S.N, J.T.S., and R.M.K. wrote the manuscript and all authors edited the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Nabel CS, Manning SA, Kohli RM. The Curious Chemical Biology of Cytosine: Deamination, Methylation, and Oxidation as Modulators of Genomic Potential. ACS Chem Biol. 2011 doi: 10.1021/cb2002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg BR, Papavasiliou FN. Beyond SHM and CSR: AID and related cytidine deaminases in the host response to viral infection. Adv Immunol. 2007;94:215–244. doi: 10.1016/S0065-2776(06)94007-3. [DOI] [PubMed] [Google Scholar]
- 3.Teperek-Tkacz M, Pasque V, Gentsch G, Ferguson-Smith AC. Epigenetic reprogramming: is deamination key to active DNA demethylation? Reproduction. 2011;142:621–632. doi: 10.1530/REP-11-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fritz EL, Papavasiliou FN. Cytidine deaminases: AIDing DNA demethylation? Genes Dev. 2010;24:2107–2114. doi: 10.1101/gad.1963010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 6.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito S, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munzel M, Globisch D, Carell T. 5-Hydroxymethylcytosine, the Sixth Base of the Genome. Angew Chem Int Ed Engl. 2011;50:6460–6468. doi: 10.1002/anie.201101547. [DOI] [PubMed] [Google Scholar]
- 9.Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25:2436–2452. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pastor WA, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hajkova P. Epigenetic reprogramming in the germline: towards the ground state of the epigenome. Philos Trans R Soc Lond B Biol Sci. 2011;366:2266–2273. doi: 10.1098/rstb.2011.0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607–620. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279:52353–52360. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 14.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He YF, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maiti A, Drohat AC. Thymine DNA Glycosylase Can Rapidly Excise 5-Formylcytosine and 5-Carboxylcytosine: POTENTIAL IMPLICATIONS FOR ACTIVE DEMETHYLATION OF CpG SITES. J Biol Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfaffeneder T, et al. The Discovery of 5-Formylcytosine in Embryonic Stem Cell DNA. Angew Chem Int Ed Engl. 2011;50:7008–7012. doi: 10.1002/anie.201103899. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L, et al. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat Chem Biol. 2012;8:328–330. doi: 10.1038/nchembio.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cortazar D, et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. 2011;470:419–423. doi: 10.1038/nature09672. [DOI] [PubMed] [Google Scholar]
- 21.Cortellino S, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conticello SG, Langlois MA, Yang Z, Neuberger MS. DNA deamination in immunity: AID in the context of its APOBEC relatives. Adv Immunol. 2007;94:37–73. doi: 10.1016/S0065-2776(06)94002-4. [DOI] [PubMed] [Google Scholar]
- 23.Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci U S A. 2003;100:4102–4107. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larijani M, et al. Methylation protects cytidines from AID-mediated deamination. Mol Immunol. 2005;42:599–604. doi: 10.1016/j.molimm.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 25.Bhutani N, et al. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Popp C, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muramatsu M, et al. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 28.Rai K, et al. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–1212. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohli RM, et al. A portable hotspot recognition loop transfers sequence preferences from APOBEC family members to activation-induced cytidine deaminase. J Biol Chem. 2009;284:22898–22904. doi: 10.1074/jbc.M109.025536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larijani M, Frieder D, Basit W, Martin A. The mutation spectrum of purified AID is similar to the mutability index in Ramos cells and in ung(−/−)msh2(−/−) mice. Immunogenetics. 2005;56:840–845. doi: 10.1007/s00251-004-0748-0. [DOI] [PubMed] [Google Scholar]
- 31.Beale RC, et al. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J Mol Biol. 2004;337:585–596. doi: 10.1016/j.jmb.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 32.Prochnow C, Bransteitter R, Klein MG, Goodman MF, Chen XS. The APOBEC-2 crystal structure and functional implications for the deaminase AID. Nature. 2007;445:447–451. doi: 10.1038/nature05492. [DOI] [PubMed] [Google Scholar]
- 33.Hansch C, et al. Aromatic” substituent constants for structure-activity correlations. J Med Chem. 1973;16:1207–1216. doi: 10.1021/jm00269a003. [DOI] [PubMed] [Google Scholar]
- 34.Pearl LH. Structure and function in the uracil-DNA glycosylase superfamily. Mutat Res. 2000;460:165–181. doi: 10.1016/s0921-8777(00)00025-2. [DOI] [PubMed] [Google Scholar]
- 35.Bennett MT, et al. Specificity of human thymine DNA glycosylase depends on N-glycosidic bond stability. J Am Chem Soc. 2006;128:12510–12519. doi: 10.1021/ja0634829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kavli B, et al. Excision of cytosine and thymine from DNA by mutants of human uracil-DNA glycosylase. EMBO J. 1996;15:3442–3447. [PMC free article] [PubMed] [Google Scholar]
- 37.Guo JU, Su Y, Zhong C, Ming GL, Song H. Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle. 2011;10:2662–2668. doi: 10.4161/cc.10.16.17093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Globisch D, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohli RM, et al. Local sequence targeting in the AID/APOBEC family differentially impacts retroviral restriction and antibody diversification. J Biol Chem. 2010;285:40956–40964. doi: 10.1074/jbc.M110.177402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424:103–107. doi: 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]
- 41.Bruniquel D, Schwartz RH. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat Immunol. 2003;4:235–240. doi: 10.1038/ni887. [DOI] [PubMed] [Google Scholar]
- 42.Martinowich K, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 43.Liu M, et al. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 44.Landry S, Narvaiza I, Linfesty DC, Weitzman MD. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011;12:444–450. doi: 10.1038/embor.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grogan BC, Parker JB, Guminski AF, Stivers JT. Effect of the thymidylate synthase inhibitors on dUTP and TTP pool levels and the activities of DNA repair glycosylases on uracil and 5-fluorouracil in DNA. Biochemistry. 2011;50:618–627. doi: 10.1021/bi102046h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.